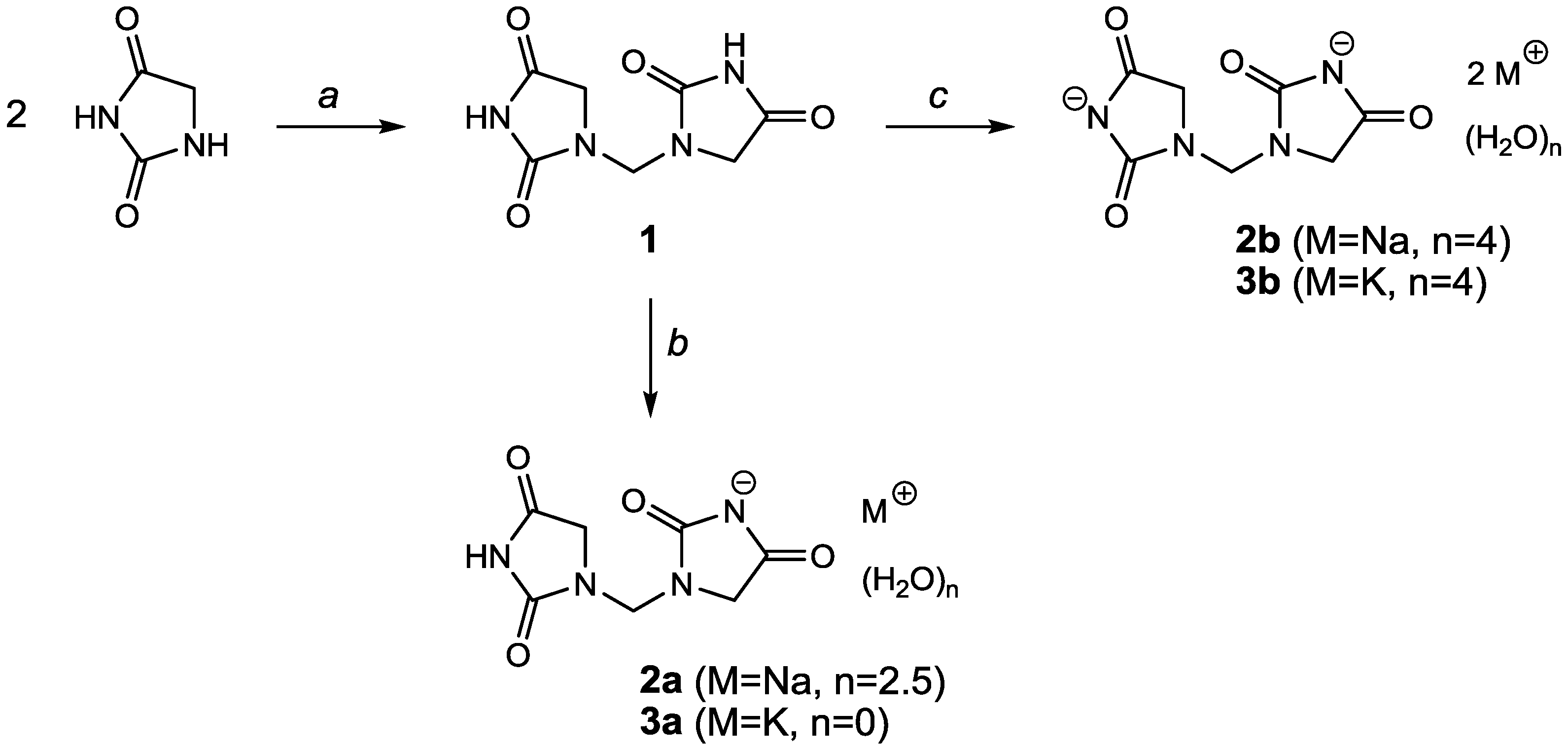
2. Results and Discussion
The preparation of the title compound
1 was essentially carried out according to the literature with minor modifications as described below. In our hands, it was not necessary to strictly limit the amount of water in the reaction mixture, in contrast to the patented method [
5]. The synthesis of the mono-valent and di-valent salts of 1,1′-methylenebis(imidazolidine-2,4-dione) was expected to be straightforward, using one or two equivalents of sodium hydroxide or potassium hydroxide, respectively (
Figure 1). However, we encountered several unexpected difficulties.
Figure 1.
Synthesis of 1,1′-methylenebis(imidazolidine-2,4-dione) and its salts. Reagents and solvents: a: CH2O/HCl/H2O; b: 1 equivalent NaOH or KOH in H2O/NH3; c: 2 equivalents NaOH or KOH in H2O.
Figure 1.
Synthesis of 1,1′-methylenebis(imidazolidine-2,4-dione) and its salts. Reagents and solvents: a: CH2O/HCl/H2O; b: 1 equivalent NaOH or KOH in H2O/NH3; c: 2 equivalents NaOH or KOH in H2O.
Reaction of the title compound with one equivalent of sodium hydroxide in the presence of ammonia (which is needed to get a clear solution) yielded
2a as an amorphous paste which was converted to a crystalline powder by prolonged stirring in ethanol. Surprisingly, this product was found by powder X-ray diffraction (PXRD) to consist of starting material
1 and disodium salt
2b. The disodium salt
2b was readily prepared by reaction of
1 with two equivalents of sodium hydroxide, but no single-crystals could be obtained. By serendipity, we found a single-crystal of the monosodium salt
2a instead in the bulk of the disodium salt. The potassium salts also posed some problems. The purity of the monopotassium salt
3a was better than that of the monosodium salt, but there was still contamination with the starting material. On the other hand, single-crystals of the dipotassium salt
3b were obtained which permitted the determination of their crystal structures. The crystallographic data and refinement details are summarized in
Table 1.
Table 1.
Crystal data and structure refinement details.
Table 1.
Crystal data and structure refinement details.
| Compound | 1 | 2a | 3b |
|---|
| CCDC Number | 950723 | 950724 | 950725 |
| Chemical formula | C7H8N4O4 | C14H24N8Na2O13 | C7H14K2N4O8 |
| Mr | 212.17 | 558.39 | 360.42 |
| Crystal size/mm3 | 0.21 × 0.13 × 0.09 | 0.43 × 0.20 × 0.08 | 0.30 × 0.30 × 0.20 |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | C2221 | C2/c | Pbcn |
| a/Å | 9.5263(3) | 29.4742(12) | 16.0906(6) |
| b/Å | 20.6048(4) | 7.8472(3) | 7.5688(3) |
| c/Å | 17.7875(6) | 10.0477(4) | 11.5890(4) |
| α/° | 90 | 90 | 90 |
| β/° | 90 | 104.849(4) | 90 |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 3491.46(17) | 2246.32(15) | 1411.38(9) |
| Z | 16 | 4 | 4 |
| Dx/g·cm−3 | 1.62 | 1.65 | 1.70 |
| μ/mm−1 | 0.14 | 0.18 | 0.72 |
| F(000)/e | 1760 | 1160 | 744 |
| T/K | 243 | 173 | 173 |
| θmax/° | 25.0 | 25.7 | 25.7 |
| h, k, l range | −10 ≤ h ≤ 11 | −35 ≤ h ≤ 27−12 | −19 ≤ h ≤ 16 |
| −24 ≤ k ≤ 24 | −9 ≤ k ≤ 9 | −12 ≤ l ≤ 11 |
| −21 ≤ l ≤ 21 | −12 ≤ l ≤ 11 | −14 ≤ l ≤ 13 |
| Measured reflections | 10711 | 6571 | 8168 |
| Independent reflections (Rint) | 1721 (0.032) | 2135 (0.027) | 1335 (0.028) |
| Observed reflections [I ≥ 2σ(I)] | 1540 | 1836 | 1226 |
| Restraints/parameters | 0/320 | 9/190 | 6/112 |
| R1/wR2 [I ≥ 2σ(I)] | 0.048/0.117 | 0.031/0.075 | 0.024/0.061 |
| R1/wR2 (all data) | 0.054/0.121 | 0.038/0.080 | 0.026/0.062 |
| Δρmax/min/e Å−3 | 0.19/−0.22 | 0.23/−0.24 | 0.26/−0.24 |
2.1. 1,1′-Methylenebis(imidazolidine-2,4-dione) (1)
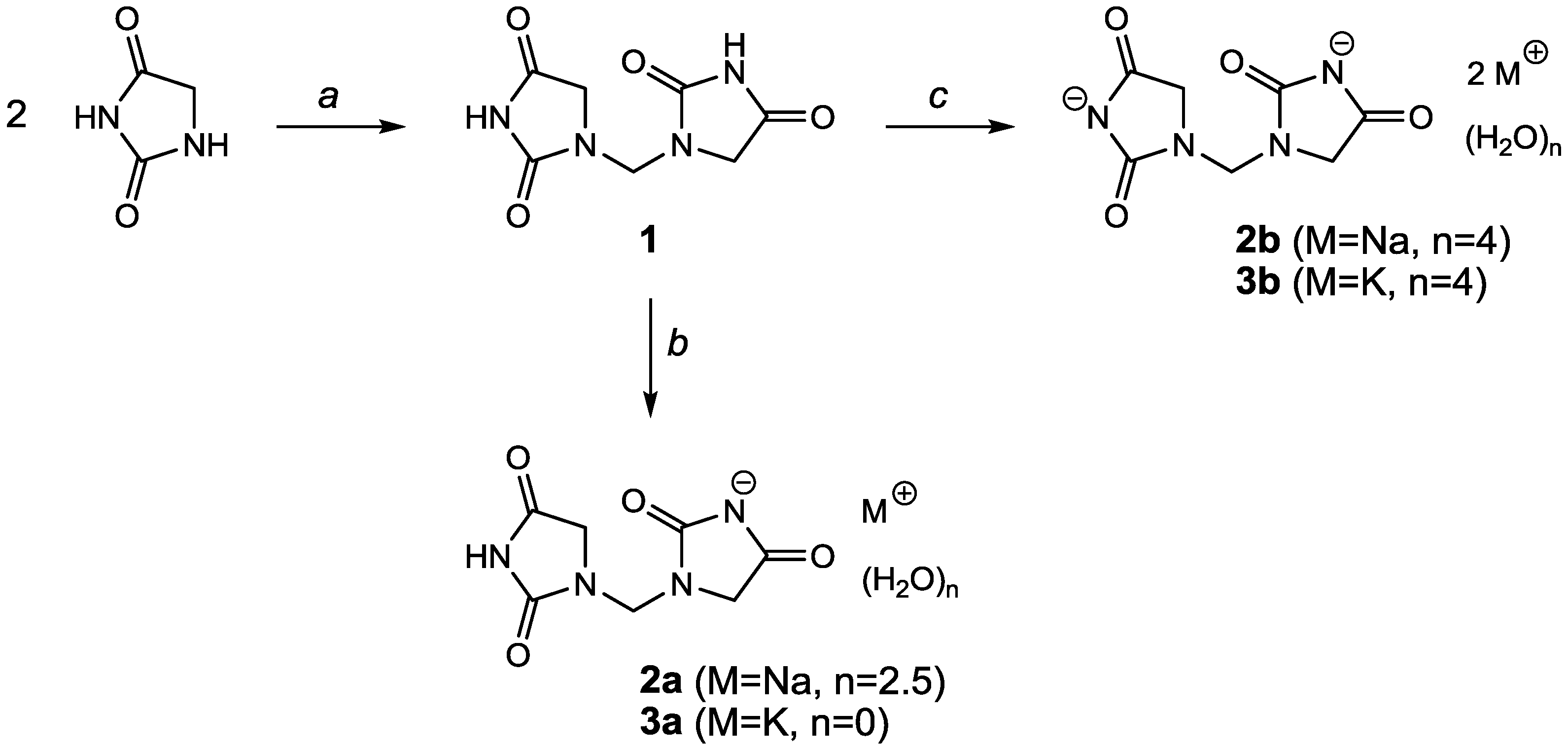
Because of the absence of significant anomalous scattering effects [Flack parameter
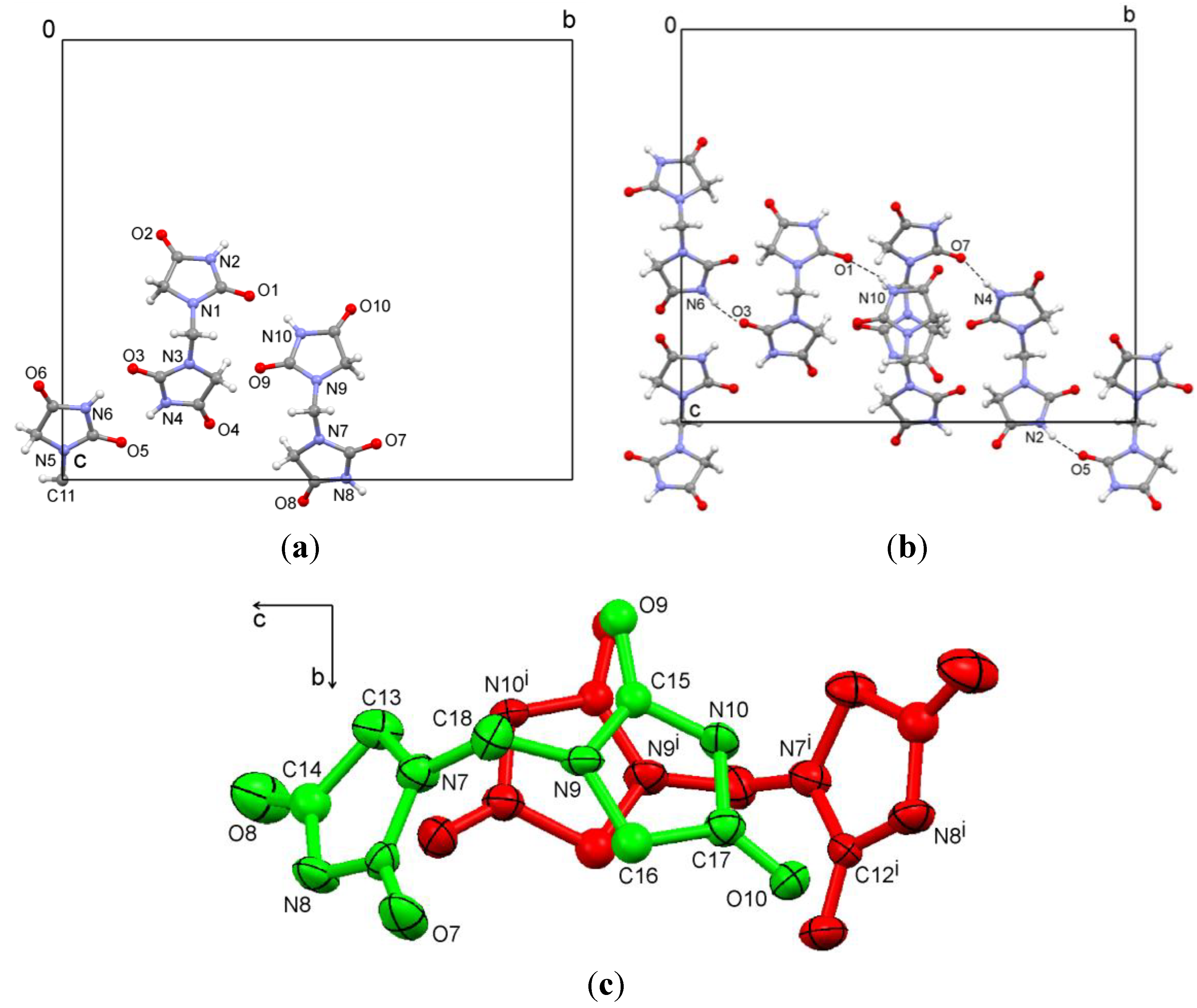
x around −2(2)], Friedel pairs were merged. All hydrogens were found, especially at nitrogen atoms, but were calculated because of a low data/parameter ratio. In the asymmetric unit, there is one complete molecule, half a molecule with C11 on a twofold rotation axis, and a disordered molecule with occupancy of 0.5 for each atom (
Figure 2a). Moderate N–H…O hydrogen bonds are observed (
Figure 2b). The disordered molecules produce a partially overlapping chain by symmetry elements of a twofold rotation axis along
a and a twofold screw axis along
c. In order to obtain reasonable bond lengths O9, C14, C15, and C16 were refined with isotropic displacement parameters (
Figure 2c). The hydrogen bond parameters are summarized in
Table 2.
Figure 2.
(a) Asymmetric unit; (b) Hydrogen bonding of 1,1′-methylenebis(imidazolidine-2,4-dione) (1); (c) View of the disordered molecule; symmetry operation (i): −x,y,3/2 − z; hydrogen atoms omitted for clarity.
Figure 2.
(a) Asymmetric unit; (b) Hydrogen bonding of 1,1′-methylenebis(imidazolidine-2,4-dione) (1); (c) View of the disordered molecule; symmetry operation (i): −x,y,3/2 − z; hydrogen atoms omitted for clarity.
Table 2.
Hydrogen bond parameters (Å,°).
Table 2.
Hydrogen bond parameters (Å,°).
| Compound | Interaction | H…A | D…A | D–H…A | Symmetry A |
|---|
| 1 | N2–H…O5 | 1.951(3) | 2.793(5) | 162.5(3) | 1/2 − x,1/2 − y,−1/2 + z |
| N4–H…O7 | 1.797(7) | 2.667(8) | 175.4(3) | 1/2 + x,−1/2 + y,z |
| N6–H…O3 | 1.897(3) | 2.750(5) | 166.2(3) | –x,y,3/2 − z |
| N10–H…O1 | 2.085(3) | 2.927(9) | 162.7(6) | x,y,z |
| 2a | N2–H…O4 | 1.84(2) | 2.707(2) | 166(2) | x,y,−1 + z |
| O7–H…N4 | 2.07(1) | 2.908(2) | 169(1) | 1 − x,y,5/2 − z |
| O6–H…O3 | 1.90(1) | 2.752(1) | 173(2) | x,−y,−1/2 + z |
| 3b | O3–H…O1 | 2.065(14) | 2.9085(16) | 172.8(19) | 1/2 − x,1/2 − y,1/2 + z |
| O3–H…N2 | 1.982(14) | 2.8085(16) | 170.0(18) | x,1 − y,1/2 + z |
| O4–H…O2 | 1.962(14) | 2.7888(15) | 172.9(17) | 1/2 − x,1/2 − y,1/2 + z |
| O4–H…O2 | 1.981(15) | 2.7756(15) | 153.4(16) | 1/2 + x,1/2 − y,1 − z |
2.2. 1,1′-Methylenebis(imidazolidine-2,4-dione), Sodium Salt, 2.5-Hydrate (2a)
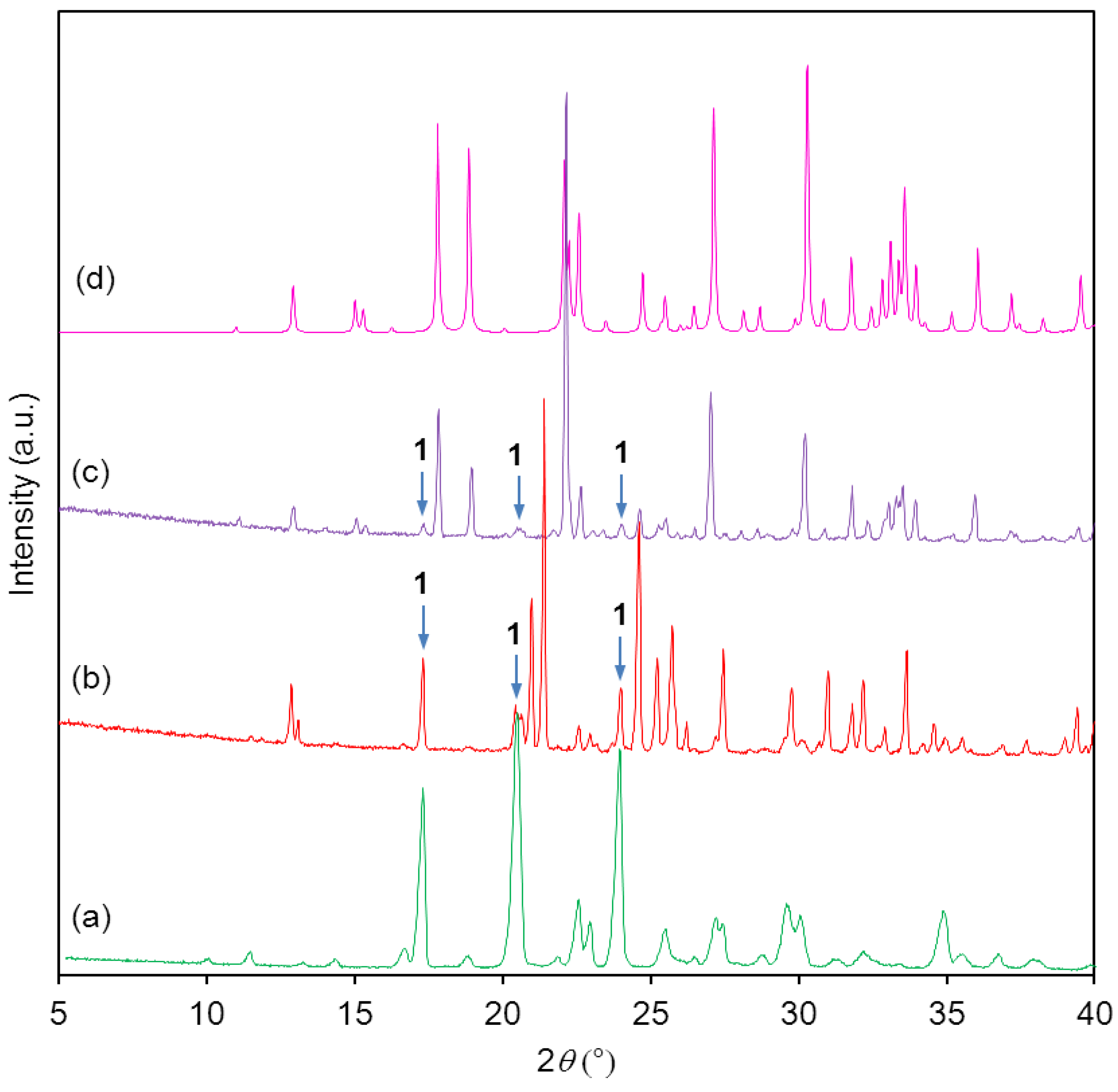
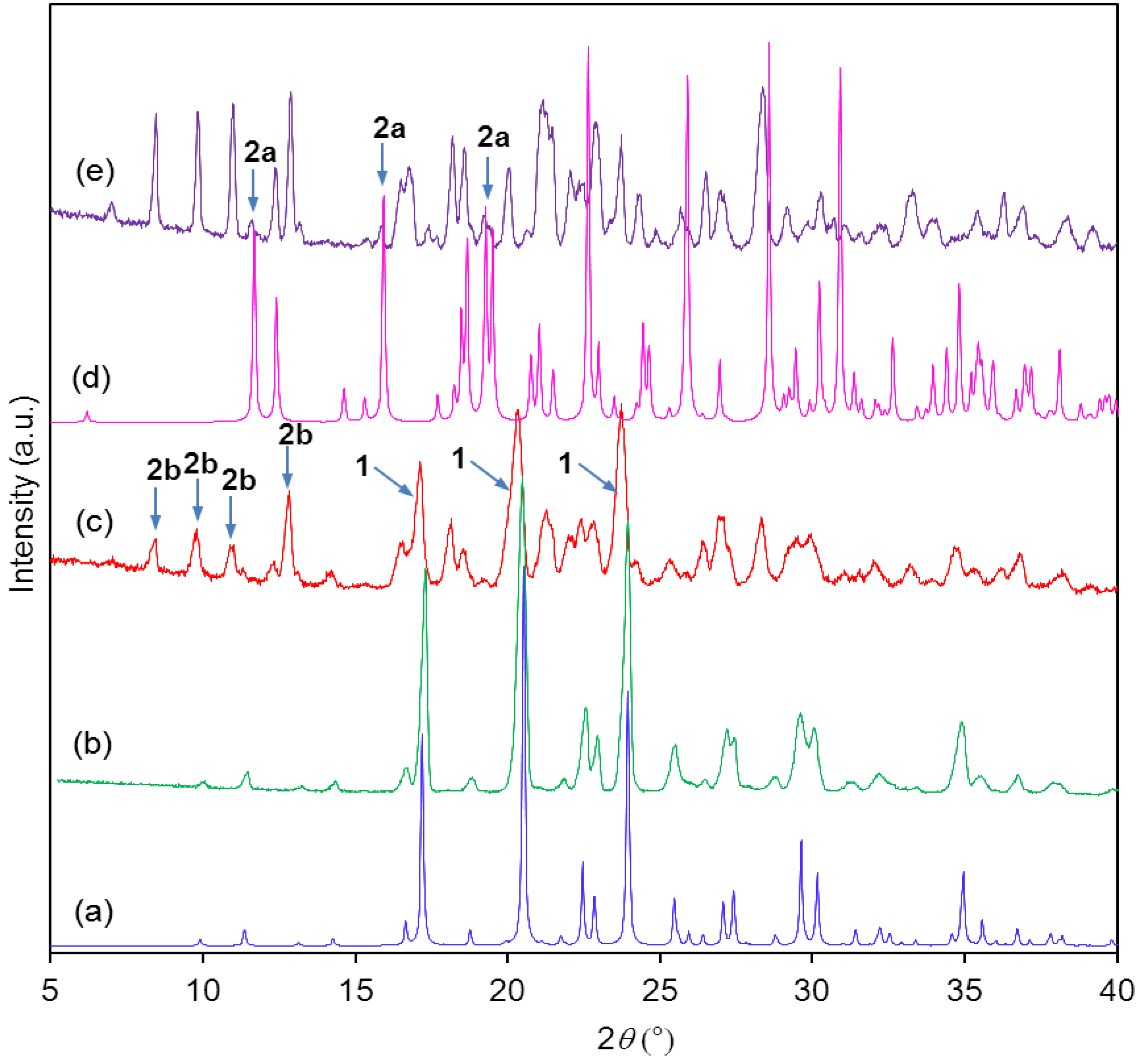
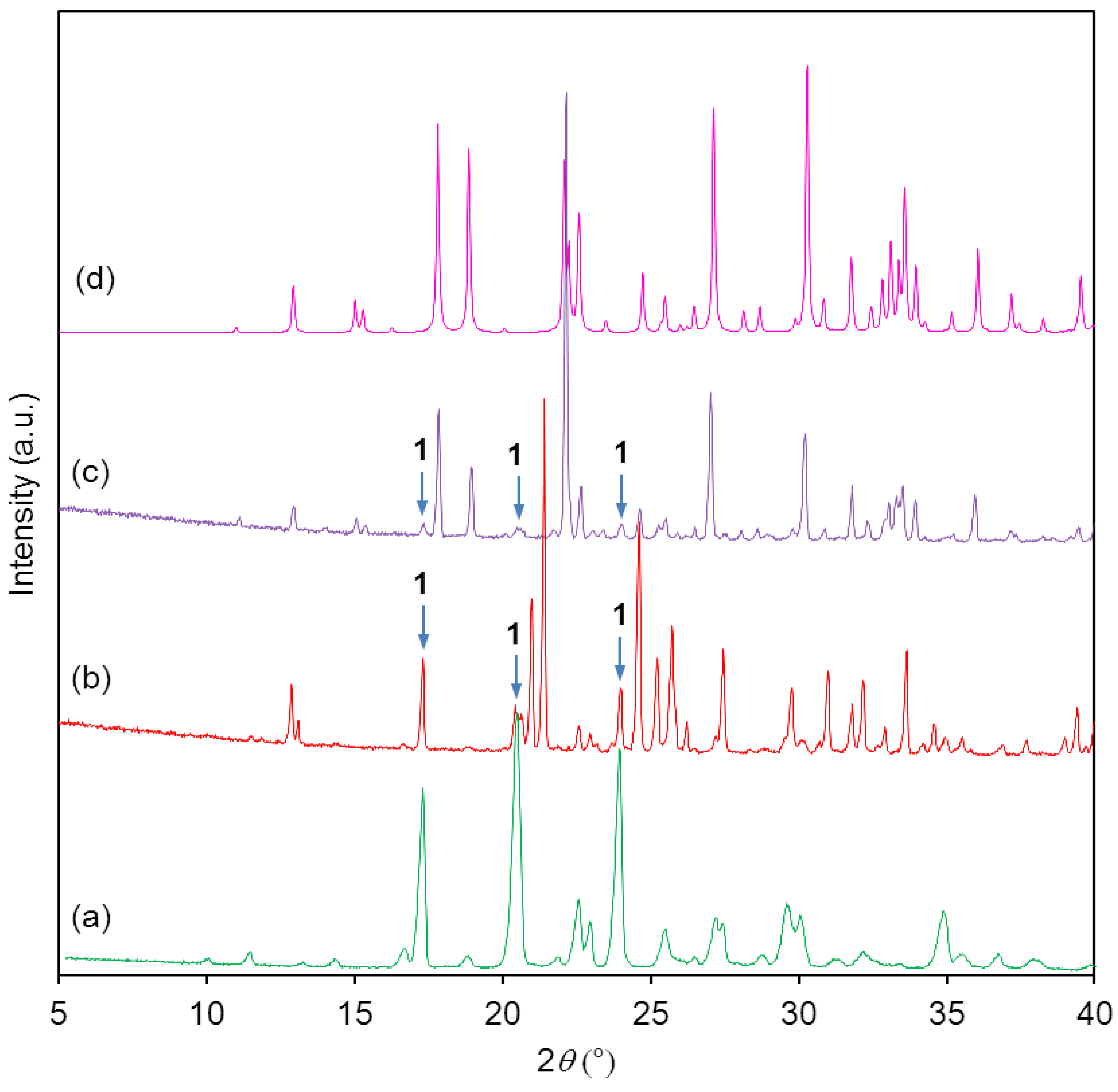
Powder X-ray diffractograms showed that the starting material was pure (
Figure 3a,b). It can be clearly seen that the attempted synthesis of the monosodium salt gave only a mixture of starting material
1 and disodium salt
2b (
Figure 3c). The disodium salt contained traces of the monosodium salt
2a (
Figure 3d,e), no single-crystals were obtained.
Figure 3.
(a) Calculated powder X-ray diffraction (PXRD) of starting material 1; (b) Experimental PXRD of starting material 1; (c) Experimental PXRD of the product from attempted synthesis of mono-sodium salt 2a; (d) Calculated PXRD of monosodium salt 2a; (e) Experimental PXRD of disodium salt 2b. Impurities are indicated by arrows.
Figure 3.
(a) Calculated powder X-ray diffraction (PXRD) of starting material 1; (b) Experimental PXRD of starting material 1; (c) Experimental PXRD of the product from attempted synthesis of mono-sodium salt 2a; (d) Calculated PXRD of monosodium salt 2a; (e) Experimental PXRD of disodium salt 2b. Impurities are indicated by arrows.
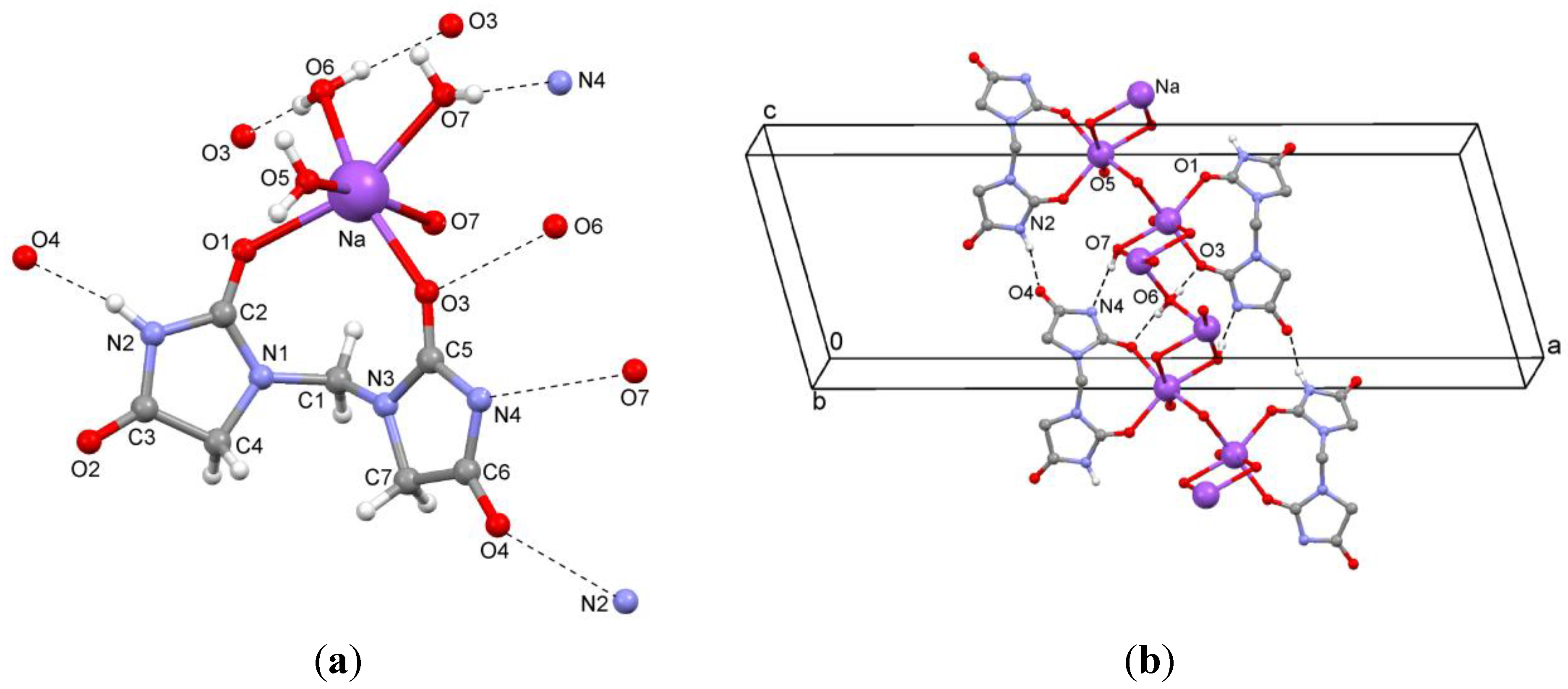
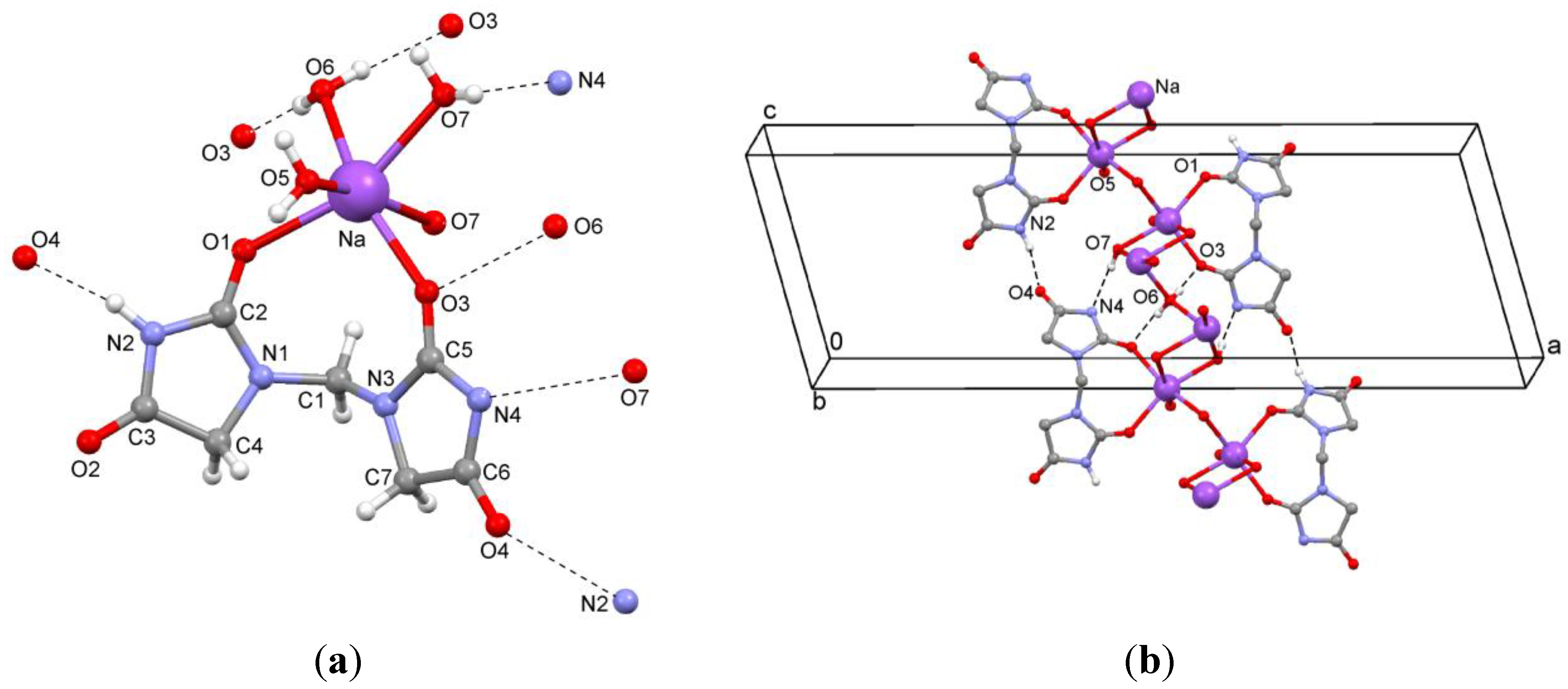
In the crystal structure of
2a, the sodium ions are coordinated to two oxygen atoms of the title ligand and four water molecules (
Figure 4a). The sodium ions are alternately linked by one (O6) or two (O7) μ
2-aqua bridges (
Figure 4b). The O6 atom of the single μ
2-aqua bridge is located on a twofold rotation axis in
b direction. An inversion center is situated in the bis(μ
2-aqua) moiety. Hydrogen bonds between ligands (N–H…O) and between ligand and water molecules (O–H…N and O–H…O) are observed (
Table 2,
Figure 4b). Sodium…oxygen contacts are summarized in
Table 3.
Figure 4.
(a) Coordination environment of the ion pair; (b) Hydrogen bonding of the sodium salt 2a.
Figure 4.
(a) Coordination environment of the ion pair; (b) Hydrogen bonding of the sodium salt 2a.
Table 3.
Metal…oxygen bonds (Å) in salts 2a and 3b.
Table 3.
Metal…oxygen bonds (Å) in salts 2a and 3b.
| Compound | Bond | M…O | Symmetry O |
|---|
| 2a | Na…O1 | 2.433(1) | x,y,z |
| Na…O3 | 2.302(1) | x,y,z |
| Na…O5 | 2.363(1) | x,y,z |
| Na…O6 | 2.381(1) | x,y,z |
| Na…O7 | 2.408(1) | x,y,z |
| Na…O7 | 2.645(1) | 1 − x,−y,2 − z |
| 3b | K…O1 | 2.782(1) | x,y,z |
| K…O1 | 2.737(1) | 1/2 − x,1/2 + y,z |
| K…O3 | 2.703(1) | x,y,z |
| K…O3 | 2.821(1) | 1/2 − x,−1/2 + y,z |
| K…O4 | 2.783(1) | x,y,z |
| K…O4 | 2.782(1) | 1/2 − x,1/2 + y,z |
2.3. 1,1′-Methylenebis(imidazolidine-2,4-dione), Dipotassium Salt, Tetrahydrate (3b)
Powder X-ray diffractograms showed that the bulk of the monopotassium salt
3a contained considerable amounts of starting material
1 (
Figure 5a,b), whereas the bulk of the dipotassium salt
3b contained only traces of
1 (
Figure 5b,c). Single-crystal and bulk material of the dipotassium salt were identical crystalline phases (
Figure 5c,d).
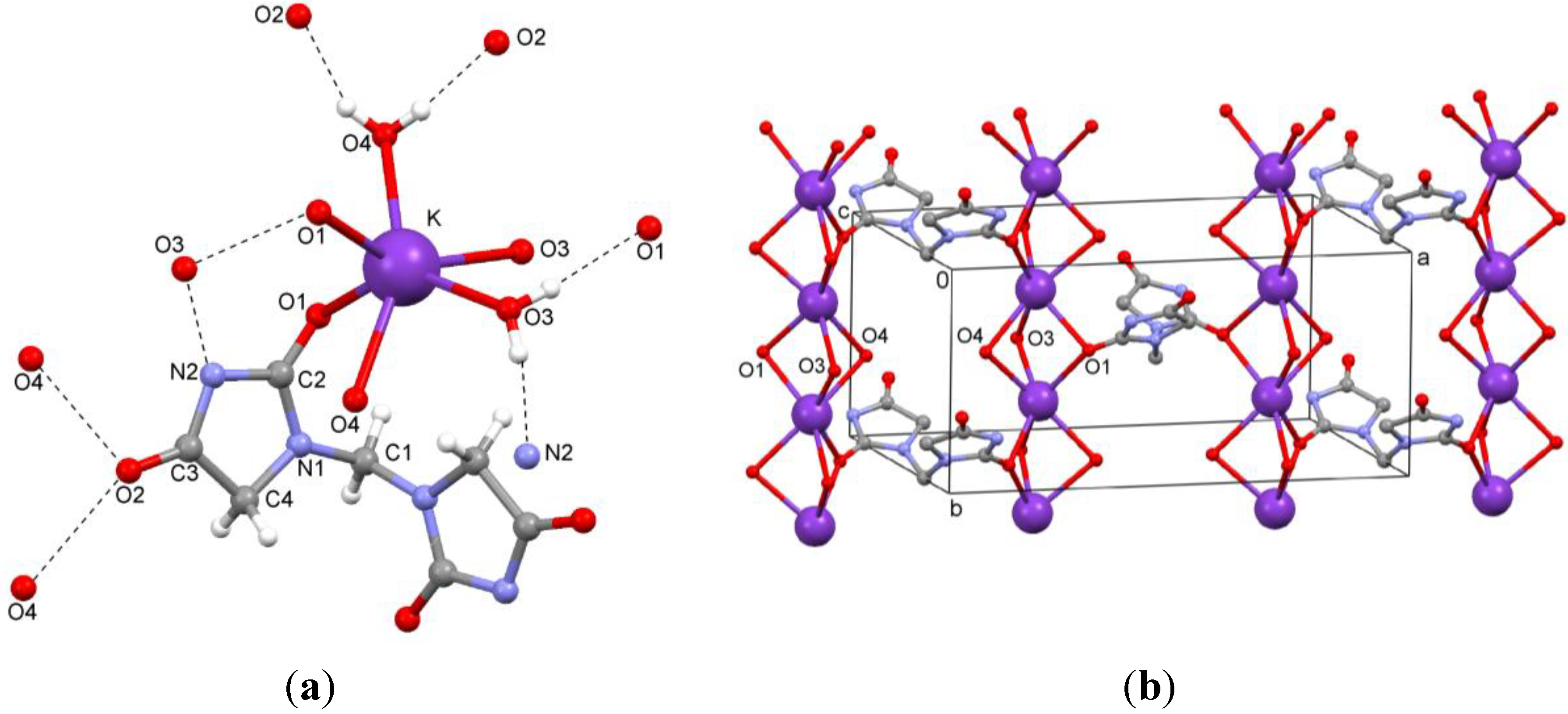
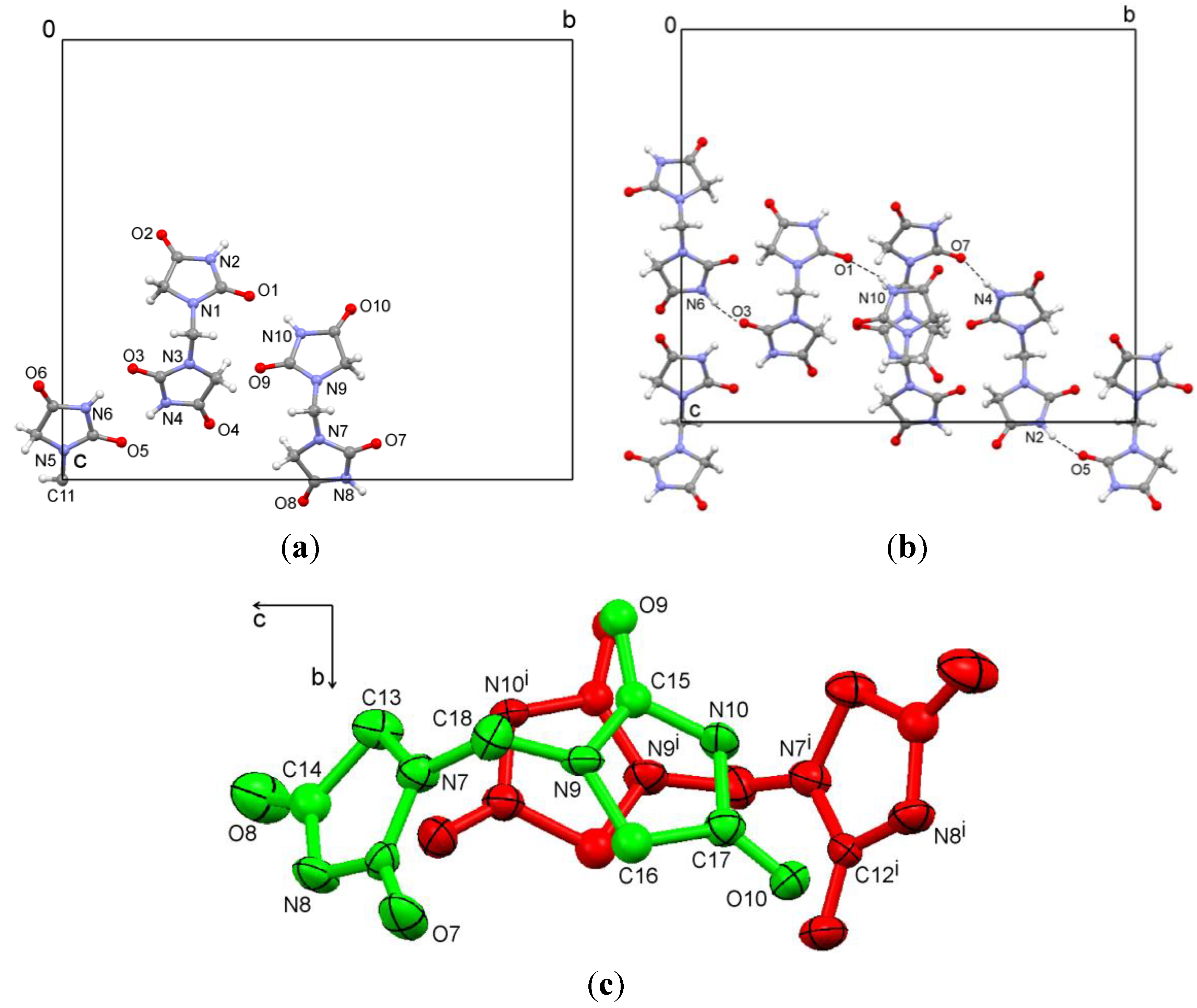
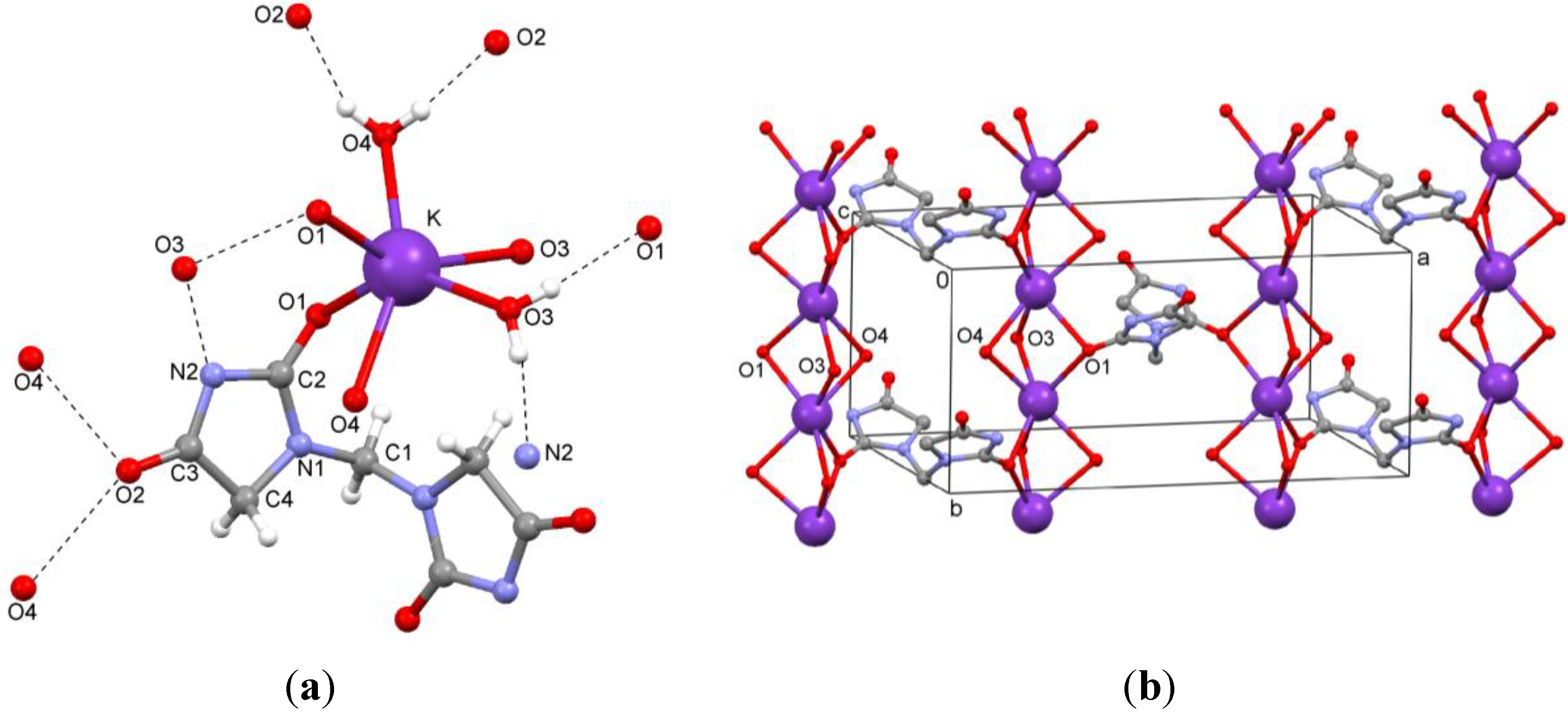
The dipotassium salt
3b is a typical coordination polymer. In the crystal structure of
3b, there is half a ligand molecule in the asymmetric unit, C1 is located on a two-fold rotation axis in the direction of the
b axis. Each potassium ion coordinates to two ligand molecules and four water molecules (
Figure 6a). The potassium atoms are arranged in columns parallel to the
b axis. They are linked by O1 of the ligand and two μ
2-aqua bridges (O3 and O4), as shown in
Figure 6b. Hydrogen bonds between ligand and water molecules (O–H…N and O–H…O) are observed (
Table 2,
Figure 6b). Potassium…oxygen contacts are summarized in
Table 3.
Figure 5.
(a) Experimental PXRD of starting material 1; (b) Experimental PXRD of mono-potassium salt 3a; (c) Experimental PXRD of dipotassium salt 3b; (d) Calculated PXRD of dipotassium salt 3b. Impurities are indicated by arrows.
Figure 5.
(a) Experimental PXRD of starting material 1; (b) Experimental PXRD of mono-potassium salt 3a; (c) Experimental PXRD of dipotassium salt 3b; (d) Calculated PXRD of dipotassium salt 3b. Impurities are indicated by arrows.
Figure 6.
(a) Coordination of the ion pair; (b) Packing of the dipotassium salt 3b.
Figure 6.
(a) Coordination of the ion pair; (b) Packing of the dipotassium salt 3b.
Comparison of experimental and calculated PXRD data (Le Bail fit) of starting material 1 and dipotassium salt 3b are available in the Supporting Information (Figures S1 and S2).
3. Experimental Section
Hydantoin (purity 99%) was purchased from Alfa Aesar (Ward Hill, MA, USA). All other chemicals were obtained from Sigma-Aldrich, St. Louis, MO, USA (European affiliate Steinheim, Germany). Nuclear magnetic resonance (NMR) spectra were recorded with Bruker Avance DPX 300 (Billerica, MA, USA) and Bruker Avance II+ 600 spectrometers. IR spectra were obtained with a Bruker Alpha FT-IR instrument. Water content was determined by coulometric Karl Fischer titration using a Mettler Toledo C20 apparatus (Greifensee, Switzerland).
The powder X-ray diffraction patterns were measured with a Bruker D8 Discover diffractometer using Cu Kα radiation. The patterns were recorded applying a step size of 0.02° with 1 s measuring time per step in the angular range of 2° to 40°.
Single-crystal diffraction intensity data were recorded by ϕ and ω scans with a Nonius KappaCCD diffractometer (Bruker) (for 1) or by ω scans with an Oxford Diffraction Gemini-R Ultra diffractometer (Agilent, Santa Clara, CA, USA) (for 2a and 3b) using Mo Kα radiation. All hydrogen atoms were identified in difference Fourier maps. Secondary CH2 (C–H = 0.99 Å) were positioned geometrically and refined using a riding model. Hydrogen atoms of NH groups as well as of the water molecules were refined with restrained distances of 0.88(1) Å (for N–H) and 0.86(1) Å (for O–H), respectively. Furthermore, an additional H…H restraint (1.35 Å) was introduced to obtain an H–O–H angle of about 104°. The isotropic displacement parameters of all H atoms were set at Uiso(H) = 1.2Ueq(C,N,O). CCDC reference numbers: 950723–950725. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
3.1. Synthesis of 1,1′-Methylenebis(imidazolidine-2,4-dione) (1)
Aqueous formaldehyde (37%, 53.1 mL, 0.510 mol), hydantoin (101.6 g, 1.020 mol), H2O (100 mL) and HCl (37%, 150 mL) were mixed and stirred for 72 h at room temperature. The resulting white precipitate was filtered off, washed with H2O (200 mL), and dried in an oven at 115 °C for 24 h (yield 96.2 g, 89%). Single-crystals were grown by slow evaporation of a solution in H2O/NH3. Melting point (M.p.) 300 °C. 1H NMR (DMSO-d6, 300 MHz): δ 3.93 (s, 4H), 4.74 (s, 2H), 10.84 (s, 2H) ppm. 13C NMR (DMSO-d6, 75 MHz): δ 48.5, 49.2, 157.4, 171.8 ppm. IR (neat): ν 3196 (m), 3074 (w), 1752 (m), 1609 (s), 1474 (m), 1454 (s), 1424 (m), 1376 (s), 1333 (m), 1263 (m), 1210 (s), 1171 (m), 1150 (s), 1098 (m), 984 (w), 961 (m), 896 (m), 843 (w), 760 (m), 714 (s), 696 (s), 639 (s), 599 (s), 572 (s), 444 (m) cm−1.
3.2. Attempted Synthesis of 1,1′-Methylenebis(imidazolidine-2,4-dione), Monosodium Salt (2a)
1,1′-Methylenebis(imidazolidine-2,4-dione) (1) (2.0 g, 9.4 mmol, prepared as described above) was dissolved in a mixture of H2O (5 mL) and ammonia (25% aqueous solution, 2.8 mL, 38 mmol). After addition of NaOH (0.38 g, 9.5 mmol) the solution was stirred for 10 min. Removal of the solvent by means of a rotary evaporator and vacuum-drying for 3 h at room temperature yielded a white paste (2.2 g). A sample of this paste (100 mg) was stirred in EtOH (3 mL) for 7 d at room temperature. The crystalline solid was filtered off and vacuum-dried to yield 81 mg (81%). Onset of decomposition: 193 °C. 1H NMR (D2O, 600 MHz): δ 4.08 (s, 4H), 4.94 (s, 2H) ppm. 13C NMR (D2O, 150 MHz): δ 51.9, 53.4, 167.0, 183.0 ppm. IR (neat): ν 3205 (w), 1753 (m), 1697 (m), 1572 (s), 1474 (m), 1455 (m), 1424 (m), 1400 (m), 1369 (m), 1332 (m), 1213 (s), 1172 (w), 1151 (m), 1099 (w), 996 (w), 962 (w), 897 (w), 857 (w), 793 (w), 764 (m), 697 (m), 672 (m), 641 (m), 626 (m), 600 (m), 575 (m), 445 (m) cm−1.
3.3. Synthesis of 1,1′-Methylenebis(imidazolidine-2,4-dione), Disodium Salt, Tetrahydrate (2b)
1,1′-Methylenebis(imidazolidine-2,4-dione) (1) (95.6 g, 0.451 mol) was suspended in H2O (250 mL), and NaOH (36.1 g, 0.903 mol) was added in portions. The mixture was stirred for 10 min, filtered, and the solvent was removed by means of a rotary evaporator. Vacuum-drying for 8 h at 50 °C yielded a clear paste which solidified after several days (124.0 g, 84%). Thermogravimetry indicated a tetrahydrate. Onset of decomposition: 189 °C. 1H NMR (D2O, 300 MHz): δ 3.93 (s, 4H), 4.90 (s, 2H) ppm. 13C NMR (D2O, 75 MHz): δ 46.4, 49.2, 170.6, 187.0 ppm. IR (neat): ν 3355 (bm), 3010 (bm), 2737 (w), 1763 (w), 1685 (m), 1569 (s), 1460 (m), 1449 (m), 1406 (m), 1265 (m), 1347 (s), 1202 (s), 1154 (s), 1113 (m), 994 (w), 964 (m), 909 (w), 855 (w), 786 (s), 760 (m), 686 (m), 617 (m), 582 (m) cm−1. Surprisingly, single-crystals of the mono-sodium salt 2.5-hydrate 2a were obtained by slow evaporation of a solution in H2O/MeOH.
3.4. Synthesis of 1,1′-Methylenebis(imidazolidine-2,4-dione), Monopotassium Salt (3a)
1,1′-Methylenebis(imidazolidine-2,4-dione) (1) (2.0 g, 9.4 mmol) was dissolved in a mixture of H2O (5 mL) and ammonia (25% aqueous solution, 2.8 mL, 38 mmol). After addition of KOH (0.53 g, 9.4 mmol) the solution was stirred for 10 min. Removal of the solvent by means of a rotary evaporator and vacuum-drying for 3 h at room temperature yielded a white solid (2.35 g, 100%). H2O: 0.42%. M.p. 257 °C (decomposition). 1H NMR (D2O, 300 MHz): δ 4.05 (s, 4H), 4.91 (s, 2H) ppm. 13C NMR (D2O, 75 MHz): δ 51.9, 53.9, 168.1, 184.1 ppm. IR (neat): ν 3208 (w), 1700 (s), 1599 (m), 1575 (m), 1474 (m), 1412 (m), 1349 (m), 1331 (m), 1283 (m), 1210 (s), 1191 (s), 1162 (m), 1117 (m), 948 (m), 902 (w), 857 (w), 790 (m), 766 (s), 717 (m), 681 (m), 648 (m), 610 (s), 589 (m), 573 (m), 440 (m) cm−1.
3.5. Synthesis of 1,1′-Methylenebis(imidazolidine-2,4-dione), Dipotassium Salt, Tetrahydrate (3b)
1,1′-Methylenebis(imidazolidine-2,4-dione) (1) (1.0 g, 0.005 mol) was suspended in H2O (5 mL), and KOH (0.5 g, 0.010 mol) was added to give a clear solution which was stirred for 10 min. The solvent was removed by means of a rotary evaporator. Vacuum-drying for 3 h at room temperature yielded a clear paste which crystallized overnight (yield 1.6 g, 93%). M.p. 65 °C (dec.). 1H NMR (D2O, 300 MHz): δ 3.95 (s, 4H), 4.91 (s, 2H) ppm. 13C NMR (D2O, 75 MHz): δ 43.8, 46.4, 166.1, 182.4 ppm. IR (neat): ν 3443 (m), 3256 (bm), 3122 (bm), 1796 (m), 1674 (m), 1598 (s), 1570 (s), 1469 (m), 1444 (s), 1423 (w), 1399 (m), 1368 (s), 1331 (s), 1267 (m), 1202 (s), 1160 (s), 996 (w), 960 (m), 896 (w), 865 (m), 792 (s), 765 (m), 700 (s), 681 (m), 616 (s), 571 (s), 494 (m), 432 (s) cm−1.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}