
What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice?


Abstract
1. Introduction
2. Materials and Methods
2.1. Crystallographic Structures
2.2. Periodic DFT Calculations
2.3. Phonon Calculations
3. Results
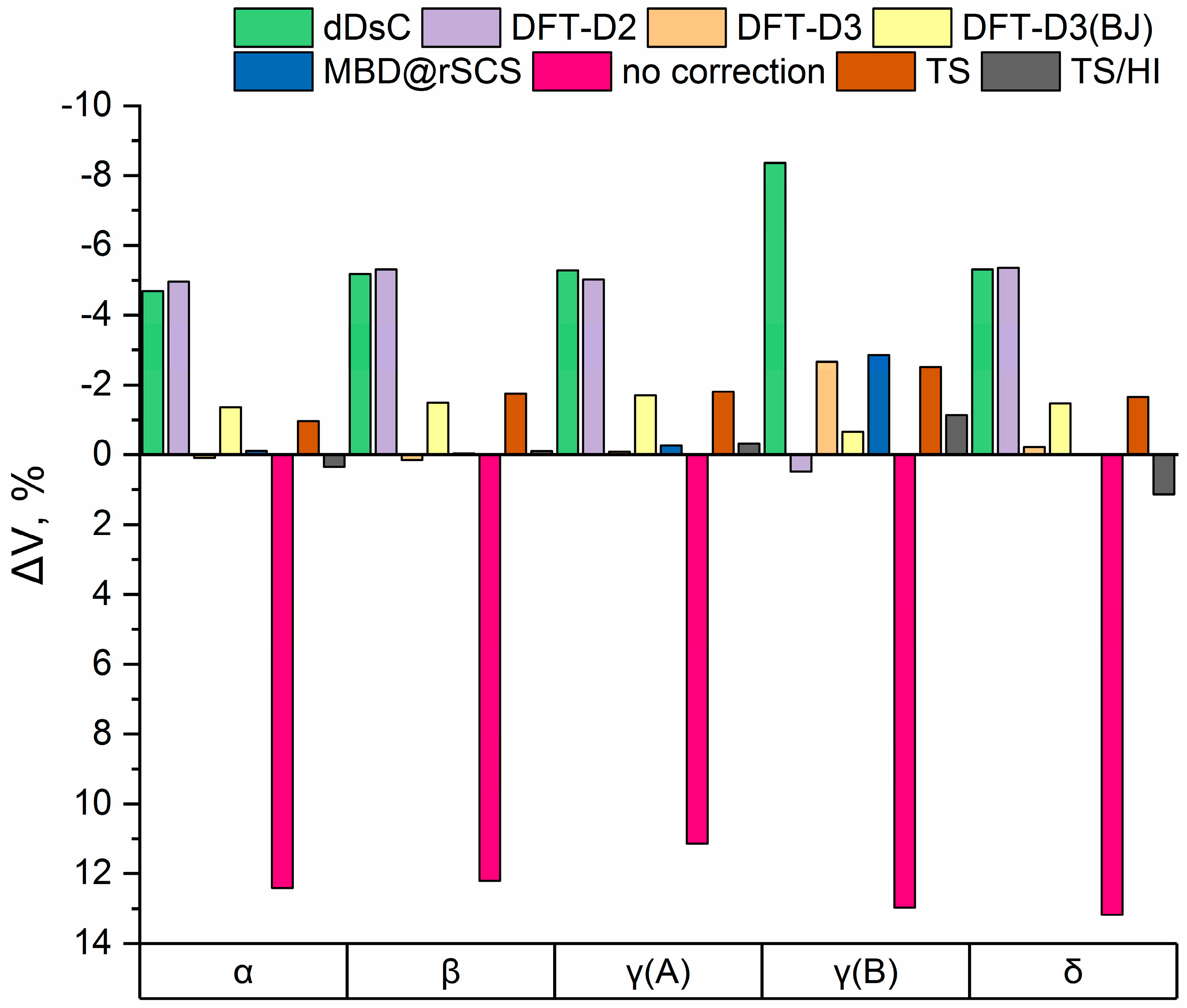
3.1. Electronic Structure Energies and Unit Cell Parameters
3.2. Gibbs Energy Calculations at 0 K
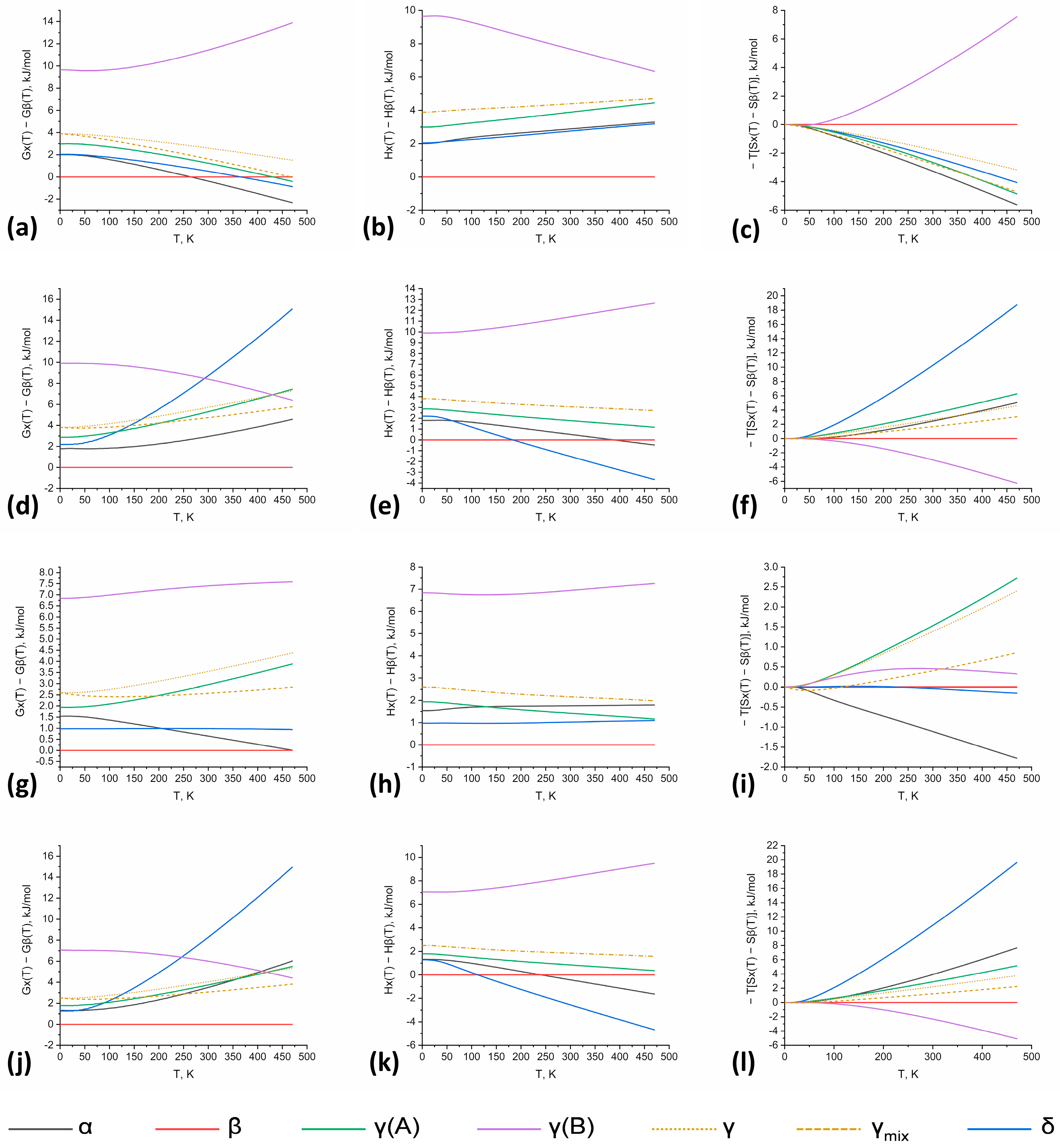
3.3. Thermodynamic Potential Calculations in the 0 K–470 K Temperature Range
4. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FD | Finite difference |
| DFPT | Density functional perturbation theory |
| DFT | Density functional theory |
| PAW | Projector augmented wave (atomic pseudopotentials) |
| D3BJ | Grimme D3 with Becke–Johnson damping (function) |
| PBE | Perdew–Burke–Ernzerhof (functional) |
| rev-vdW-DF2 | Revised van der Waals density (functional) |
| AM05 | Armiento–Mattsson (functional) |
| PBEsol | Revised PBE for solids (functional) |
| PW91 | Perdew–Wang (functional) |
| revPBE | Revised PBE from Zhang and Yang (functional) |
| RPBE | Revised PBE from Hammer et al. (functional) |
| dDsC | dDsC dispersion correction method (dDsC) |
| DFT-D2 | DFT-D2 method of Grimme (dispersion correction) |
| DFT-D3 | DFT-D3 method of Grimme with zero-damping function (dispersion correction) |
| DFT-D3BJ | DFT-D2 method of Grimme with Becke–Johnson damping (dispersion correction) |
| TS | Tkatchenko–Scheffler method (dispersion correction) |
| TS/HI | Tkatchenko–Scheffler method with iterative Hirshfeld partitioning (dispersion correction) |
| MBD@rsSCS | Many-body dispersion energy method (dispersion correction) |
References
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, What It Is and How to Identify It: A Systematic Review. RSC Adv. 2013, 3, 16905. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and Fictions about Polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef] [PubMed]
- Kersten, K.; Kaur, R.; Matzger, A. Survey and Analysis of Crystal Polymorphism in Organic Structures. IUCrJ 2018, 5, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J. Polymorphism of Pharmaceuticals. In Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2020; pp. 342–375. [Google Scholar]
- Tandon, R.; Tandon, N.; Gupta, N.; Gupta, R. Art of Synthesis of Desired Polymorphs: A Review. Asian J. Chem. 2018, 30, 5–14. [Google Scholar] [CrossRef]
- Ainurofiq, A.; Dinda, K.E.; Pangestika, M.W.; Himawati, U.; Wardhani, W.D.; Sipahutar, Y.T. The Effect of Polymorphism on Active Pharmaceutical Ingredients: A Review. Int. J. Res. Pharm. Sci. 2020, 11, 1621–1630. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Feeder, N.; Davey, R.J. Open Questions in Organic Crystal Polymorphism. Commun. Chem. 2020, 3, 142. [Google Scholar] [CrossRef]
- Braga, D.; Casali, L.; Grepioni, F. The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools? Int. J. Mol. Sci. 2022, 23, 9013. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ito, S.; Itai, S.; Yamamoto, K. Physicochemical Properties and Bioavailability of Carbamazepine Polymorphs and Dihydrate. Int. J. Pharm. 2000, 193, 137–146. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Xiao, Y.; Wang, T.; Huang, X. The Effects of Polymorphism on Physicochemical Properties and Pharmacodynamics of Solid Drugs. Curr. Pharm. Des. 2018, 24, 2375–2382. [Google Scholar] [CrossRef]
- Llinàs, A.; Box, K.J.; Burley, J.C.; Glen, R.C.; Goodman, J.M. A New Method for the Reproducible Generation of Polymorphs: Two Forms of Sulindac with Very Different Solubilities. J. Appl. Crystallogr. 2007, 40, 379–381. [Google Scholar] [CrossRef]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Estimation of the Solubility of Metastable Polymorphs: A Critical Review. Cryst. Growth Des. 2018, 18, 7228–7237. [Google Scholar] [CrossRef]
- McGregor, L.; Rychkov, D.A.D.A.; Coster, P.L.P.L.; Day, S.; Drebushchak, V.A.V.A.; Achkasov, A.F.A.F.; Nichol, G.S.G.S.; Pulham, C.R.C.R.; Boldyreva, E.V.E.V. A New Polymorph of Metacetamol. CrystEngComm 2015, 17, 6183–6192. [Google Scholar] [CrossRef]
- Bonilha Dezena, R.M. Ritonavir Polymorphism: Analytical Chemistry Approach to Problem Solving in the Pharmaceutical Industry. Braz. J. Anal. Chem. 2020, 7, 12–17. [Google Scholar] [CrossRef]
- Anwar, J.; Zahn, D. Polymorphic Phase Transitions: Macroscopic Theory and Molecular Simulation. Adv. Drug Deliv. Rev. 2017, 117, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, A.M.; Lampronti, G.I.; Cruz-Cabeza, A.J.; Hunter, C.A.; Sanders, J.K.M. Solvation and Surface Effects on Polymorph Stabilities at the Nanoscale. Chem. Sci. 2016, 7, 6617–6627. [Google Scholar] [CrossRef]
- Kras, W.; Carletta, A.; Montis, R.; Sullivan, R.A.; Cruz-Cabeza, A.J. Switching Polymorph Stabilities with Impurities Provides a Thermodynamic Route to Benzamide Form III. Commun. Chem. 2021, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Brits, M.; Liebenberg, W.; de Villiers, M.M. Characterization of Polymorph Transformations That Decrease the Stability of Tablets Containing the WHO Essential Drug Mebendazole. J. Pharm. Sci. 2010, 99, 1138–1151. [Google Scholar] [CrossRef]
- Ho, R.; Shin, Y.; Chen, Y.; Poloni, L.; Chen, S.; Sheikh, A.Y. Multiscale Assessment of Api Physical Properties in the Context of Materials Science Tetrahedron Concept. In Chemical Engineering in the Pharmaceutical Industry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019; pp. 689–712. ISBN 9781119600800. [Google Scholar]
- Drebushchak, V.A.; McGregor, L.; Rychkov, D.A. Cooling Rate “Window” in the Crystallization of Metacetamol Form II. J. Therm. Anal. Calorim. 2017, 127, 1807–1814. [Google Scholar] [CrossRef]
- Mishra, M.K.; Ramamurty, U.; Desiraju, G.R. Solid Solution Hardening of Molecular Crystals: Tautomeric Polymorphs of Omeprazole. J. Am. Chem. Soc. 2015, 137, 1794–1797. [Google Scholar] [CrossRef]
- Bag, P.P.; Chen, M.; Sun, C.C.; Reddy, C.M. Direct Correlation among Crystal Structure, Mechanical Behaviour and Tabletability in a Trimorphic Molecular Compound. CrystEngComm 2012, 14, 3865. [Google Scholar] [CrossRef]
- Ghosh, S.; Reddy, C.M. Elastic and Bendable Caffeine Cocrystals: Implications for the Design of Flexible Organic Materials. Angew. Chem. Int. Ed. 2012, 51, 10319–10323. [Google Scholar] [CrossRef]
- Raju, K.B.; Ranjan, S.; Vishnu, V.S.; Bhattacharya, M.; Bhattacharya, B.; Mukhopadhyay, A.K.; Reddy, C.M. Rationalizing Distinct Mechanical Properties of Three Polymorphs of a Drug Adduct by Nanoindentation and Energy Frameworks Analysis: Role of Slip Layer Topology and Weak Interactions. Cryst. Growth Des. 2018, 18, 3927–3937. [Google Scholar] [CrossRef]
- Masunov, A.E.; Wiratmo, M.; Dyakov, A.A.; Matveychuk, Y.V.; Bartashevich, E.V. Virtual Tensile Test for Brittle, Plastic, and Elastic Polymorphs of 4-Bromophenyl 4-Bromobenzoate. Cryst. Growth Des. 2020, 20, 6093–6100. [Google Scholar] [CrossRef]
- Domalski, E.S.; Hearing, E.D. Heat Capacities and Entropies of Organic Compounds in the Condensed Phase. Volume III. J. Phys. Chem. Ref. Data 1996, 25, 1. [Google Scholar] [CrossRef]
- De Wit, H.G.; Van Miltenburg, J.; De Kruif, C. Thermodynamic Properties of Molecular Organic Crystals Containing Nitrogen, Oxygen, and Sulphur 1. Vapour Pressures and Enthalpies of Sublimation. J. Chem. Thermodyn. 1983, 15, 651–663. [Google Scholar] [CrossRef]
- De Wit, H.G.; De Kruif, C.; Van Miltenburg, J. Thermodynamic Properties of Molecular Organic Crystals Containing Nitrogen, Oxygen, and Sulfur II. Molar Heat Capacities of Eight Compounds by Adiabatic Calorimetry. J. Chem. Thermodyn. 1983, 15, 891–902. [Google Scholar] [CrossRef]
- De Wit, H.G.M.; Offringa, J.C.A.; De Kruif, C.G.; Van Miltenburg, J.C. Thermodynamic Properties of Molecular Organic Crystals Containing Nitrogen, Oxygen and Sulfur. III. Molar Heat Capacities Measured by Differential Scanning Calorimetry. Thermochim. Acta 1983, 65, 43–51. [Google Scholar] [CrossRef]
- Schnieders, M.J.; Baltrusaitis, J.; Shi, Y.; Chattree, G.; Zheng, L.; Yang, W.; Ren, P. The Structure, Thermodynamics, and Solubility of Organic Crystals from Simulation with a Polarizable Force Field. J. Chem. Theory Comput. 2012, 8, 1721–1736. [Google Scholar] [CrossRef]
- Palmer, D.S.; McDonagh, J.L.; Mitchell, J.B.O.; van Mourik, T.; Fedorov, M.V. First-Principles Calculation of the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules. J. Chem. Theory Comput. 2012, 8, 3322–3337. [Google Scholar] [CrossRef]
- Dybeck, E.C.; Schieber, N.P.; Shirts, M.R. Effects of a More Accurate Polarizable Hamiltonian on Polymorph Free Energies Computed Efficiently by Reweighting Point-Charge Potentials. J. Chem. Theory Comput. 2016, 12, 3491–3505. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Grimme, S. Accurate Modeling of Organic Molecular Crystals by Dispersion-Corrected Density Functional Tight Binding (DFTB). J. Phys. Chem. Lett. 2014, 5, 1785–1789. [Google Scholar] [CrossRef] [PubMed]
- Červinka, C.; Fulem, M.; Stoffel, R.P.; Dronskowski, R. Thermodynamic Properties of Molecular Crystals Calculated within the Quasi-Harmonic Approximation. J. Phys. Chem. A 2016, 120, 2022–2034. [Google Scholar] [CrossRef]
- Bidault, X.; Chaudhuri, S. Improved Predictions of Thermomechanical Properties of Molecular Crystals from Energy and Dispersion Corrected DFT. J. Chem. Phys. 2021, 154, 164105. [Google Scholar] [CrossRef]
- Kapil, V.; Engel, E.A. A Complete Description of Thermodynamic Stabilities of Molecular Crystals. Proc. Natl. Acad. Sci. USA 2022, 119, e2111769119. [Google Scholar] [CrossRef]
- Hunnisett, L.M.; Nyman, J.; Francia, N.; Abraham, N.S.; Adjiman, C.S.; Aitipamula, S.; Alkhidir, T.; Almehairbi, M.; Anelli, A.; Anstine, D.M.; et al. The Seventh Blind Test of Crystal Structure Prediction: Structure Generation Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2024, 80, 517–547. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Reilly, A.M. Sixth Blind Test of Organic Crystal-Structure Prediction Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 776–777. [Google Scholar] [CrossRef]
- Bardwell, D.A.; Adjiman, C.S.; Arnautova, Y.A.; Bartashevich, E.; Boerrigter, S.X.M.; Braun, D.E.; Cruz-Cabeza, A.J.; Day, G.M.; Della Valle, R.G.; Desiraju, G.R.; et al. Towards Crystal Structure Prediction of Complex Organic—A Report on the Fifth Blind Test. Acta Crystallogr. Sect. B Struct. Sci. 2011, 67, 535–551. [Google Scholar] [CrossRef]
- Hoja, J.; Reilly, A.M.; Tkatchenko, A. First-Principles Modeling of Molecular Crystals: Structures and Stabilities, Temperature and Pressure. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1294. [Google Scholar] [CrossRef]
- Hasan, S.; Rulis, P.; Ching, W.Y. First-Principles Calculations of the Structural, Electronic, Optical, and Mechanical Properties of 21 Pyrophosphate Crystals. Crystals 2022, 12, 1139. [Google Scholar] [CrossRef]
- Dubok, A.S.; Rychkov, D. Deformcell: A Python Script to Simplify and Fasten Mechanical Properties Calculations of Molecular Crystals in VASP Package for Research and Teaching Purposes. J. Struct. Chem. 2024, 65, 132571. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density Functional Theory Is Straying from the Path toward the Exact Functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Kamencek, T.; Wieser, S.; Kojima, H.; Bedoya-Martínez, N.; Dürholt, J.P.; Schmid, R.; Zojer, E. Evaluating Computational Shortcuts in Supercell-Based Phonon Calculations of Molecular Crystals: The Instructive Case of Naphthalene. J. Chem. Theory Comput. 2020, 16, 2716–2735. [Google Scholar] [CrossRef]
- Duong, T.C.; Paulson, N.H.; Stan, M.; Chaudhuri, S. An Efficient Approximation of the Supercell Approach to the Calculation of the Full Phonon Spectrum. Calphad 2021, 72, 102215. [Google Scholar] [CrossRef]
- Shang, H.; Carbogno, C.; Rinke, P.; Scheffler, M. Lattice Dynamics Calculations Based on Density-Functional Perturbation Theory in Real Space. Comput. Phys. Commun. 2017, 215, 26–46. [Google Scholar] [CrossRef]
- Running Phonon Calculations. Available online: https://www.tcm.phy.cam.ac.uk/castep/Phonons_Guide/2-sec:examples.html#sec:dfpt-gamma (accessed on 10 March 2025).
- Sholl, D.S.; Steckel, J.A. Density Functional Theory; Wiley: Hoboken, NJ, USA, 2009; ISBN 9780470373170. [Google Scholar]
- Lee, J.G. Computational Materials Science, 2nd ed.; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2016; ISBN 9781315368429. [Google Scholar]
- Martin, R.M. Electronic Structure; Cambridge University Press: Cambridge, UK, 2020; ISBN 9781108555586. [Google Scholar]
- Takaki, Y.; Sasada, Y.; Watanabé, T. The Crystal Structure of α-Pyrazinamide. Acta Crystallogr. 1960, 13, 693–702. [Google Scholar] [CrossRef]
- Rø, G.; Sørum, H. The Crystal and Molecular Structure of β-Pyrazinecarboxamide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 991–998. [Google Scholar] [CrossRef]
- Tamura, C.; Kuwano, H. Crystallographic Data of Carboxylic Acids and Carboxyamides of Picoline and Pyrazine Derivatives. Acta Crystallogr. 1961, 14, 693–694. [Google Scholar] [CrossRef]
- Rø, G.; Sørum, H. The Crystal and Molecular Structure of δ-Pyrazinecarboxamide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 1677–1684. [Google Scholar] [CrossRef]
- Cherukuvada, S.; Thakuria, R.; Nangia, A. Pyrazinamide Polymorphs: Relative Stability and Vibrational Spectroscopy. Cryst. Growth Des. 2010, 10, 3931–3941. [Google Scholar] [CrossRef]
- Castro, R.A.E.; Maria, T.M.R.; Évora, A.O.L.; Feiteira, J.C.; Silva, M.R.; Beja, A.M.; Canotilho, J.; Eusébio, M.E.S. A New Insight into Pyrazinamide Polymorphic Forms and Their Thermodynamic Relationships. Cryst. Growth Des. 2010, 10, 274–282. [Google Scholar] [CrossRef]
- Borba, A.; Albrecht, M.; Gómez-Zavaglia, A.; Suhm, M.A.; Fausto, R. Low Temperature Infrared Spectroscopy Study of Pyrazinamide: From the Isolated Monomer to the Stable Low Temperature Crystalline Phase. J. Phys. Chem. A 2010, 114, 151–161. [Google Scholar] [CrossRef]
- Rajalakshmi, G.; Hathwar, V.R.; Kumaradhas, P. Intermolecular Interactions, Charge-Density Distribution and the Electrostatic Properties of Pyrazinamide Anti-TB Drug Molecule: An Experimental and Theoretical Charge-Density Study. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Ciochoń, P.; Petriĉek, V.; Madsen, A.Ø. Polymorph Stability Prediction: On the Importance of Accurate Structures: A Case Study of Pyrazinamide. Cryst. Growth Des. 2014, 14, 381–388. [Google Scholar] [CrossRef]
- Hoser, A.A.; Rekis, T.; Madsen, A.Ø. Dynamics and Disorder: On the Stability of Pyrazinamide Polymorphs. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2022, 78, 416–424. [Google Scholar] [CrossRef]
- David, S.; Nidhin, P.V.; Srinivasan, P. Ab Initio Prediction of the Polymorphic Structures of Pyrazinamide: A Validation Study. J. Serbian Chem. Soc. 2016, 81, 763–776. [Google Scholar] [CrossRef]
- Li, K.; Gbabode, G.; Vergé-Depré, M.; Robert, B.; Barrio, M.; Itié, J.-P.; Tamarit, J.-L.; Rietveld, I.B. The Pressure–temperature Phase Diagram of Tetramorphic Pyrazinamide. CrystEngComm 2022, 24, 5041–5051. [Google Scholar] [CrossRef]
- Dubok, A.S.; Rychkov, D.A. Relative Stability of Pyrazinamide Polymorphs Revisited: A Computational Study of Bending and Brittle Forms Phase Transitions in a Broad Temperature Range. Crystals 2023, 13, 617. [Google Scholar] [CrossRef]
- Nangia, A.; Srinivasulu, A. CSD Communication (Private Communication); CCDC: Boston, MA, USA, 2005. [Google Scholar] [CrossRef]
- Björkman, T. CIF2Cell: Generating Geometries for Electronic Structure Programs. Comput. Phys. Commun. 2011, 182, 1183–1186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Hamada, I. Van Der Waals Density Functional Made Accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Armiento, R.; Mattsson, A.E. Functional Designed to Include Surface Effects in Self-Consistent Density Functional Theory. Phys. Rev. B 2005, 72, 085108. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Armiento, R.; Paier, J.; Kresse, G.; Wills, J.M.; Mattsson, T.R. The AM05 Density Functional Applied to Solids. J. Chem. Phys. 2008, 128, 084714. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Armiento, R. Implementing and Testing the AM05 Spin Density Functional. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 155101. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Corminboeuf, C. A Generalized-Gradient Approximation Exchange Hole Model for Dispersion Coefficients. J. Chem. Phys. 2011, 134, 044117. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Corminboeuf, C. Comprehensive Benchmarking of a Density-Dependent Dispersion Correction. J. Chem. Theory Comput. 2011, 7, 3567–3577. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Tkatchenko, A.; DiStasio, R.A.; Car, R.; Scheffler, M. Accurate and Efficient Method for Many-Body van Der Waals Interactions. Phys. Rev. Lett. 2012, 108, 236402. [Google Scholar] [CrossRef]
- Ambrosetti, A.; Reilly, A.M.; DiStasio, R.A.; Tkatchenko, A. Long-Range Correlation Energy Calculated from Coupled Atomic Response Functions. J. Chem. Phys. 2014, 140, 18A508. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Bučko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Improved Density Dependent Correction for the Description of London Dispersion Forces. J. Chem. Theory Comput. 2013, 9, 4293–4299. [Google Scholar] [CrossRef] [PubMed]
- Bučko, T.; Lebègue, S.; Ángyán, J.G.; Hafner, J. Extending the Applicability of the Tkatchenko-Scheffler Dispersion Correction via Iterative Hirshfeld Partitioning. J. Chem. Phys. 2014, 141, 034114. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Togo, A. First-Principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Borioni, J.L.; Puiatti, M.; Vera, D.M.A.; Pierini, A.B. In Search of the Best DFT Functional for Dealing with Organic Anionic Species. Phys. Chem. Chem. Phys. 2017, 19, 9189–9198. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Abburu, S.; Alsberg, B.K. Can Chemometrics Be Used to Guide the Selection of Suitable DFT Functionals? Chemom. Intell. Lab. Syst. 2015, 142, 87–94. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Grimme, S. Organic Crystal Polymorphism: A Benchmark for Dispersion-Corrected Mean-Field Electronic Structure Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 502–513. [Google Scholar] [CrossRef]
- Terentjev, A.V.; Constantin, L.A.; Pitarke, J.M. Dispersion-Corrected PBEsol Exchange-Correlation Functional. Phys. Rev. B 2018, 98, 214108. [Google Scholar] [CrossRef]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the Performance of Recent Density Functionals for Bulk Solids. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 155107. [Google Scholar] [CrossRef]
- Beran, G.J.O. Modeling Polymorphic Molecular Crystals with Electronic Structure Theory. Chem. Rev. 2016, 116, 5567–5613. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.; Day, G.M. Static and Lattice Vibrational Energy Differences between Polymorphs. CrystEngComm 2015, 17, 5154–5165. [Google Scholar] [CrossRef]
- Fedorov, A.Y.; Rychkov, D.A.; Losev, E.A.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V. Effect of Pressure on Two Polymorphs of Tolazamide: Why No Interconversion? CrystEngComm 2017, 19, 2243–2252. [Google Scholar] [CrossRef]
- Dolgonos, G.A.; Hoja, J.; Boese, A.D. Revised Values for the X23 Benchmark Set of Molecular Crystals. Phys. Chem. Chem. Phys. 2019, 21, 24333–24344. [Google Scholar] [CrossRef]
- O’Connor, D.; Bier, I.; Hsieh, Y.-T.; Marom, N. Performance of Dispersion-Inclusive Density Functional Theory Methods for Energetic Materials. J. Chem. Theory Comput. 2022, 18, 4456–4471. [Google Scholar] [CrossRef]
- Li, K.; Gbabode, G.; Barrio, M.; Tamarit, J.-L.; Vergé-Depré, M.; Robert, B.; Rietveld, I.B. The Phase Relationship between the Pyrazinamide Polymorphs α and γ. Int. J. Pharm. 2020, 580, 119230. [Google Scholar] [CrossRef]
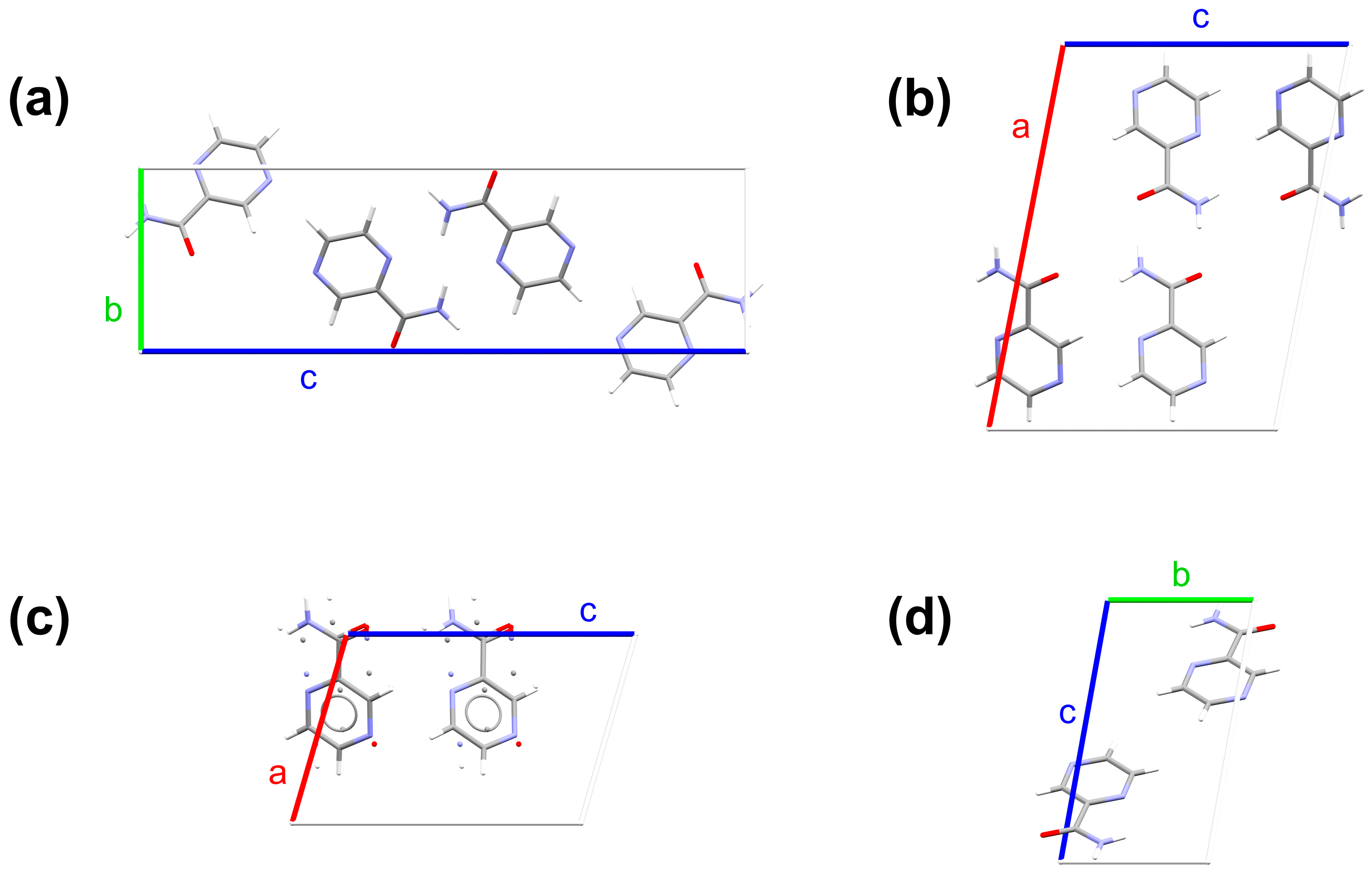


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | VASP (5.4.4/6.4.3) | CASTEP (23.1.1) | Quantum Espresso (7.3) | ABINIT (10.2.3) |
|---|---|---|---|---|
| DFPT implementation | q = 0 | q = 0 and q ≠ 0 | q = 0 and q ≠ 0 | q = 0 and q ≠ 0 |
| Calculation Method | α | β | γ(A) | γ(B) | γ | δ |
|---|---|---|---|---|---|---|
| PBE-D3BJ, kJ/mol * | 2.7 (III) | 0 (I) | 3.1 | 10.0 | 4.1 (IV) | 2.1 (II) |
| rev-vdW-DF2, kJ/mol * | 2.1 (III) | 0 (I) | 2.0 | 7.0 | 2.6 (IV) | 1.1 (II) |
| Calculation Method | A | Β | γ(A) | γ(B) | γ | δ |
|---|---|---|---|---|---|---|
| FD/PBE-D3BJ supercell, kJ/mol | 2.0 (II–III) | 0 (I) | 3.1 | 9.6 | 4.0 (IV) | 2.0 (II–III) |
| FD/PBE-D3BJ primitive cell, kJ/mol | 1.8 (II) | 0 (I) | 2.9 | 9.9 | 3.8 (IV) | 2.2 (III) |
| DFPT/rev-vdW-DF2 supercell, kJ/mol | 1.5 (III) | 0 (I) | 1.9 | 6.8 | 2.6 (IV) | 1.0 (II) |
| DFPT/rev-vdW-DF2 primitive cell, kJ/mol | 1.3 (II–III) | 0 (I) | 1.8 | 7.1 | 2.5 (IV) | 1.3 (II–III) |
| FD/rev-vdW-DF2 primitive cell, kJ/mol | 1.3(II) | 0 (I) | 1.9 | 7.2 | 2.6 (IV) | 1.5 (III) |
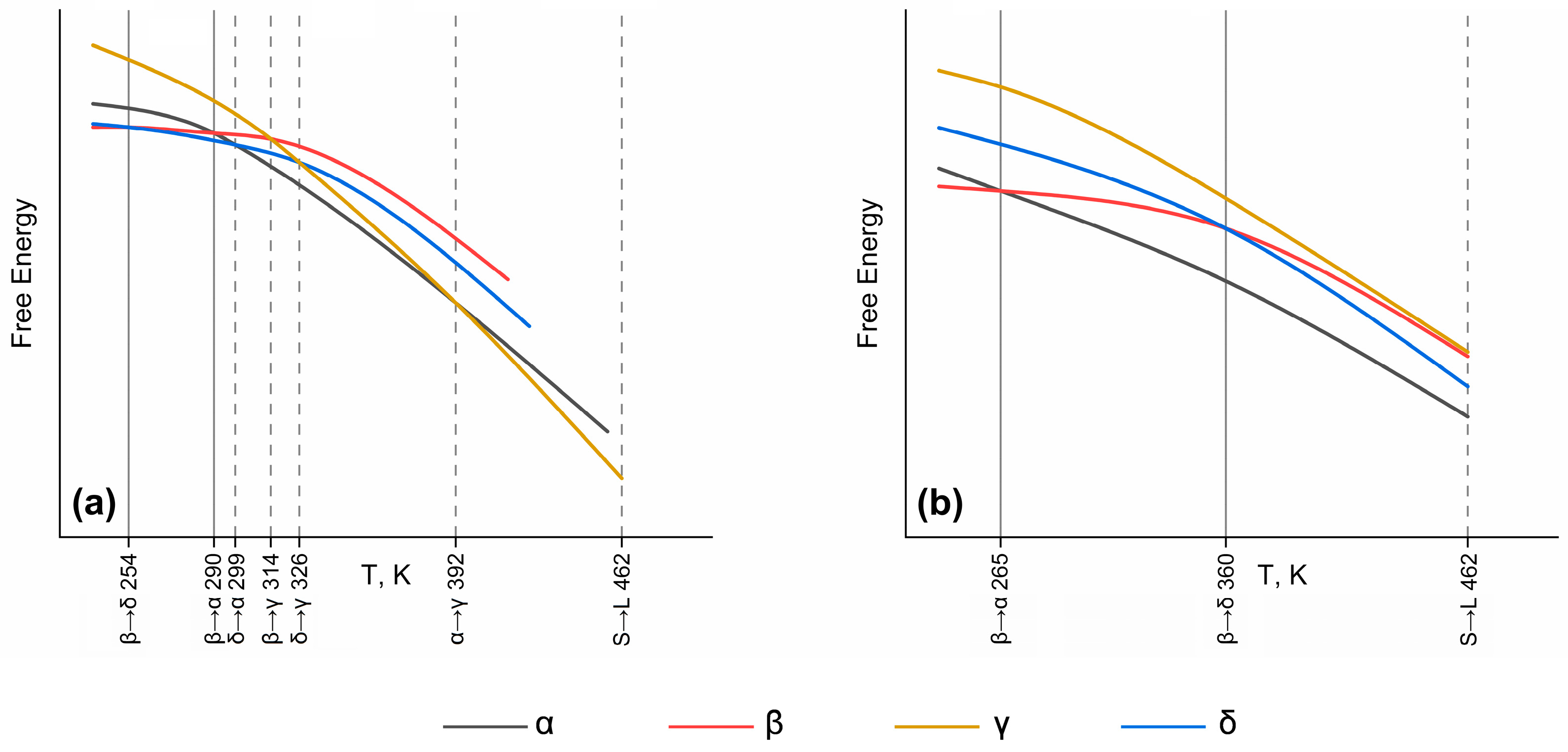
| Method | Δ → α | Β → γ | Δ → γ | A → γ |
|---|---|---|---|---|
| 1 Ttrans, K | 299 | 314 | 326 | 399 |
| 1 Exp. ΔH, kJ/mol | 0.4 | 2.2 | 2.1 | 1.6 |
| FD/PBE-D3BJ supercell, kJ/mol | 0.2 | –4.5 | 1.7 | 1.6 |
| FD/PBE-D3BJ primitive cell, kJ/mol | 2.0 | 3.0 | 4.9 | 2.9 |
| DFPT/rev-vdW-DF2 supercell, kJ/mol | 0.7 | 2.1 | 1.1 | 0.3 |
| DFPT/rev-vdW-DF2 primitive cell, kJ/mol | 2.1 | 1.8 | 4.7 | 2.8 |
| FD/rev-vdW-DF2 primitive cell, kJ/mol | 1.8 | 1.9 | 4.6 | 3.1 |
| FD/PBE-D3BJ Supercell | FD/PBE-D3BJ Primitive Cell | DFPT/Rev-Vdw-DF2 Supercell | DFPT/Rev-vdW-DF2 Primitive Cell | FD/Rev-Vdw-DF2 Primitive Cell | |
| FD/PBE-D3BJ supercell | 1 | 0.19 | 0.85 | −0.26 | −0.16 |
| FD/PBE-D3BJ primitive cell | 0.19 | 1 | 0.18 | 0.90 | 0.91 |
| DFPT/rev-vdW-DF2 supercell | 0.85 | 0.18 | 1 | −0.24 | −0.24 |
| DFPT/rev-vdW-DF2 primitive cell | −0.26 | 0.90 | −0.24 | 1 | 0.98 |
| FD/rev-vdW-DF2 primitive cell | −0.16 | 0.91 | −0.24 | 0.98 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubok, A.S.; Rychkov, D.A. What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals 2025, 15, 274. https://doi.org/10.3390/cryst15030274
Dubok AS, Rychkov DA. What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals. 2025; 15(3):274. https://doi.org/10.3390/cryst15030274
Chicago/Turabian StyleDubok, Aleksandr S., and Denis A. Rychkov. 2025. "What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice?" Crystals 15, no. 3: 274. https://doi.org/10.3390/cryst15030274
APA StyleDubok, A. S., & Rychkov, D. A. (2025). What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals, 15(3), 274. https://doi.org/10.3390/cryst15030274