Polarization Rotation and Monoclinic Distortion in Ferroelectric (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals under Electric Fields

Abstract
:
1. Introduction
2. Experiments
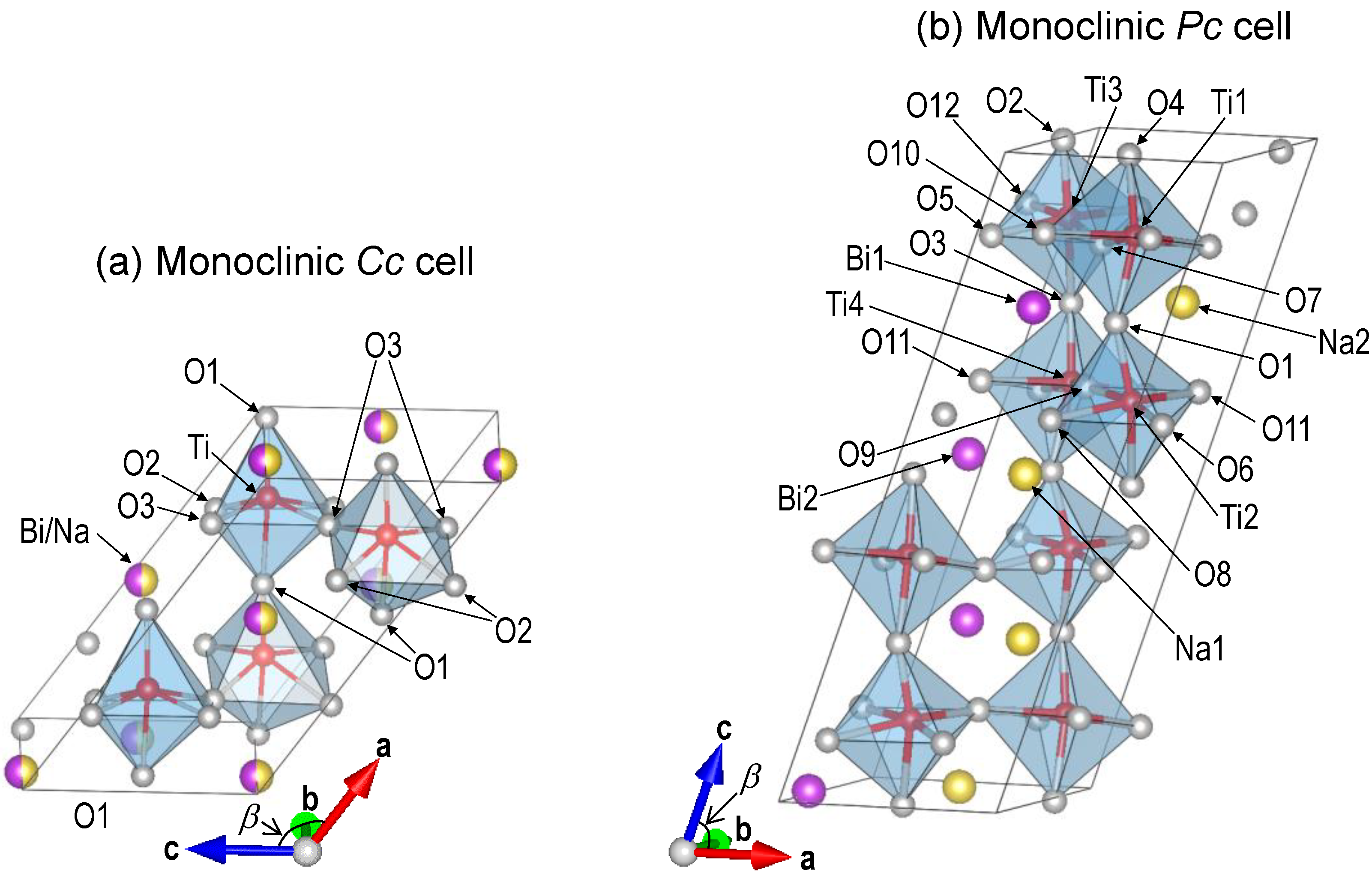


3. Method of DFT Calculations
4. Results and Discussion
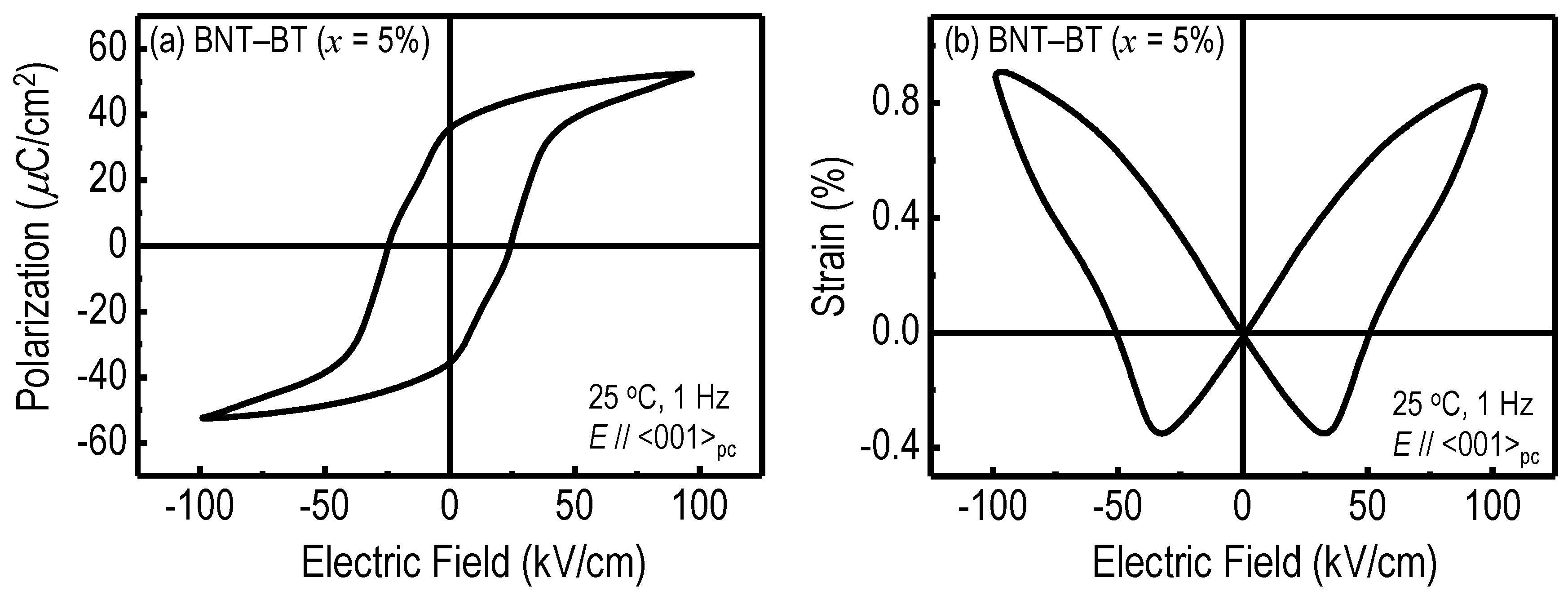
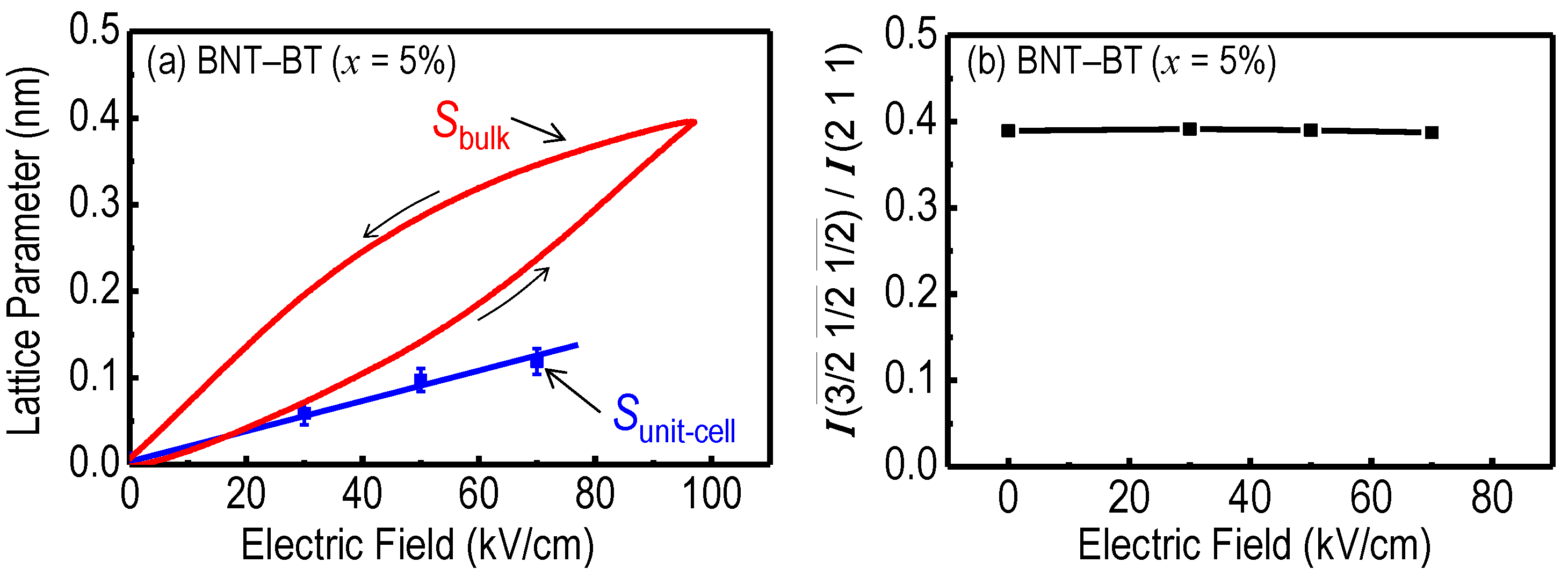
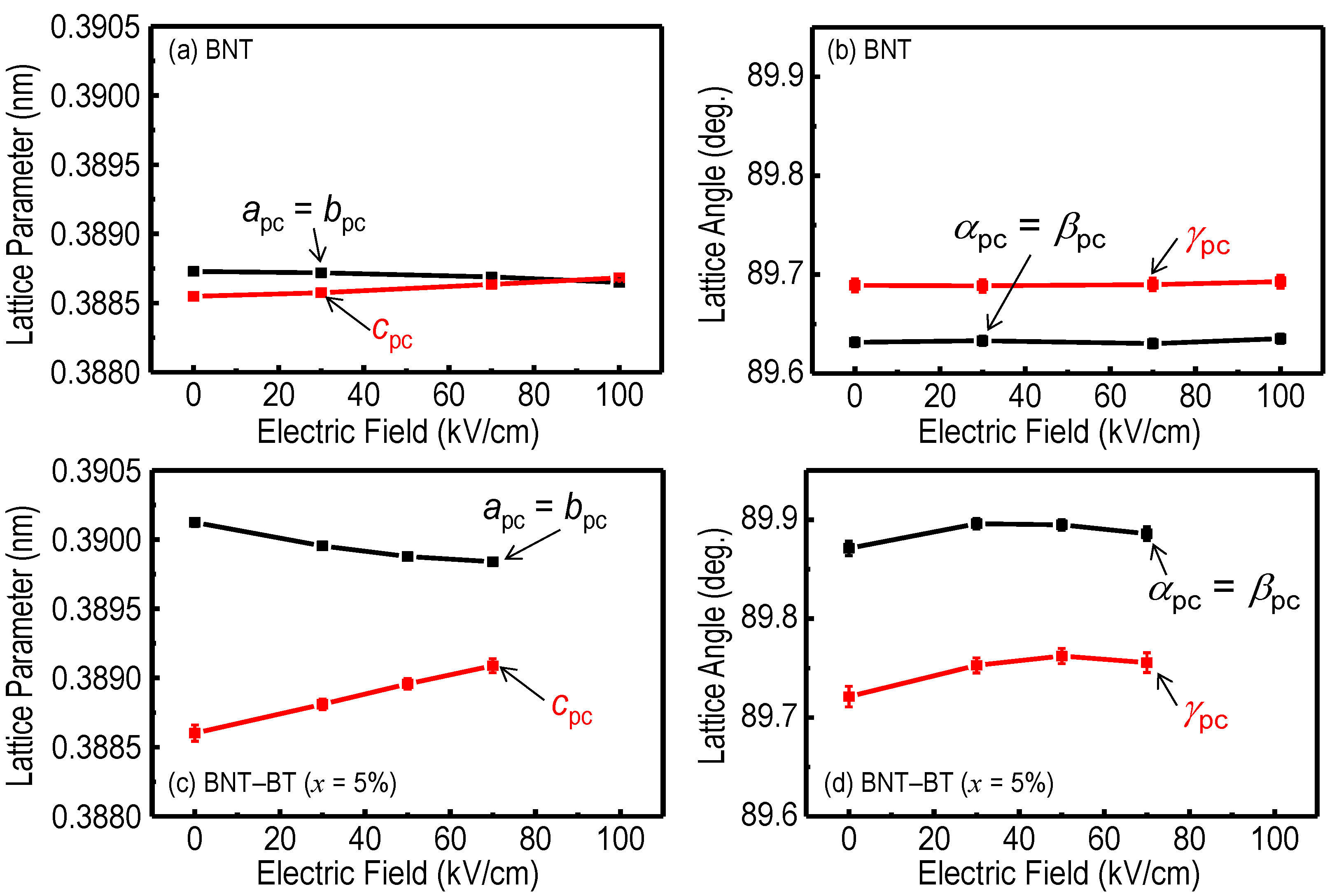

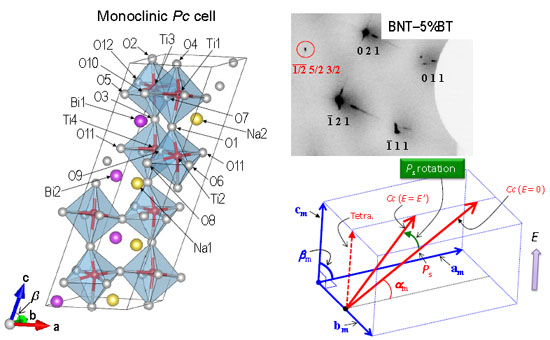{kind=link}
{kind=link}
{kind=link}
{kind=link}
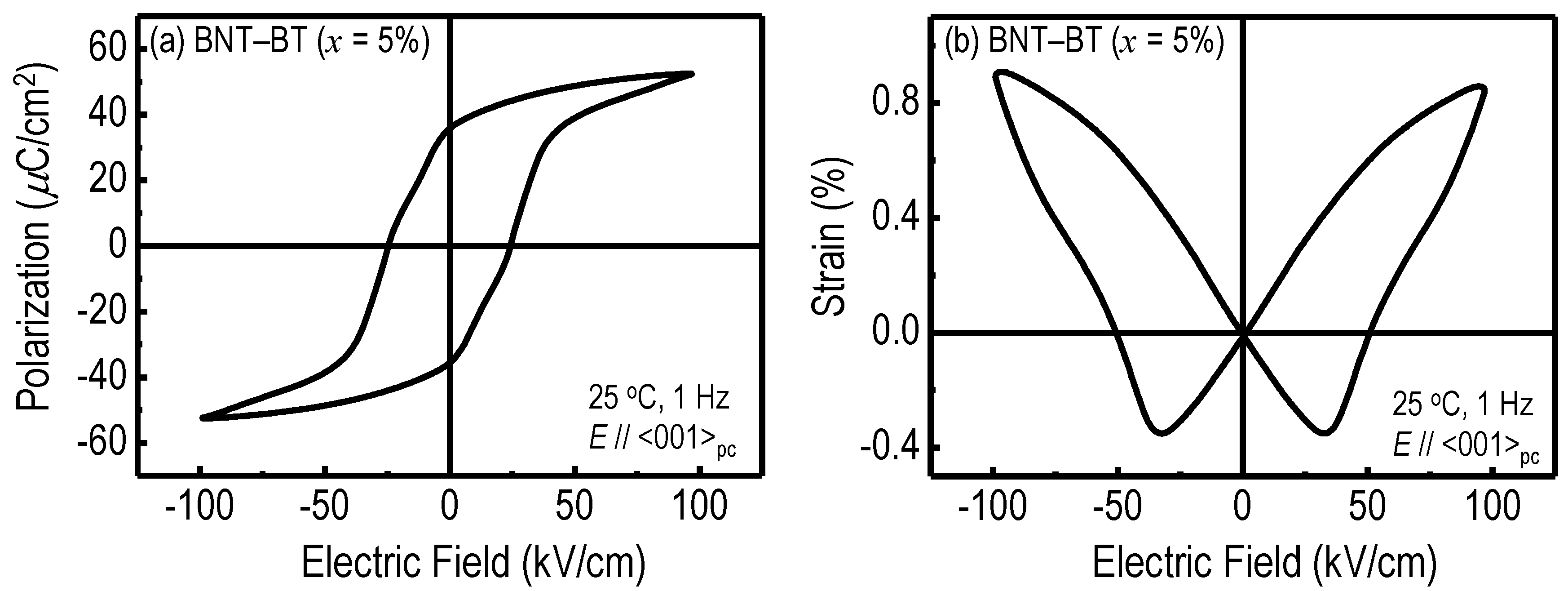{kind=link}
{kind=link}
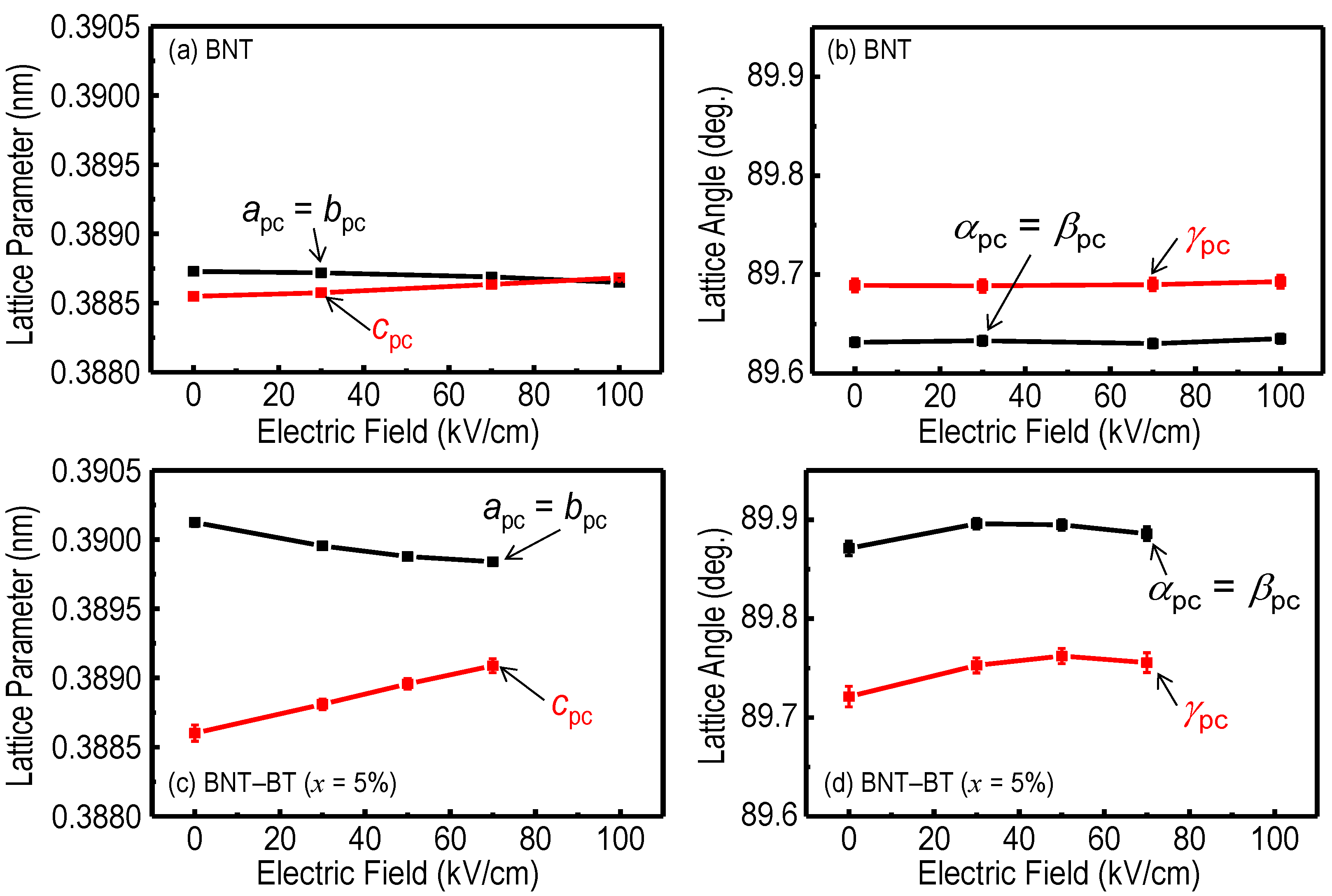{kind=link}
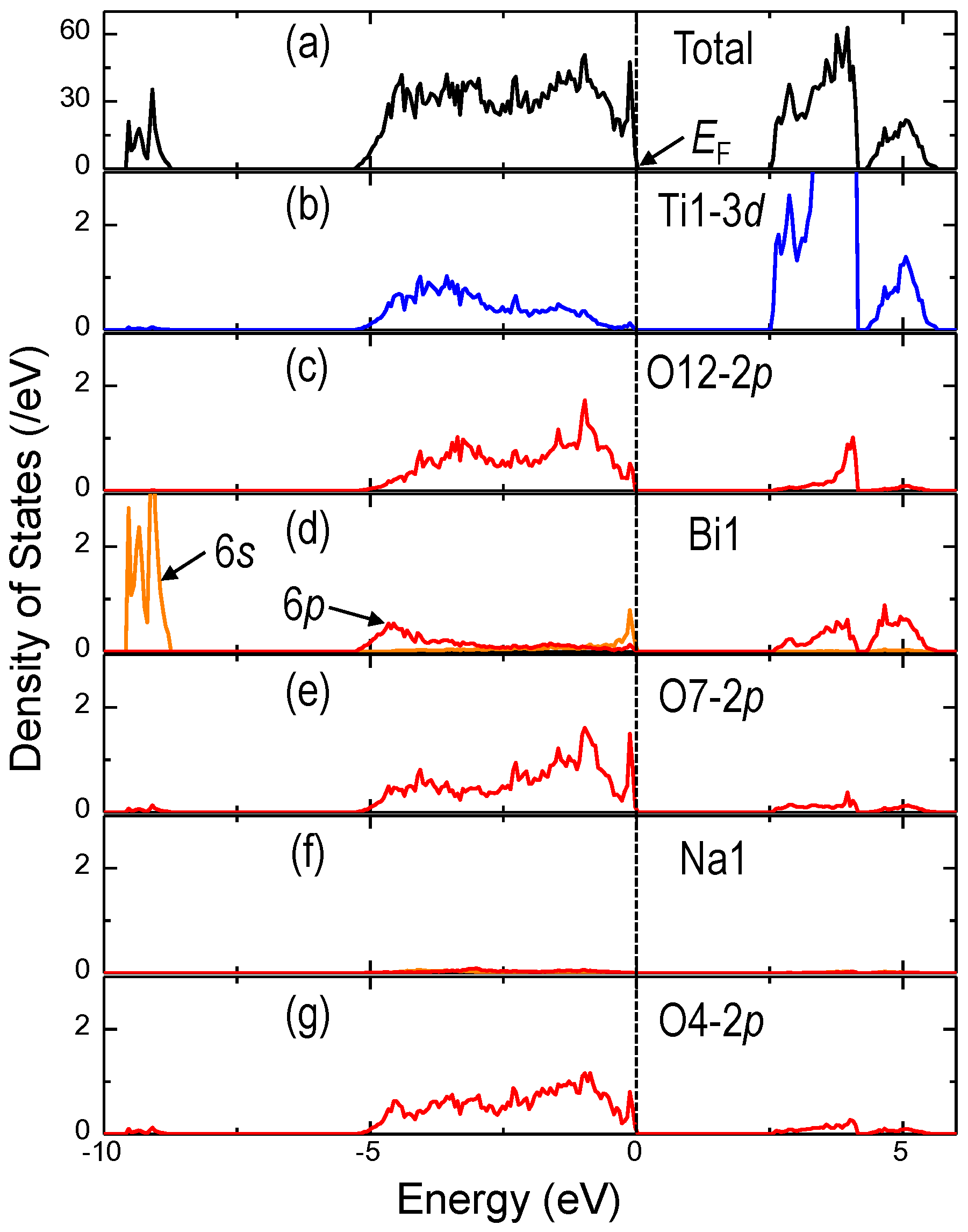{kind=link}
{kind=link}
| Bond length (nm) | BNT b | BNT (Ein-situ = 0 kV/cm) | BNT–5%BT (Ein-situ = 0 kV/cm) |
|---|---|---|---|
| Ti1–O12 | 0.1812 | 0.1813 | 0.1812 |
| Ti1–O5 | 0.1861 | 0.1864 | 0.1863 |
| Ti1–O4 | 0.1894 | 0.1893 | 0.1900 |
| Ti1–O1 | 0.2069 | 0.2085 | 0.2080 |
| Ti1–O10 | 0.2120 | 0.2116 | 0.2133 |
| Ti1–O7 | 0.2179 | 0.2178 | 0.2193 |
| BVS a | 4.02 | 3.99 | 3.95 |
| Ti2–O11 | 0.1869 | 0.1876 | 0.1874 |
| Ti2–O6 | 0.1875 | 0.1880 | 0.1882 |
| Ti2–O1 | 0.1905 | 0.1904 | 0.1915 |
| Ti2–O2 | 0.2048 | 0.2069 | 0.2064 |
| Ti2–O8 | 0.2075 | 0.2072 | 0.2083 |
| Ti2–O9 | 0.2076 | 0.2071 | 0.2082 |
| BVS a | 4.02 | 3.98 | 3.93 |
| Ti3–O2 | 0.1859 | 0.1856 | 0.1859 |
| Ti3–O7 | 0.1906 | 0.1908 | 0.1909 |
| Ti3–O10 | 0.1945 | 0.1953 | 0.1954 |
| Ti3–O5 | 0.2006 | 0.2005 | 0.2014 |
| Ti3–O12 | 0.2020 | 0.2022 | 0.2030 |
| Ti3–O3 | 0.2069 | 0.2089 | 0.2087 |
| BVS a | 4.05 | 4.01 | 3.97 |
| Ti4–O3 | 0.1848 | 0.1845 | 0.1847 |
| Ti4–O8 | 0.1851 | 0.1854 | 0.1851 |
| Ti4–O9 | 0.1869 | 0.1871 | 0.1870 |
| Ti4–O6 | 0.2102 | 0.2104 | 0.2118 |
| Ti4–O4 | 0.2117 | 0.2136 | 0.2133 |
| Ti4–O11 | 0.2128 | 0.2125 | 0.2145 |
| BVS a | 4.02 | 3.99 | 3.96 |
| Bond length (nm) | BNT b | BNT (Ein-situ = 0 kV/cm) | BNT–5%BT (Ein-situ = 0 kV/cm) |
|---|---|---|---|
| Bi1–O7 | 0.2271 | 0.2274 | 0.2270 |
| Bi1–O1 | 0.2298 | 0.2302 | 0.2293 |
| Bi1–O10 | 0.2358 | 0.2349 | 0.2352 |
| Bi1–O9 | 0.2403 | 0.2422 | 0.2426 |
| Bi1–O3 | 0.2457 | 0.2461 | 0.2473 |
| Bi1–O5 | 0.2472 | 0.2473 | 0.2495 |
| Bi1–O8 | 0.3074 | 0.3095 | 0.3100 |
| Bi1–O3 | 0.3082 | 0.3096 | 0.3107 |
| Bi1–O12 | 0.3120 | 0.3120 | 0.3135 |
| Bi1–O11 | 0.3199 | 0.3208 | 0.3207 |
| Bi1–O6 | 0.3245 | 0.3252 | 0.3252 |
| Bi1–O1 | 0.3298 | 0.3277 | 0.3302 |
| BVS a | 3.16 | 3.13 | 3.10 |
| Bi2–O4 | 0.2277 | 0.2277 | 0.2270 |
| Bi2–O6 | 0.2332 | 0.2329 | 0.2327 |
| Bi2–O11 | 0.2355 | 0.2350 | 0.2354 |
| Bi2–O2 | 0.2419 | 0.2431 | 0.2444 |
| Bi2–O10 | 0.2433 | 0.2451 | 0.2452 |
| Bi2–O8 | 0.2464 | 0.2470 | 0.2490 |
| Bi2–O7 | 0.3064 | 0.3084 | 0.3078 |
| Bi2–O9 | 0.3089 | 0.3102 | 0.3123 |
| Bi2–O2 | 0.3109 | 0.3118 | 0.3128 |
| Bi2–O5 | 0.3134 | 0.3139 | 0.3138 |
| Bi2–O12 | 0.3152 | 0.3158 | 0.3160 |
| Bi2–O4 | 0.3316 | 0.3298 | 0.3321 |
| BVS a | 3.14 | 3.10 | 3.07 |
| Bond length (nm) | BNT b | BNT (Ein-situ = 0 kV/cm) | BNT–5%BT (Ein-situ = 0 kV/cm) |
|---|---|---|---|
| Na1–O4 | 0.2423 | 0.2439 | 0.2454 |
| Na1–O12 | 0.2430 | 0.2451 | 0.2463 |
| Na1–O6 | 0.2447 | 0.2456 | 0.2479 |
| Na1–O8 | 0.2508 | 0.2516 | 0.2519 |
| Na1–O9 | 0.2510 | 0.2515 | 0.2518 |
| Na1–O2 | 0.2582 | 0.2589 | 0.2590 |
| Na1–O2 | 0.2997 | 0.2974 | 0.2990 |
| Na1–O11 | 0.3065 | 0.3061 | 0.3072 |
| Na1–O5 | 0.3071 | 0.3093 | 0.3095 |
| Na1–O4 | 0.3108 | 0.3109 | 0.3116 |
| Na1–O10 | 0.3124 | 0.3129 | 0.3128 |
| Na1–O7 | 0.3178 | 0.3178 | 0.3177 |
| BVS a | 1.14 | 1.11 | 1.09 |
| Na2–O1 | 0.2432 | 0.2446 | 0.2461 |
| Na2–O7 | 0.2434 | 0.2442 | 0.2465 |
| Na2–O11 | 0.2434 | 0.2454 | 0.2465 |
| Na2–O5 | 0.2530 | 0.2539 | 0.2542 |
| Na2–O12 | 0.2554 | 0.2559 | 0.2560 |
| Na2–O3 | 0.2565 | 0.2568 | 0.2577 |
| Na2–O3 | 0.3023 | 0.3003 | 0.3011 |
| Na2–O9 | 0.3026 | 0.3022 | 0.3016 |
| Na2–O8 | 0.3058 | 0.3052 | 0.3046 |
| Na2–O6 | 0.3081 | 0.3088 | 0.3089 |
| Na2–O1 | 0.3084 | 0.3087 | 0.3096 |
| Na2–O10 | 0.3096 | 0.3108 | 0.3123 |
| BUS a | 1.14 | 1.11 | 1.08 |
| Atom | xx | yy | zz | xy | xz | yx | yz | zx | zy |
|---|---|---|---|---|---|---|---|---|---|
| Bi1 | 4.18 | 5.05 | 5.03 | 0.29 | −0.64 | −0.35 | 0.31 | −0.41 | −0.33 |
| Bi2 | 4.41 | 5.08 | 5.09 | 0.28 | −0.44 | −0.24 | 0.33 | −0.66 | −0.37 |
| Na1 | 1.17 | 1.18 | 1.11 | −0.03 | 0.02 | 0.02 | −0.04 | −0.03 | 0.03 |
| Na2 | 1.19 | 1.20 | 1.14 | −0.02 | −0.03 | 0.03 | −0.02 | 0.04 | 0.04 |
| Ti1 | 6.58 | 5.29 | 5.77 | 0.33 | −0.78 | −0.10 | 0.67 | −0.44 | −0.38 |
| Ti2 | 5.42 | 7.21 | 6.48 | −0.36 | −0.98 | 0.34 | −0.51 | −1.38 | 0.54 |
| Ti3 | 5.07 | 8.13 | 6.62 | −0.07 | −0.37 | 0.52 | 0.08 | −0.49 | 0.10 |
| Ti4 | 6.03 | 6.05 | 5.97 | −0.02 | −0.08 | 0.00 | 0.08 | 0.07 | 0.06 |
| O1 | −2.77 | −1.32 | −5.02 | 0.02 | 0.46 | −0.16 | −0.17 | 0.59 | −0.24 |
| O2 | −1.15 | −2.90 | −5.44 | 0.23 | −0.06 | 0.10 | 0.30 | 0.04 | 0.56 |
| O3 | −1.27 | −2.88 | −5.22 | 0.11 | 0.36 | 0.09 | 0.36 | 0.17 | 0.51 |
| O4 | −3.02 | −1.25 | −4.69 | 0.08 | 0.87 | −0.09 | −0.22 | 0.60 | −0.24 |
| O6 | −3.31 | −3.91 | −2.08 | −1.46 | −0.10 | −1.49 | 0.96 | −0.14 | 0.73 |
| O7 | −2.96 | −3.70 | −2.14 | −1.22 | 0.06 | −1.27 | 0.81 | 0.16 | 0.76 |
| O9 | −2.89 | −4.08 | −2.19 | −1.49 | 0.32 | −1.59 | −0.66 | 0.59 | −0.55 |
| O10 | −2.76 | −3.98 | −2.97 | 1.26 | 0.09 | 1.10 | −0.01 | 0.07 | 0.09 |
| O11 | −2.99 | −4.06 | −2.17 | 1.54 | −0.29 | 1.44 | −0.55 | −0.34 | −0.73 |
| O12 | −3.37 | −3.96 | −1.42 | −2.05 | −0.05 | −2.15 | −0.07 | 0.05 | 0.17 |
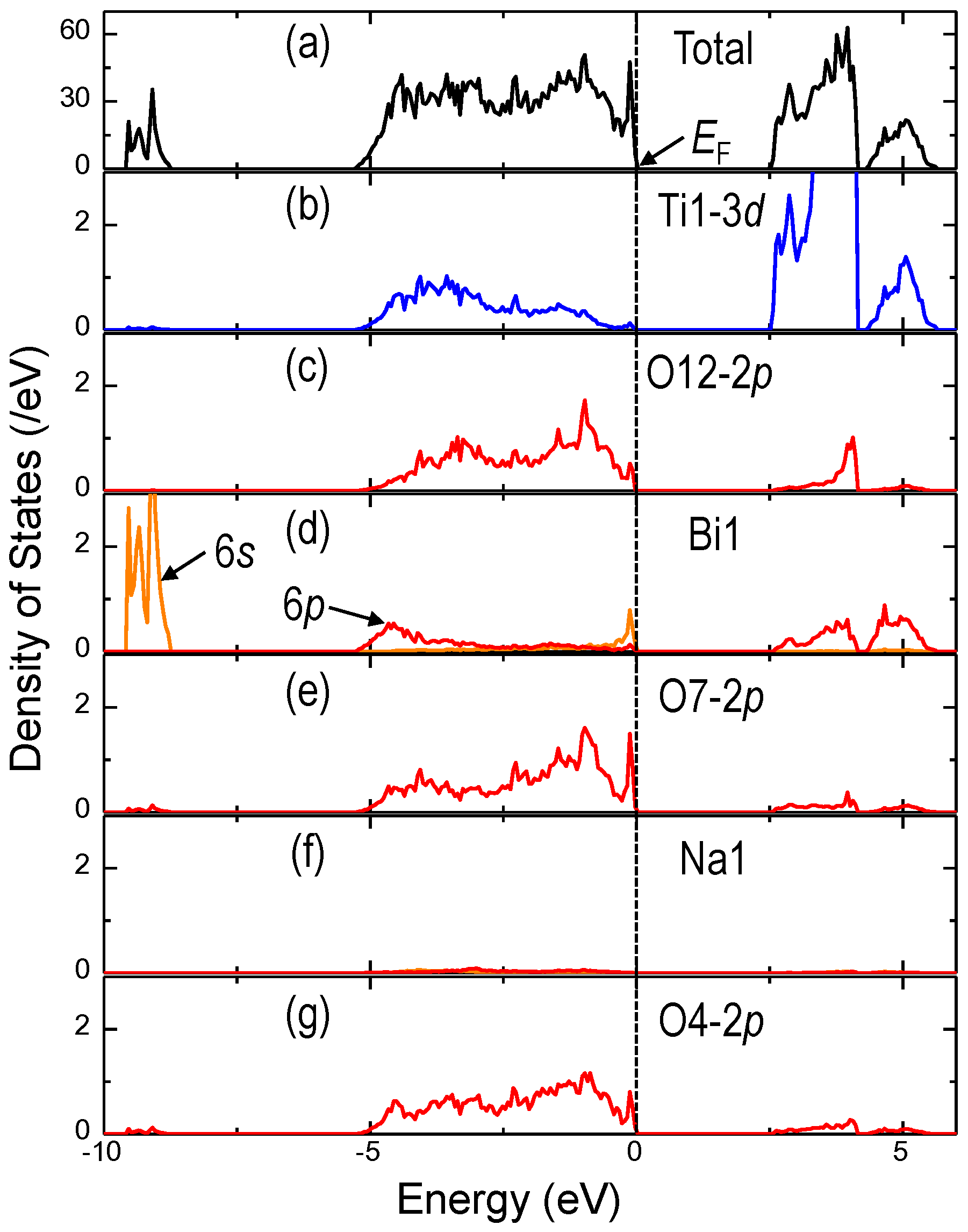
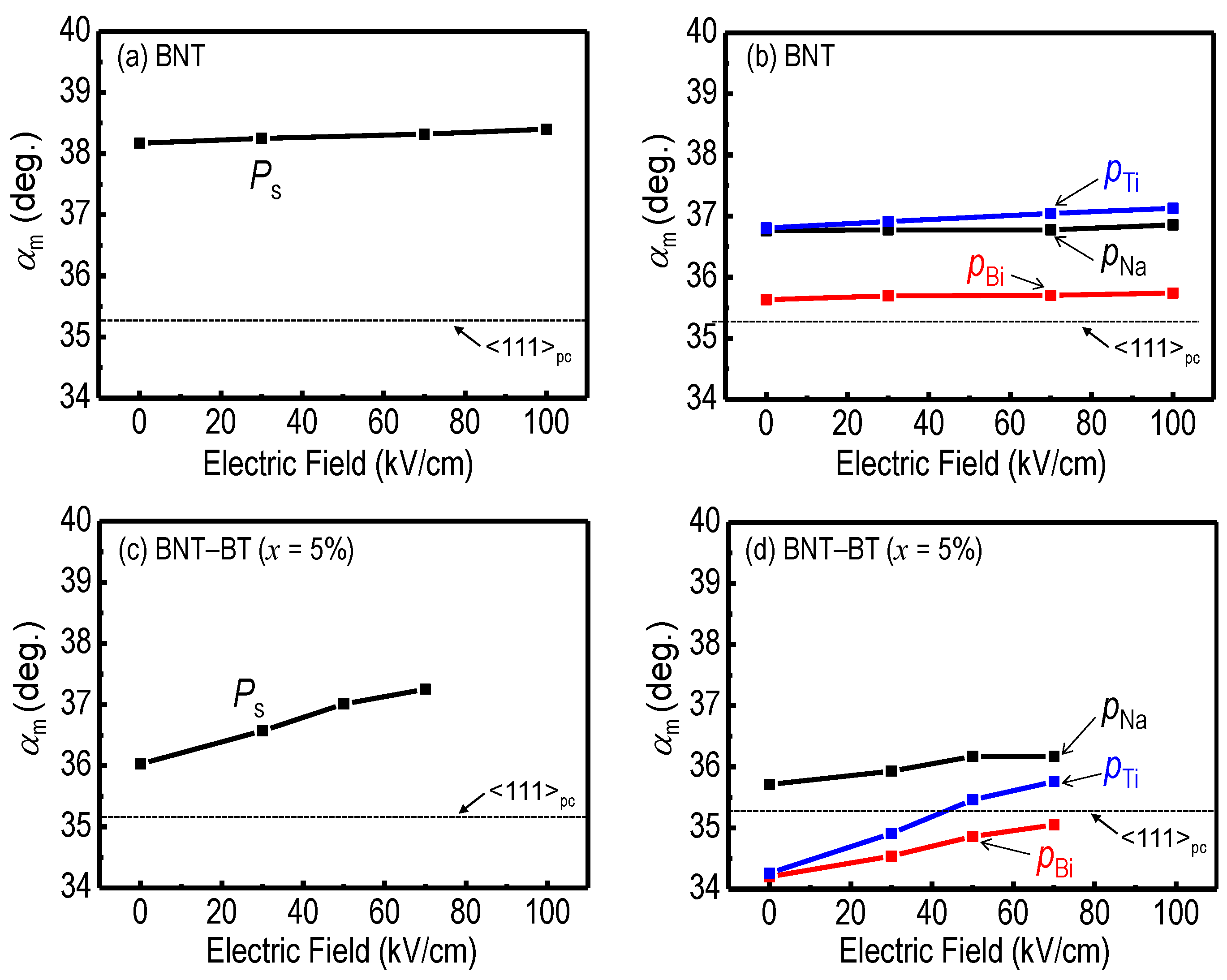
| Atom | x (−) | y (−) | z (−) | ∆x (nm) a | ∆z (nm) a | pa b (μC/cm2) | pc b (μC/cm2) | αpatom c (deg) | [u | u | v] d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bi1 | 0.2726 | 0.7478 | 0.2566 | 0.03296 | 0.02772 | 8.24 | 8.64 | 35.96 | 1 | 1 | 1.03 |
| Bi2 | 0.0203 | 0.2544 | 0.0077 | 0.03172 | 0.02944 | 8.71 | 8.85 | 35.32 | 1 | 1 | 1.00 |
| Na1 | 0.4986 | 0.7498 | 0.0034 | 0.01976 | 0.02231 | 1.63 | 1.66 | 35.36 | 1 | 1 | 1.00 |
| Na2 | 0.7470 | 0.2538 | 0.2529 | 0.01885 | 0.02162 | 1.48 | 1.74 | 38.19 | 1 | 1 | 1.11 |
| Ti1 | 0.6218 | 0.7584 | 0.3732 | 0.01876 | 0.01381 | 7.73 | 4.90 | 26.08 | 1 | 1 | 0.69 |
| Ti2 | 0.8627 | 0.7524 | 0.1224 | 0.01371 | 0.01237 | 4.27 | 4.20 | 34.69 | 1 | 1 | 0.98 |
| Ti3 | 0.0977 | 0.2557 | 0.3733 | 0.00544 | 0.01394 | 1.54 | 6.15 | 57.76 | 1 | 1 | 2.24 |
| Ti4 | 0.3670 | 0.2498 | 0.1247 | 0.01609 | 0.01628 | 6.56 | 6.74 | 35.56 | 1 | 1 | 1.01 |
| O1 | 0.6980 | 0.8128 | 0.2421 | −0.00814 | 0.00362 | 1.66 | −1.58 | −52.90 | 1 | 1 | −1.87 |
| O2 | 0.9758 | 0.6910 | −0.0088 | 0.00719 | 0.00222 | −0.58 | −0.81 | 41.79 | 1 | 1 | 1.26 |
| O3 | 0.2194 | 0.1891 | 0.2410 | 0.00362 | 0.00190 | −0.27 | −0.64 | 51.02 | 1 | 1 | 1.75 |
| O4 | 0.4440 | 0.3116 | −0.0093 | −0.01037 | 0.00144 | 2.24 | −0.89 | −23.40 | 1 | 1 | −0.61 |
| O5 | 0.8876 | 0.5307 | 0.3518 | 0.02746 | −0.02158 | −8.30 | 3.77 | −26.80 | 1 | 1 | −0.71 |
| O6 | 0.1304 | −0.0168 | 0.0979 | 0.02352 | −0.02803 | −5.15 | 3.78 | −42.63 | 1 | 1 | −1.30 |
| O7 | 0.3762 | 0.4831 | 0.3456 | 0.02119 | −0.03195 | −4.43 | 4.93 | −59.32 | 1 | 1 | −2.38 |
| O8 | 0.6378 | 0.0294 | 0.1000 | 0.02754 | −0.02457 | −8.59 | 5.22 | −35.77 | 1 | 1 | −1.02 |
| O9 | 0.5488 | 0.5333 | 0.1308 | −0.02147 | 0.02631 | 4.84 | −4.83 | −54.92 | 1 | 1 | −2.01 |
| O10 | 0.2898 | 0.9599 | 0.3780 | −0.02645 | 0.02170 | 5.13 | −4.54 | −49.99 | 1 | 1 | −1.69 |
| O11 | 0.0448 | 0.4676 | 0.1279 | −0.02367 | 0.02158 | 4.43 | −2.66 | −35.39 | 1 | 1 | −1.00 |
| O12 | 0.8008 | 0.0293 | 0.3814 | −0.02041 | 0.02734 | 4.63 | −2.73 | −34.78 | 1 | 1 | −0.98 |
| Atom | x (−) | y (−) | z (−) | ∆x (nm) a | ∆z (nm) a | pa b (μC/cm2) | pc b (μC/cm2) | αpatom c (deg) | [u | u | v] d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bi1 | 0.2739 | 0.7477 | 0.2565 | 0.03420 | 0.02729 | 8.61 | 8.46 | 34.76 | 1 | 1 | 0.98 |
| Bi2 | 0.0222 | 0.2542 | 0.0074 | 0.03328 | 0.02880 | 9.20 | 8.56 | 33.65 | 1 | 1 | 0.94 |
| Na1 | 0.4994 | 0.7495 | 0.0032 | 0.02066 | 0.02188 | 1.70 | 1.62 | 34.18 | 1 | 1 | 0.96 |
| Na2 | 0.7472 | 0.2539 | 0.2528 | 0.01940 | 0.02112 | 1.53 | 1.70 | 37.28 | 1 | 1 | 1.08 |
| Ti1 | 0.6232 | 0.7589 | 0.3731 | 0.01998 | 0.01334 | 8.31 | 4.67 | 23.98 | 1 | 1 | 0.63 |
| Ti2 | 0.8635 | 0.7520 | 0.1220 | 0.01464 | 0.01165 | 4.66 | 3.79 | 30.98 | 1 | 1 | 0.85 |
| Ti3 | 0.0979 | 0.2557 | 0.3731 | 0.00601 | 0.01344 | 1.76 | 5.90 | 55.97 | 1 | 1 | 2.09 |
| Ti4 | 0.3688 | 0.2496 | 0.1246 | 0.01757 | 0.01595 | 7.18 | 6.62 | 33.49 | 1 | 1 | 0.94 |
| O1 | 0.6960 | 0.8121 | 0.2422 | −0.00887 | 0.00373 | 1.80 | −1.64 | −51.08 | 1 | 1 | −1.75 |
| O2 | 0.9750 | 0.6913 | −0.0088 | 0.00716 | 0.00207 | −0.57 | −0.76 | 40.68 | 1 | 1 | 1.22 |
| O3 | 0.2194 | 0.1894 | 0.2410 | 0.00406 | 0.00166 | −0.31 | −0.55 | 46.05 | 1 | 1 | 1.47 |
| O4 | 0.4429 | 0.3104 | −0.0092 | −0.01058 | 0.00140 | 2.28 | −0.88 | −22.81 | 1 | 1 | −0.59 |
| O5 | 0.8864 | 0.5309 | 0.3521 | 0.02726 | −0.02123 | −8.23 | 3.72 | −26.64 | 1 | 1 | −0.71 |
| O6 | 0.1289 | −0.0165 | 0.0982 | 0.02313 | −0.02769 | −5.06 | 3.73 | −42.74 | 1 | 1 | −1.31 |
| O7 | 0.3741 | 0.4837 | 0.3459 | 0.02048 | −0.03148 | −4.28 | 4.85 | −59.92 | 1 | 1 | −2.44 |
| O8 | 0.6373 | 0.0297 | 0.1001 | 0.02781 | −0.02452 | −8.66 | 5.23 | −35.55 | 1 | 1 | −1.01 |
| O9 | 0.5488 | 0.5326 | 0.1310 | −0.02114 | 0.02637 | 4.77 | −4.82 | −55.30 | 1 | 1 | −2.04 |
| O10 | 0.2893 | 0.9609 | 0.3781 | −0.02640 | 0.02163 | 5.12 | −4.52 | −49.84 | 1 | 1 | −1.68 |
| O11 | 0.0452 | 0.4690 | 0.1279 | −0.02314 | 0.02126 | 4.32 | −2.62 | −35.70 | 1 | 1 | −1.02 |
| O12 | 0.8013 | 0.0287 | 0.3812 | −0.01976 | 0.02680 | 4.48 | −2.67 | −35.16 | 1 | 1 | −1.00 |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shrout, T.R. Characteristics of relaxor-based piezoelectric single crystals for ultrasonic transducers. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 1997, 44, 1140–1147. [Google Scholar] [CrossRef]
- Noheda, B.; Zhong, Z.; Cox, D.; Shirane, G.; Park, S.-E.; Rehrig, P. Electric-field-induced phase transitions in rhombohedral Pb(Zn1/3Nb2/3)1−xTixO3. Phys. Rev. B 2002, 65. [Google Scholar] [CrossRef]
- Fu, H.; Cohen, R. Polarization rotation mechanism for ultrahigh electromechanical response in single-crystal piezoelectrics. Nature 2000, 403, 281–283. [Google Scholar] [CrossRef]
- Damjanovic, D. Comments on origins of enhanced piezoelectric properties in ferroelectrics. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2009, 56, 1574–1585. [Google Scholar] [CrossRef]
- Takenaka, T.; Maruyama, K.; Sakata, K. (Bi1/2Na1/2)TiO3-BaTiO3 system for lead-free piezoelectric ceramics. Jpn. J. Appl. Phys. 1991, 30, 2236–2239. [Google Scholar] [CrossRef]
- Guo, Y.; Kakimoto, K.; Ohsato, H. (Na0.5K0.5)NbO3–LiTaO3 lead-free piezoelectric ceramics. Mater. Lett. 2005, 59, 241–244. [Google Scholar] [CrossRef]
- Maeder, M.D.; Damjanovic, D.; Setter, N. Lead free piezoelectric materials. J. Electroceram. 2004, 13, 385–392. [Google Scholar] [CrossRef]
- Nagata, H.; Yoshida, M.; Makiuchi, Y.; Takenaka, T. Large piezoelectric constant and high curie temperature of lead-free piezoelectric ceramic ternary system based on bismuth sodium titanate-bismuth potassium titanate-barium titanate near the morphotropic phase boundary. Jpn. J. Appl. Phys. 2003, 42, 7401–7403. [Google Scholar] [CrossRef]
- Shrout, T.R.; Zhang, S.J. Lead-free piezoelectric ceramics: Alternatives for PZT? J. Electroceram. 2007, 19, 113–126. [Google Scholar] [CrossRef]
- Vakhrushev, S.B.; Isupov, V.A.; Kvyatkovsky, B.E.; Okuneva, N.M.; Pronin, I.P.; Smolensky, G.A.; Syrnikov, P.P. Phase transitions and soft modes in sodium bismuth titanate. Ferroelectrics 1985, 63, 153–160. [Google Scholar] [CrossRef]
- Takenaka, T.; Sakata, K. Dielectric, piezoelectric and pyroelectric properties of (BiNa)1/2TiO3-based ceramics. Ferroelectrics 1989, 95, 153–156. [Google Scholar] [CrossRef]
- Sasaki, A.; Chiba, T.; Mamiya, Y.; Otsuki, E. Dielectric and piezoelectric properties of (Bi0.5Na0.5)TiO3–(Bi0.5K0.5)TiO3 systems. Jpn. J. Appl. Phys. 1999, 38, 5564–5567. [Google Scholar] [CrossRef]
- Roleder, K.; Franke, I.; Glazer, A.M.; Thomas, P.A.; Miga, S.; Suchanicz, J. The piezoelectric effect in Na0.5Bi0.5TiO3 ceramics. J. Phys. Condens. Matter 2002, 14, 5399–5406. [Google Scholar] [CrossRef]
- Jones, G.O.; Thomas, P.A. The tetragonal phase of Na0.5Bi0.5TiO3—A new variant of the perovskite structure. Acta Crystallogr. Sect. B Struct. Sci. 2000, 56, 426–430. [Google Scholar] [CrossRef]
- Elkechai, O.; Manier, M.; Mercurio, J.P. Na0.5Bi0.5TiO3–K0.5Bi0.5TiO3 (NBT-KBT) system: A structural and electrical study. Phys. Status Solidi 1996, 157, 499–506. [Google Scholar] [CrossRef]
- Kreisel, J.; Glazer, A.M.; Jones, G.; Thomas, P.A.; Abello, L.; Lucazeau, G. An X-ray diffraction and Raman spectroscopy investigation of A-site substituted perovskite compounds: The (Na1−xKx)0.5Bi0.5TiO3 (0 ≤ x ≤ 1) solid solution. J. Phys. Condens. Matter 2000, 12, 3267–3280. [Google Scholar]
- Jones, G.O.; Kreisel, J.; Jennings, V.; Geday, M.A.; Thomas, P.A.; Glazer, A.M. Investigation of a peculiar relaxor ferroelectric: Na0.5Bi0.5TiO3. Ferroelectrics 2002, 270, 191–196. [Google Scholar] [CrossRef]
- Aleksandrova, I.P.; Sukhovsky, A.A.; Ivanov, Y.N.; Yablonskaya, Y.E.; Vakhrushev, S.B. Local and average structure of relaxor Na1/2Bi1/2TiO3 from the point of view of NMR. Ferroelectrics 2009, 378, 16–22. [Google Scholar] [CrossRef]
- Aksel, E.; Forrester, J.S.; Jones, J.L.; Thomas, P.A.; Page, K.; Suchomel, M.R. Monoclinic crystal structure of polycrystalline Na0.5Bi0.5TiO3. Appl. Phys. Lett. 2011, 98. [Google Scholar] [CrossRef]
- Picht, G.; Töpfer, J.; Hennig, E. Structural properties of (Bi0.5Na0.5)1−xBaxTiO3 lead-free piezoelectric ceramics. J. Eur. Ceram. Soc. 30, 2010, 3445–3453. [Google Scholar]
- Jones, G.O.; Thomas, P.A. Investigation of the structure and phase transitions in the novel A-site substituted distorted perovskite compound Na0.5Bi0.5TiO3. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 168–178. [Google Scholar] [CrossRef]
- Gorfman, S.; Glazer, A.M.; Noguchi, Y.; Miyayama, M.; Luo, H.; Thomas, P.A. Observation of a low-symmetry phase in Na0.5Bi0.5TiO3 crystals by optical birefringence microscopy. J. Appl. Crystallogr. 2012, 45, 444–452. [Google Scholar] [CrossRef]
- Chiang, Y.-M.; Farrey, G.W.; Soukhojak, A.N. Lead-free high-strain single-crystal piezoelectrics in the alkaline–bismuth–titanate perovskite family. Appl. Phys. Lett. 1998, 73, 10–1063. [Google Scholar]
- Jo, W.; Daniels, J.E.; Jones, J.L.; Tan, X.; Thomas, P.A.; Damjanovic, D.; Rödel, J. Evolving morphotropic phase boundary in lead-free (Bi1/2Na1/2)TiO3–BaTiO3 piezoceramics. J. Appl. Phys. 2011, 109. [Google Scholar] [CrossRef]
- Cernea, M.; Trupina, L.; Dragoi, C.; Vasile, B.S.; Trusca, R. Structural and piezoelectric characteristics of BNT–BT0.05 thin films processed by sol-gel technique. J. Alloys Compd. 2012, 515, 166–170. [Google Scholar] [CrossRef]
- Cordero, F.; Craciun, F.; Trequattrini, F.; Mercadelli, E.; Galassi, C. Phase transitions and phase diagram of the ferroelectric perovskite (Na0.5Bi0.5)1−xBaxTiO3 by anelastic and dielectric measurements. Phys. Rev. B 2010, 81. [Google Scholar] [CrossRef]
- Ma, C.; Guo, H.; Tan, X. A New Phase Boundary in (Bi1/2Na1/2)TiO3−BaTiO3 Revealed via a Novel Method of Electron Diffraction Analysis. Adv. Funct. Mater. 2013, 23, 5261–5266. [Google Scholar] [CrossRef]
- Ma, C.; Guo, H.; Beckman, S.P.; Tan, X. Creation and destruction of morphotropic phase boundaries through electrical poling: A case study of lead-free (Bi1/2Na1/2)TiO3–BaTiO3 piezoelectrics. Phys. Rev. Lett. 2012, 109. [Google Scholar] [CrossRef]
- Yao, J.; Yan, L.; Ge, W.; Luo, L.; Li, J.; Viehland, D.; Zhang, Q.; Luo, H. Evolution of domain structures in Na1/2Bi1/2TiO3 single crystals with BaTiO3. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef]
- Kling, J.; Tan, X.; Jo, W.; Kleebe, H.-J.; Fuess, H.; Rödel, J. In situ transmission electron microscopy of electric field-triggered reversible domain formation in bi-based lead-free piezoceramics. J. Am. Ceram. Soc. 2010, 93, 2452–2455. [Google Scholar] [CrossRef]
- Kreisel, J.; Glazer, A.M. Estimation of the compressibility of Na0.5Bi0.5TiO3 and related perovskite-type titanates. J. Phys. Condens. Matter 2000, 12, 9689–9698. [Google Scholar] [CrossRef]
- Kreisel, J.; Glazer, A.; Bouvier, P.; Lucazeau, G. High-pressure raman study of a relaxor ferroelectric: The Na0.5Bi0.5TiO3 perovskite. Phys. Rev. B 2001, 63. [Google Scholar] [CrossRef]
- Kreisel, J.; Bouvier, P.; Dkhil, B.; Thomas, P.; Glazer, A.; Welberry, T.; Chaabane, B.; Mezouar, M. High-pressure X-ray scattering of oxides with a nanoscale local structure: Application to Na1/2Bi1/2TiO3. Phys. Rev. B 2003, 68. [Google Scholar] [CrossRef]
- Kreisel, J.; Bouvier, P.; Dkhil, B.; Chaabane, B.; Glazer, A.M.; Thomas, P.A.; Welberry, T.R. Effect of high pressure on the relaxor ferroelectrics Na1/2Bi1/2TiO3 (NBT) and PbMg1/3Nb2/3O3 (PMN). Ferroelectrics 2004, 302, 293–298. [Google Scholar] [CrossRef]
- Thomas, P.A.; Trujillo, S.; Boudard, M.; Gorfman, S.; Kreisel, J. Diffuse X-ray scattering in the lead-free piezoelectric crystals Na1/2Bi1/2TiO3 and Ba-doped Na1/2Bi1/2TiO3. Solid State Sci. 2010, 12, 311–317. [Google Scholar] [CrossRef]
- Ge, W.; Cao, H.; Li, J.; Viehland, D.; Zhang, Q.; Luo, H. Influence of dc-bias on phase stability in Mn-doped Na0.5Bi0.5TiO3–5.6 at.%BaTiO3 single crystals. Appl. Phys. Lett. 2009, 95. [Google Scholar] [CrossRef]
- Daniels, J.E.; Jo, W.; Rödel, J.; Rytz, D.; Donner, W. Structural origins of relaxor behavior in a 0.96(Bi1/2Na1/2)TiO3–0.04BaTiO3 single crystal under electric field. Appl. Phys. Lett. 2011, 98. [Google Scholar] [CrossRef]
- Keeble, D.S.; Barney, E.R.; Keen, D.A.; Tucker, M.G.; Kreisel, J.; Thomas, P.A. Bifurcated polarization rotation in bismuth-based piezoelectrics. Adv. Funct. Mater. 2013, 23, 185–190. [Google Scholar] [CrossRef]
- Oh, T.; Kim, M.-H. Phase relation and dielectric properties in (Bi1/2Na1/2)1−xBaxTiO3 lead-free ceramics. Mater. Sci. Eng. B Solid State Mater. Adv. Technol. 2006, 132, 239–246. [Google Scholar] [CrossRef]
- Xu, C.; Lin, D.; Kwok, K.W. Structure, electrical properties and depolarization temperature of (Bi0.5Na0.5)TiΟ–BaTiΟ3 lead-free piezoelectric ceramics. Solid State Sci. 2008, 10, 934–940. [Google Scholar] [CrossRef]
- Maurya, D.; Pramanick, A.; An, K.; Priya, S. Enhanced piezoelectricity and nature of electric-field induced structural phase transformation in textured lead-free piezoelectric Na0.5Bi0.5TiO3–BaTiO3 ceramics. Appl. Phys. Lett. 2012, 100. [Google Scholar] [CrossRef]
- Rödel, J.; Jo, W.; Seifert, K.T.P.; Anton, E.-M.; Granzow, T.; Damjanovic, D. Perspective on the Development of Lead-free Piezoceramics. J. Am. Ceram. Soc. 2009, 92, 1153–1177. [Google Scholar] [CrossRef]
- Chen, M.; Xu, Q.; Kim, B.H.; Ahn, B.K.; Ko, J.H.; Kang, W.J.; Nam, O.J. Structure and electrical properties of (Na0.5Bi0.5)1−xBaxTiO3 piezoelectric ceramics. J. Eur. Ceram. Soc. 2008, 28, 843–849. [Google Scholar] [CrossRef]
- Roleder, K.; Suchanicz, J.; Kania, A. Time dependence of electric permittivity in Na0.5Bi0.5TiO3 single crystals. Ferroelectrics 1989, 89, 1–5. [Google Scholar] [CrossRef]
- Suchanicz, J.; Roleder, K.; Kania, A.; Hańaderek, J. Electrostrictive strain and pyroeffect in the region of phase coexistence in Na0.5Bi0.5TiO3. Ferroelectrics 1988, 77, 107–110. [Google Scholar] [CrossRef]
- Onozuka, H.; Kitanaka, Y.; Noguchi, Y.; Miyayama, M. Crystal Growth and characterization of (Bi0.5Na0.5)TiO3–BaTiO3 single crystals obtained by a top-seeded solution growth method under high-pressure oxygen atmosphere. Jpn. J. Appl. Phys. 2011, 50. [Google Scholar] [CrossRef]
- Kitanaka, Y.; Onozuka, H.; Noguchi, Y.; Miyayama, M. High-performance ferroelectric Bi0.5Na0.5TiO3 Single crystals grown by top-seeded solution growth method under high-pressure oxygen atmosphere. Ferroelectrics 2011, 414, 24–29. [Google Scholar] [CrossRef]
- Kitanaka, Y.; Noguchi, Y.; Miyayama, M. High-Performance ferroelectric Bi4Ti3O12 single crystals grown by top-seeded solution growth method under high-pressure oxygen atmosphere. Jpn. J. Appl. Phys. 2010, 49. [Google Scholar] [CrossRef]
- Hirano, K.; Onozuka, H.; Kitanaka, Y.; Noguchi, Y.; Miyayama, M. Leakage current and polarization properties of (Bi0.5Na0.5)TiO3–BaTiO3 single crystals. Key Eng. Mater. 2014, 582, 96–99. [Google Scholar]
- Moriyoshi, C.; Hiramoto, S.; Ohkubo, H.; Kuroiwa, Y.; Osawa, H.; Sugimoto, K.; Kimura, S.; Takata, M.; Kitanaka, Y.; Noguchi, Y.; et al. Synchrotron Radiation study on time-resolved tetragonal lattice strain of BaTiO3 under electric field. Jpn. J. Appl. Phys. 2011, 50. [Google Scholar] [CrossRef]
- Kitanaka, Y.; Yanai, K.; Noguchi, Y.; Miyayama, M.; Kagawa, Y.; Moriyoshi, C.; Kuroiwa, Y. Non-180° polarization rotation of ferroelectric (Bi0.5Na0.5)TiO3 single crystals under electric field. Phys. Rev. B 2014, 89. [Google Scholar] [CrossRef]
- Yanai, K.; Kitanaka, Y.; Noguchi, Y.; Miyayama, M.; Moriyoshi, C.; Kuroiwa, Y.; Kurushima, K.; Mori, S.; Kagawa, Y. Enhanced polarization switching in ferroelectric Bi0.5Na0.5TiO3 single crystals by defect control. Phys. Status Solidi 2013, 443, 791–795. [Google Scholar]
- Kitanaka, Y.; Noguchi, Y.; Miyayama, M.; Kagawa, Y.; Moriyoshi, C.; Kuroiwa, Y. Synchrotron radiation analyses of domain switching and lattice strain behaviors for ferroelectric (Bi0.5Na0.5)TiO3 single crystals under electric fields. Ferroelectrics 2013, 443, 1–7. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Wang, F.; Wang, Y.; Lin, D.; Zhao, X.; Luo, H.; Ge, W.; Viehland, D. Enhanced piezoelectric and ferroelectric properties in Mn-doped Na0.5Bi0.5TiO3–BaTiO3 single crystals. Appl. Phys. Lett. 2009, 95. [Google Scholar] [CrossRef]
- Aksel, E.; Forrester, J.S.; Nino, J.C.; Page, K.; Shoemaker, D.P.; Jones, J.L. Local atomic structure deviation from average structure of Na0.5Bi0.5TiO3: Combined X-ray and neutron total scattering study. Phys. Rev. B 2013, 87. [Google Scholar] [CrossRef]
- Langreth, D.; Perdew, J. Theory of nonuniform electronic systems. I. Analysis of the gradient approximation and a generalization that works. Phys. Rev. B 1980, 21, 5469–5493. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.; Ruzsinszky, A.; Csonka, G.; Vydrov, O.; Scuseria, G.; Constantin, L.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef]
- Grüning, M.; Marini, A.; Gonze, X. Implementation and testing of Lanczos-based algorithms for random-phase approximation eigenproblems. Comput. Mater. Sci. 2011, 50, 2148–2156. [Google Scholar] [CrossRef]
- Glazer, A.M. The classification of tilted octahedra in perovskites. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 3384–3392. [Google Scholar] [CrossRef]
- Stokes, H.T.; Kisi, E.H.; Hatch, D.M.; Howard, C.J. Group-theoretical analysis of octahedral tilting in ferroelectric perovskites. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 934–938. [Google Scholar] [CrossRef]
- Bubesh Babu, J.; He, M.; Zhang, D.F.; Chen, X.L.; Dhanasekaran, R. Enhancement of ferroelectric properties of Na1/2Bi1/2TiO3–BaTiO3 single crystals by Ce dopings. Appl. Phys. Lett. 2007, 90. [Google Scholar] [CrossRef]
- Ge, W.; Liu, H.; Zhao, X.; Fang, B.; Li, X.; Wang, F.; Zhou, D.; Yu, P.; Pan, X.; Lin, D.; et al. Crystal growth and high piezoelectric performance of 0.95Na0.5Bi0.5TiO3–0.05BaTiO3 lead-free ferroelectric materials. J. Phys. D Appl. Phys. 2008, 41. [Google Scholar] [CrossRef]
- Suzuki, M.; Morishita, A.; Kitanaka, Y.; Noguchi, Y.; Miyayama, M. Polarization and piezoelectric properties of high performance bismuth sodium titanate single crystals grown by high-oxygen-pressure flux method. Jpn. J. Appl. Phys. 2010, 49. [Google Scholar] [CrossRef]
- Bethe, K.; Welz, F. Preparation and properties of (Ba,Sr)TiO3 single crystals. Mater. Res. Bull. 1971, 6, 209–217. [Google Scholar] [CrossRef]
- Bell, A.J. Phenomenologically derived electric field-temperature phase diagrams and piezoelectric coefficients for single crystal barium titanate under fields along different axes. J. Appl. Phys. 2001, 89. [Google Scholar] [CrossRef]
- Li, Y.L.; Cross, L.E.; Chen, L.Q. A phenomenological thermodynamic potential for BaTiO3 single crystals. J. Appl. Phys. 2005, 98. [Google Scholar] [CrossRef]
- Uesu, Y.; Matsuda, M.; Yamada, Y.; Fujishiro, K.; Cox, D.E.; Noheda, B.; Shirane, G. Symmetry of high-piezoelectric pb-based complex perovskites at the morphotropic phase boundary: I. Neutron diffraction study on Pb(Zn1/3Nb2/3)O3–9%PbTiO3. J. Phys. Soc. Jpn. 2002, 71, 960–965. [Google Scholar] [CrossRef]
- Yamada, Y.; Uesu, Y.; Matsuda, M.; Fujishiro, K.; Cox, D.E.; Noheda, B.; Shirane, G. Symmetry of high-piezoelectric pb based complex perovskites at the morphotropic phase boundary: II. Theoretical treatment. J. Phys. Soc. Jpn. 2002, 71, 966–970. [Google Scholar] [CrossRef]
- Ohwada, K.; Hirota, K.; Rehrig, P.; Fujii, Y.; Shirane, G. Neutron diffraction study of field-cooling effects on the relaxor ferroelectric Pb[(Zn1/3Nb2/3)0.92Ti0.08]O3. Phys. Rev. B 2003, 67. [Google Scholar] [CrossRef]
- Rajan, K.K.; Shanthi, M.; Chang, W.S.; Jin, J.; Lim, L.C. Dielectric and piezoelectric properties of [001] and [011]-poled relaxor ferroelectric PZN–PT and PMN–PT single crystals. Sens. Actuators A Phys. 2007, 133, 110–116. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B Struct. Sci. 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Sayers, D.; Stern, E.; Lytle, F. New Technique for Investigating noncrystalline structures: Fourier analysis of the extended X-ray—Absorption fine structure. Phys. Rev. Lett. 1971, 27, 1204–1207. [Google Scholar] [CrossRef]
- Zhao, X.; Vanderbilt, D. Phonons and lattice dielectric properties of zirconia. Phys. Rev. B 2002, 65. [Google Scholar] [CrossRef]
- Zhong, W.; Vanderbilt, D. Phase transitions in BaTiO3 from first principles. Phys. Rev. Lett. 1994, 73, 1861–1864. [Google Scholar] [CrossRef]
- Bagayoko, D.; Zhao, G.L.; Fan, J.D.; Wang, J.T. Ab initio calculations of the electronic structure and optical properties of ferroelectric tetragonal. J. Phys. Condens. Matter 1998, 10, 5645–5655. [Google Scholar] [CrossRef]
- Gou, G.Y.; Bennett, J.W.; Takenaka, H.; Rappe, A.M. Post density functional theoretical studies of highly polar semiconductive Pb(Ti1−xNix)O3−x solid solutions: Effects of cation arrangement on band gap. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef]
- Miyazawa, H.; Natori, E.; Miyashita, S.; Shimoda, T.; Ishii, F.; Oguchi, T. Electronic states of perovskite-type oxides and ferroelectricity. Jpn. J. Appl. Phys. 2000, 39, 5679–5682. [Google Scholar] [CrossRef]
- Kitanaka, Y.; Ogino, M.; Hirano, K.; Noguchi, Y.; Miyayama, M.; Kagawa, Y.; Moriyoshi, C.; Kuroiwa, Y.; Torii, S.; Kamiyama, T. Crystal Structural analyses of ferrielectric tetragonal (Bi1/2Na1/2)TiO3–7%BaTiO3 powders and single crystals. Jpn. J. Appl. Phys. 2013, 52. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ogino, M.; Noguchi, Y.; Kitanaka, Y.; Miyayama, M.; Moriyoshi, C.; Kuroiwa, Y. Polarization Rotation and Monoclinic Distortion in Ferroelectric (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals under Electric Fields. Crystals 2014, 4, 273-295. https://doi.org/10.3390/cryst4030273
Ogino M, Noguchi Y, Kitanaka Y, Miyayama M, Moriyoshi C, Kuroiwa Y. Polarization Rotation and Monoclinic Distortion in Ferroelectric (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals under Electric Fields. Crystals. 2014; 4(3):273-295. https://doi.org/10.3390/cryst4030273
Chicago/Turabian StyleOgino, Motohiro, Yuji Noguchi, Yuuki Kitanaka, Masaru Miyayama, Chikako Moriyoshi, and Yoshihiro Kuroiwa. 2014. "Polarization Rotation and Monoclinic Distortion in Ferroelectric (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals under Electric Fields" Crystals 4, no. 3: 273-295. https://doi.org/10.3390/cryst4030273
APA StyleOgino, M., Noguchi, Y., Kitanaka, Y., Miyayama, M., Moriyoshi, C., & Kuroiwa, Y. (2014). Polarization Rotation and Monoclinic Distortion in Ferroelectric (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals under Electric Fields. Crystals, 4(3), 273-295. https://doi.org/10.3390/cryst4030273