Abstract
We have synthesized novel cobalt(II) and nickel(II) pincer ligand complexes containing novel tridentate ligand precursors that coordinate via oxygen, nitrogen, and oxygen donor atoms. The novel tridentate ONO ligands, which are neutral, incorporate a carbonyl-substituted imidazole functionality and contain R groups of ethyl, isopropyl, or tert-butyl. The ligand precursors were thoroughly characterized using NMR spectroscopy, ESI-MS, and IR spectroscopy. The metal complexes were thoroughly characterized using single crystal X-ray diffraction, elemental analysis, ESI-MS, and cyclic voltammetry. The nickel(II) and cobalt(II) complexes with ethyl, isopropyl, and t-butyl wingtip groups had a pseudo-octahedral geometry about the metal center. The nickel(II) complex with R = isopropyl had a monoclinic lattice with C121 space group (a = 21.7639(8); b = 11.0649(5); c = 10.9225(4); alpha = 90.0 degrees; beta = 90.609(3) degrees; gamma = 90.0 degrees). The cobalt(II) complex with R = ethyl had a monoclinic lattice with P21/n space group (a = 17.7907(7); b = 21.5278(6); c = 21.8597(7); alpha = 90.0 degrees; beta = 95.063(3) degrees; gamma = 90.0 degrees). The cobalt(II) complexes were paramagnetic with μeff = 1.59 BM (R = ethyl) and 6.67 BM (R = t-butyl). The nickel(II) complex was paramagnetic with μeff = 2.59 BM. The ligand precursors and metal complexes are redox-active.
1. Introduction
Pincer ligands, coined by van Koten in 1989, are tridentate ligands that occupy adjacent binding sites of a metal center and adopt a meridional or facial geometry [1,2]. Complexes with pincer ligands have been prepared with a variety of metal centers to be utilized as catalysts [3,4]. Pincer ligands are suitable for catalysis because they are likely to prevent dimerization of the complexes, which can slow down the catalytic capability of a complex [5]. Carbon, oxygen, nitrogen, phosphorus, and sulfur are all common donor atoms for pincer ligands [1].
We have previously prepared pincer ligand precursors with SNS donor atoms (sulfur, nitrogen, and sulfur donors), shown in Figure 1, due to their increased complex stability [5,6,7]. The SNS ligands precursors have modeled the structure and reactivity of the zinc active site in LADH, liver alcohol dehydrogenase [6,7,8,9]. The pincer ligands were then metalated with zinc(II) chloride to prepare zinc(II) model complexes of LADH [6,7]. The complex replicated the active site of LADH as two flanking sulfur atoms replicate the coordination to two cysteine sulfur atoms and the nitrogen replicates a histidine coordination in LADH. Similar pincer ligand precursors have since been metalated with copper(II), nickel(II), and cobalt(II) to form three and five coordinate complexes [10,11,12,13,14,15,16] (Figure 2). These complexes have shown reduction activity and it is hypothesized that the redox capability of the ligand may aid during electrocatalysis.
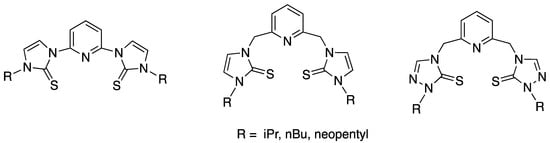
Figure 1.
The SNS pincer ligand precursors previously prepared [6,7].
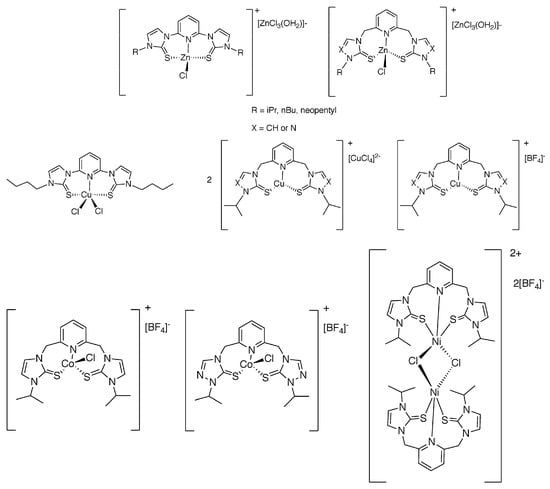
Figure 2.
Previously prepared SNS zinc(II), copper(II), copper(I), cobalt(II), and nickel(II) complexes [6,7,9,10,11,12,13,14,15,16].
We are now interested in utilizing oxygen rather than sulfur donor atoms in the preparation of the pincer ligand precursor. This substitution comes from hard-soft chemistry, as oxygen is smaller and harder than sulfur, so it is more likely to coordinate with the harder metal centers of the first-row transition metals. This change in coordination could also affect the catalytic activity of the metal complex since the metal–ligand bond may be stronger. Others have reported ONO pincer complexes previously. Most of the pincer ligand precursors have anionic charges [17,18,19,20,21,22,23,24,25,26,27,28,29]. One example of a vanadium complex with a neutral ONO pincer ligand has been reported [30].
We are interested in gaining knowledge in the coordination chemistry of ONO pincer ligands and how variability in the wingtip groups may affect the coordination geometry about the metal center. Variations in the coordination chemistry could produce differences in the catalytic ability of these complexes. In this article, we report the preparation and characterization of ONO pincer ligand precursors and metal complexes containing ethyl, isopropyl, and t-butyl wingtip groups.
2. Materials and Methods
2.1. General Procedures
Reagents were purchased from commercial sources and were used as received, except where otherwise noted. Tetrahydrofuran was dried using a MBraun Solvent Purification system. The following compounds were purchased from Acros Organics or Fisher Scientific: sodium hydride, isopropyl isocyanate, t-butyl isocyanate, ethyl isocyanate, ethyl acetate, methanol, hydrochloric acid (12 M), and aminoacetaldehyde diethyl acetal. The following reactant was purchased from TCI America: 2,6 bis(bromo)methylpyridine. Acetonitrile used in electrochemical experiments was freshly degassed before use. Tetrabutylammonium tetrafluoroborate [NBu4][BF4], purchased from Acros Organics, was used as received before electrochemical experiments.
NMR spectra were recorded at 20 °C on a JEOL spectrometer at 400 MHz (1H NMR) and 100 MHz (13C{1H} NMR) and referenced to CDCl3 (δ in ppm, J in Hz). Elemental analyses were performed by Robertson Microlit Laboratories (Ledgewood, NJ, USA). Electrospray ionization mass spectrometry (ESI-MS) was performed using a direct flow injection of 5 μL on an Agilent 6550A QTOF instrument in both positive and negative ion modes. Optimized ESI-MS conditions were 3000 kV capillary, 10 V cone, and 120 °C source temperature. The concentrations of sample used for ESI-MS were 3 to 5 mg/mL. UV-Visible spectra were collected on a Cary 100 UV-Visible Spectrometer. IR spectra were collected using a Bruker Alpha FT-IR with an ATR accessory. The Evans method was used for magnetic susceptibility measurements [31]. Cyclic voltammetry measurements were obtained using a Gamry Electronics Potentiostat.
Single crystal X-ray analyses were collected on an XtaLAB Synergy diffractometer or Rigaku MicroMax-007HF diffractometer. The data were refined and solved using Olex 2 and ShelXT [32,33,34].
Full details of the X-ray structure determination are included in the CIF as Supporting Information. The Cambridge Structure Database Deposition numbers are 2402484 and 2402485. The X-ray structure data for one complex, with Deposition number 2402486, is reported in CSD Communications [35]. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif (accessed on 9 February 2025). Additional characterization data is presented in the Supporting Information (SI).
2.2. General Procedures for Ligand Syntheses and Metal Complexes
The synthesis of each metal complex begins with a three-step synthesis of the ONO ligand precursor, as shown in Scheme 1. In step 1, amino acetaldehyde diethyl acetal is reacted with an isocyanate of a varying R group. In step 2, the urea-like product of step 1 is then cyclized under acidic conditions [36]. The step 2 product is reacted with the strong base sodium hydride and 2,6-dibromometylpyridine to create the final ligand precursor. After each step, the product is characterized using 1H, 13C{1H}, HSQC, and COSY NMR spectroscopy. Once the ligand precursor has been synthesized, the ligand is metalated with various first-row transition metals to create the desired metal complex.
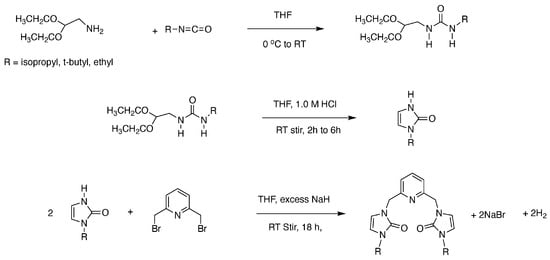
Scheme 1.
Preparation of ONO pincer ligand precursors.
2.3. Synthesis of Ligand Precursors
2.3.1. Preparation of 1-(2,2-Diethoxyethyl)-3-isopropylurea
A two-necked 50 mL round-bottomed flask was acquired and 10 mL of dry THF, obtained from the solvent system, was added. Next, aminoacetaldehyde diethyl acetal (7.189 g, 53.98 mmol) was massed and then added to the round-bottomed flask via syringe and left to stir for 5 min in an ice bath to cool it to 0 °C. Once the solution had become thoroughly mixed, isopropyl isocyanate (4.603 g, 54.09 mmol) was dissolved in 15 mL of THF in a pear-shaped flask and then added dropwise via syringe to the round-bottomed flask while stirring. Finally, the pear-shaped flask was washed with 2 mL of dry THF, transferred to the two-neck flask, and removed from the ice bath to continue reacting at room temperature. After 15 min, the reaction was complete and transferred to a one-neck round-bottomed flask and the solvent was evaporated under reduced pressure. This was analyzed via 1H, 13C{1H} HSQC, and COSY NMR. Mass of product: 11.74 g; Percent yield: 99.63%.
1H NMR (CDCl3, 400 MHz) δ 4.840 (broad s, 1H, NH); 4.770 (m, 1H, NH); 4.469 (m, 1H, CH); 3.833 (septet, 1H, isopropyl CH); 3.694 (m, 2H, CH2), 3.538 (m, 2H, CH2); 3.264 (m, 2H, CH2); 1.199 (m, 6H, CH3); 1.124 (m, 6H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 158.077 (C=O); 102.123 (CH); 63.174 (CH2); 43.410 (CH2); 42.250 (CH, isopropyl); 23.521 (CH3); 15.457 (CH3).
ESI-MS (Positive Ion Mode) m/z: 241.15 (M + Na)
ATR-IR (frequency, intensity): 3356.13 cm−1 (w); 3320.19 cm−1 (W); 2975.99 cm−1 (m); 2930.07 cm−1 (w); 2874.54 cm−1 (w); 1625.12 cm−1 (s); 1561.37 cm−1 (s); 1456.79 cm−1 (w); 1428.47 cm−1 (w); 1372.45 cm−1 (m); 1339.45 cm−1 (w); 1321.44 cm−1 (w); 1247.07 cm−1 (m); 1195.78 cm−1 (m); 1123.83 cm−1 (s); 1089.73 cm−1 (w); 1057.66 cm−1 (vs); 1024.92 cm−1 (m); 963.02 cm−1 (w); 926.41 cm−1 (w); 902.98 cm−1 (w); 850.61 cm−1 (w); 818.55 cm−1 (w); 653.29 cm−1 (m); 561.16 cm−1 (w); 523.20 cm−1 (w); 477.63 cm−1 (w); 451.40 cm−1 (w); 418.22 cm−1 (w).
2.3.2. Preparation of 1-(2,2-Diethoxyethyl)-3-t-butylurea
A two-necked 50 mL round-bottomed flask was acquired and aminoacetaldehyde diethyl acetal (6.362 g, 47.76 mmol) was added in 10 mL of THF, then cooled. Once the solution was cooled to 0 °C, t-butyl isocyanate (4.341 g, 43.78 mmol) was dissolved in 15 mL of THF in a pear-shaped flask and then added dropwise via syringe to the round-bottomed flask while stirring. Finally, the pear-shaped flask was washed with 2 mL of dry THF and the two-neck flask was removed from the ice bath to continue reacting at room temperature. After 15 min, the reaction was complete and transferred to a one-neck round-bottomed flask. The solvent was then evaporated under reduced pressure. Mass of product: 9.982 g; Percent yield: 98.14%
1H NMR (CDCl3, 400 MHz) δ 5.242 (m, 2H, NH); 4.383 (m, 1H, CH); 3.609 (m, 2H, CH2); 3.454 (m, 2H, CH2); 3.158 (m, 2H, CH2); 1.214 (m, 9H, CH3); 1.110 (m, 6H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 158.12 (C=O); 102.083 (CH2); 67.855 (CH); 62.834 (CH2); 49.935 (CH2); 42.854 (t-butyl C); 29.476 (CH3); 15.284 (CH3).
ESI-MS (Positive Ion Mode) m/z: 255.1684 (M + Na)
ATR-IR (frequency, intensity): 3334.29 cm−1 (w); 2976.89 cm−1 (w); 2962.38 cm−1 (w); 2932.99 cm−1 (w); 2869.78 cm−1 (w); 1627.32 cm−1 (s); 1560.88 cm−1 (vs); 1482.91 cm−1 (w); 1450.54 cm−1 (w); 1424.06 cm−1 (w); 1388.78 cm−1 (w); 1360.72 cm−1 (w); 1343.62 cm−1 (w); 1280.88 cm−1 (s); 1244.07 cm−1 (w); 1219.54 cm−1 (m); 1133.16 cm−1 (s); 1061.80 cm−1 (vs); 1019.03 cm−1 (s); 948.07 cm−1 (w); 928.70 cm−1 (w); 903.15 cm−1 (w); 845.29 cm−1 (w); 777.17 cm−1 (w); 626.77 cm−1 (m); 562.37 cm−1 (w); 515.95 cm−1 (w); 487.05 cm−1 (w); 425.40 cm−1 (w); 407.15 cm−1 (w).
2.3.3. Preparation of 1-(2,2-Diethoxyethyl)-3-ethylurea
A two-necked 50 mL round-bottomed flask was acquired and aminoacetaldehyde diethyl acetal (8.895 g, 66.78 mmol) was added in 10 mL of THF, then cooled. Once the solution was cooled to 0 °C, ethyl isocyanate (4.90 g, 63.2 mmol) was dissolved in 15 mL of THF in a pear-shaped flask and then added dropwise via syringe to the round-bottomed flask while stirring. Finally, the pear-shaped flask was washed with 2 mL of dry THF and the two-neck flask was removed from the ice bath to continue reacting at room temperature. After 15 min, the reaction was complete and transferred to a one-neck round-bottomed flask. The solvent was then evaporated under reduced pressure. Mass of product: 12.92 g; Percent yield: 100.0%
1H NMR (CDCl3, 400 MHz) δ 5.203 (m, 2H, NH); 4.446 (m, 1H, CH); 3.664 (m, 2H, CH2); 3.513 (m, 2H, CH2); 3.233 (m, 2H, CH2); 3.161 (m, 2H, CH2); 1.184 (m, 6H, CH3); 1.071 (m, 3H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 158.843 (C=O); 102.070 (CH2); 63.073 (CH); 62.834 (CH2); 43.180 (CH2); 35.135 (CH2); 15.509 (CH3); 15.366 (CH3).
ESI-MS (Positive Ion Mode) m/z: 227.1385 (M + Na)
ATR-IR (frequency, intensity): 3326.71 cm−1 (m); 2973.10 cm−1 (m); 2930.29 cm−1(w); 2896.86 cm−1 (w); 1624.88 cm−1 (s); 1585.81 cm−1 (s); 1458.87 cm−1 (w); 1442.87 cm−1 (w); 1377.29 cm−1 (m); 1354.94 cm−1 (w); 1251.81 cm−1 (m); 1132.48 cm−1 (s); 1089.72 cm−1 (w); 1060.29 cm−1 (vs); 1025.08 cm−1 (w); 911.49 cm−1 (m); 883.12 cm−1 (w); 818.64 cm−1 (w); 768.61 cm−1 (w); 639.67 cm−1 (br); 562.47 cm−1 (m); 438.60 cm−1 (w).
2.3.4. Preparation of 1-Isopropyl-1,3-dihydro-2H-imidazol-2-one
Taking a 50 mL round-bottomed flask, 10 mL of THF was added and cooled to 0 °C by placing the flask in the ice bath. The 1-(2,2-diethoxyethyl)-3-isopropylurea (2.914 g, 13.35 mmol) was then added to the flask and stirred until dissolution, which was about 2 min. Next, 16.0 mL of 1.0 M HCl (16.0 mmol) was added dropwise via syringe, and the reaction was left to stir for 4 h. Upon completion, 1.0 M NaOH was added to neutralize the solution to a pH of 7.0. The reaction was then transferred to a separatory funnel, the product was extracted with 50 mL of chloroform 3 times, and the organic layer was collected. The product in the organic layer was then dried with magnesium sulfate to eliminate any remaining water and then filtered via gravity filtration. Finally, the solvent was removed under reduced pressure. Mass of product: 1.382 g; Percent yield: 82.14%
1H NMR (CDCl3, 400 MHz) δ 10.991 (s, 1H, NH); 6.309 (m, 1H, CH); 6.238 (m, 1H, CH); 4.382 (m, 1H, CH); 1.290 (m, 6H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 154.138 (C=O); 108.706 (CH); 107.614 (CH); 44.282 (isopropyl CH); 22.289 (CH3).
ESI-MS (Positive Ion Mode) m/z: 127.0858 (M + H)
ATR-IR (intensity, frequency) 3142.76 cm−1 (w); 2975.11 cm−1(m); 2936.87 cm−1 (w); 2877.11 cm−1 (w); 1651.82 cm−1 (vs); 1433.62 cm−1 (w); 1389.06 cm−1 (w); 1368.62 cm−1 (w);
1326.48 cm−1 (w); 1265.99 cm−1 (w); 1247.79 cm−1 (w); 1176.66 cm−1 (w);
1131.76 cm−1 (w); 1099.22 cm−1 (w); 1033.71 cm−1 (w); 913.41 cm−1 (w);
878.42 cm−1 (w); 777.37 cm−1 (w); 669.67 cm−1 (w); 597.54 cm−1 (w); 420.22 cm−1 (w).
2.3.5. Preparation of 1-t-Butyl-1,3-dihydro-2H-imidazol-2-one
A 50 mL round-bottomed flask was charged with 10 mL of THF. This solvent was cooled to 0 °C and the 1-(2,2-diethoxyethyl)-3-t-butylurea (5.169 g, 22.25 mmol) was then added to the flask and stirred until dissolution, which took about 2 min. Next, 26.7 mL of 1.0 M HCl (26.7 mmol) was added dropwise via syringe, and the reaction was left to stir for 4 h. Upon completion, 1.0 M NaOH was added to neutralize the solution to a pH of 7.0. The reaction was then transferred to a separatory funnel, the product was extracted with 50 mL of chloroform 3 times, and the organic layer was collected. The product in the organic layer was then dried with magnesium sulfate to eliminate any remaining water and then filtered via gravity filtration. Finally, the solvent was removed under reduced pressure, and the product was a white crystalline solid. Mass of product: 2.84 g; Percent yield: 91.2%
1H NMR (CDCl3, 400 MHz) δ 10.820 (s, 1H, NH); 6.314 (m, 2H, CH); 1.544 (s, 9H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 154.263 (C=O); 109.339 (CH); 107.758 (CH); 55.024 (t-butyl C); 28.403 (CH3).
ESI-MS (Positive Ion Mode) m/z: 141.1024 (M + H)
ATR-IR (frequency, intensity) 3139.57 cm−1 (w); 2971.95 cm−1 (m); 2912.38 cm−1 (w); 2802.17 cm−1 (w); 1659.18 cm−1 (vs); 1578.71 cm−1 (w); 1515.92 cm−1 (w); 1478.47 cm−1 (w); 1460.68 cm−1 (w); 1434.72 cm−1 (w); 1415.69 cm−1 (m); 1395.26 cm−1 (w); 1364.01 cm−1 (m); 1284.42 cm−1 (w); 1249.35 cm−1 (vs); 1230.24 cm−1 (w); 1193.44 cm−1 (w); 1157.88 cm−1 (m); 1113.18 cm−1 (w); 1062.50 cm−1 (w); 1022.96 cm−1 (w); 905.17 cm−1 (m); 844.64 cm−1 (m); 825.85 cm−1 (w); 812.13 cm−1 (w); 798.09 cm−1 (w); 759.56 cm−1 (s); 679.09 cm−1 (s); 654.59 cm−1 (vs); 622.31 cm−1 (m); 600.88 cm−1 (w); 547.85 cm−1 (w); 430.00 cm−1 (w).
2.3.6. Preparation of 1-Ethyl-1,3-dihydro-2H-imidazol-2-one
A 50 mL round-bottomed flask was charged with 10 mL of THF. This solvent was cooled to 0 °C and the 1-(2,2-diethoxyethyl)-3-ethyl-urea (3.362 g, 16.45 mmol) was then added to the flask and stirred until dissolution, which took about 2 min. Next, 20.0 mL of 1.0 M HCl (20.0 mmol) was added dropwise via syringe, and the reaction was left to stir for 2.75 h. Upon completion, 1.0 M NaOH was added to neutralize the solution to a pH of 7.0. The reaction was then transferred to a separatory funnel, the product was extracted with 50 mL of chloroform 3 times, and the organic layer was collected. The product in the organic layer was then dried with magnesium sulfate to eliminate any remaining water and then filtered via gravity filtration. Finally, the solvent was removed under reduced pressure, and the product was a white crystalline solid. Mass of product: 1.343 g; Percent yield: 72.80%
1H NMR (CDCl3, 400 MHz) δ 10.967 (s, 1H, NH); 6.32 (m, 1H, CH); 6.208 (m, 1H, CH); 3.679 (m, 2H, CH2); 1.291 (m, 3H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 154.330 (C=O); 110.944 (CH); 108.625 (CH); 38.134 (CH2); 14.901 (CH3).
ESI-MS (Positive Ion Mode) m/z: 113.0720 (M + H)
ATR-IR (frequency, intensity) 3135.82 cm−1 (w); 2969.10 cm−1 (m); 2799.08 cm−1 (w); 1655.12 cm−1 (vs); 1575.54 cm−1 (w); 1510.60 cm−1 (w); 1463.94 cm−1 (w); 1429.93 cm−1 (m); 1383.56 cm−1 (w); 1352.53 cm−1 (w); 1263.09 cm−1 (s); 1202.97 cm−1 (w); 1128.75 cm−1 (w); 1115.85 cm−1 (w); 1093.99 cm−1 (s); 1046.11 cm−1 (w); 965.31 cm−1 (w); 908.30 cm−1 (m); 839.01 cm−1 (m); 794.56 cm−1 (m); 769.88 cm−1 (m); 753.34 cm−1 (m); 680.95 cm−1 (vs); 667.81 cm−1 (w); 598.62 cm−1 (w); 537.62 cm−1 (m).
2.3.7. Preparation of 3,3′-(Pyridine-2,6-diylbis(methylene))bis(1-isopropyl-1,3-dihydro-2H-imidazole-2-one
Sodium hydride (0.2414 g, 10.06 mmol) was added to a two-necked round-bottomed flask. The sodium hydride was washed in 5 mL of pentane two times to remove any impurities and removed via syringe. Once the pentane was removed, 5 mL of dry THF was added to the round-bottomed flask. Once the sodium hydride had been dissolved, 1-isopropyl-1,3-dihydro-2H-imidazol-2-one (0.5100 g, 4.043 mmol) was dissolved in 5 mL of dry THF in a pear-shaped flask. This solution was then added to the round-bottomed flask via syringe and left to stir for 5 min. Finally, 2,6-dibromomethylpyridine (0.5506 g, 2.078 mmol) was dissolved in 5 mL of dry THF in a pear-shaped flask and then added to the round-bottomed flask dropwise via syringe. The reaction was left for 18 h at room temperature. Silica column chromatography with a 5:1 mixture of ethyl acetate and methanol was used to purify the product. Mass of product: 0.530 g; Percent yield: 21.9%
1H NMR (CDCl3, 400 MHz) δ 7.625 (m, 1H, CH); 7.103 (m, 2H, CH); 6.315 (m, 2H, imidazole CH); 6.251(m, 2H, imidazole CH); 4.923 (m, 4H, CH2); 4.408 (m, 2H, CH); 1.295 (m, 12H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 156.883 (C=O); 152.595 (C pyridine); 138.097 (CH); 120.800 (CH); 110.767 (CH); 107.010 (CH); 48.757 (CH2); 44.770 (isopropyl CH); 22.270 (CH3).
ESI-MS (Positive Ion Mode) m/z: 378.1870 (M + Na)
ATR-IR (intensity, frequency) 3096.10 cm−1 (w); 2973.14 cm−1 (w); 2923.29 cm−1 (w); 1663.12 cm−1 (vs); 1590.78 cm−1 (w); 1573.57 cm−1 (w); 1513.41 cm−1 (w); 1446.34 cm−1 (s); 1416.76 cm−1 (w); 1408.27 cm−1 (w); 1393.73 cm−1 (w); 1371.63 cm−1 (w); 1360.11 cm−1 (w); 1245.27 cm−1 (w); 1225.87 cm−1 (s); 1175.12 cm−1 (w); 1147.00 cm−1 (w); 1133.49 cm−1 (w); 1095.88 cm−1 (w); 1067.35 cm−1 (w); 1019.78 cm−1 (w); 991.10 cm−1 (w); 947.23 cm−1 (w); 917.22 cm−1 (w); 872.75 cm−1 (w); 815.72 cm−1 (w); 785.70 cm−1 (w); 762.25 cm−1 (s); 717.29 cm−1 (w); 671.50 cm−1 (vs); 595.30 cm−1 (w); 563.73 cm−1 (w); 540.05 cm−1 (w); 515.96 cm−1 (w); 491.48 cm−1 (w); 471.64 cm−1 (w); 447.78 cm−1 (w); 427.16 cm−1 (w).
2.3.8. Preparation of 3,3′-(Pyridine-2,6-diylbis(methylene))bis(1-t-butyl-1,3-dihydro-2H-immidazol-2-one Product
The synthesis of the ligand precursor with the t-butyl wingtip group began with sodium hydride (0.3553 g, 14.81 mmol), which was washed in 5 mL of pentane twice to remove any impurities. Once the pentane was removed via syringe, 5 mL of dry THF was added to the round-bottomed flask. Once the sodium hydride had been dissolved, the cyclized product (0.5053 g, 3.605 mmol) was dissolved in 5 mL of dry THF in a pear-shaped flask. This solution was then added to the round-bottomed flask via syringe to deprotonate and left to stir for 5 min. Finally, 2,6-dibromomethylpyridine (0.5174 g, 1.953 mmol) was dissolved in 5 mL of dry THF in a pear-shaped flask and then added to the round-bottomed flask dropwise via syringe. The reaction was left to stir and warm up to room temperature for 3 h. Silica column chromatography with a 5:1 mixture of ethyl acetate and methanol was used to purify the product. Mass of product: 0.682 g; Percent yield: 98.6%
1H NMR (CDCl3, 400 MHz) δ 7.564 (m, 1H, CH); 7.023 (m, 2H, CH); 6.300 (m, 2H, CH); 6.213 (m, 2H, CH); 4.853 (m, 4H, CH2); (s, 18H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 156.883 (C=O); 152.863 (C pyridine); 138.060 (CH); 120.033 (CH); 109.550 (CH imidazole); 108.266 (CH imidazole); 55.043 (C(CH3)3); 48.536 (CH2); 28.355 (CH3).
ESI-MS (Positive Ion Mode) m/z: 384.2423 (M + H)
ATR-IR (frequency, intensity) 3168.04 cm−1 (w); 3131.00 cm−1 (w); 2979.30 cm−1 (w); 2966.24 cm−1 (w); 2925.20 cm−1 (w); 1671.02 cm−1 (vs); 1593.94 cm−1 (m); 1574.91 cm−1 (m); 1504.52 cm−1 (w); 1478.42 cm−1 (w); 1452.58 cm−1 (s); 1441.88 cm−1 (m); 1406.93 cm−1 (w); 1367.92 cm−1 (m); 1354.58 cm−1 (m); 1278.93 cm−1 (s); 1239.40 cm−1 (m); 1195.75 cm−1 (w); 1151.85 cm−1 (w); 1092.24 cm−1 (w); 1001.43 cm−1 (m); 946.82 cm−1 (w); 910.56 cm−1 (w); 841.29 cm−1 (m); 814.77 cm−1 (w); 782.32 cm−1 (w); 760.75 cm−1 (w); 750.32 cm−1 (w); 713.99 cm−1 (w); 664.99 cm−1 (s); 648.88 cm−1 (s); 588.47 cm−1 (w); 566.78 cm−1 (w); 558.22 cm−1 (w); 436.30 cm−1 (w); 407.61 cm−1 (w).
2.3.9. Preparation of 3,3′-(Pyridine-2,6-diylbis(methylene))bis(1-ethyl-1,3-dihydro-2H-immidazol-2-one Product
The synthesis of the ligand precursor with the ethyl wingtip group began with sodium hydride (0.3450 g, 14.38 mmol), which was washed in 5 mL of pentane twice to remove any impurities. Once the pentane was removed via syringe, 5 mL of dry THF was added to the round-bottomed flask. Once the sodium hydride had been dissolved, the cyclized product (0.5034 g, 4.489 mmol) was dissolved in 5 mL of dry THF in a pear-shaped flask. This solution was then added to the round-bottomed flask via syringe to deprotonate and left to stir for 5 min. Finally, 2,6-dibromomethylpyridine (0.554 g, 2.09 mmol) was dissolved in 5 mL of dry THF in a pear-shaped flask and then added to the round-bottomed flask dropwise via syringe. The reaction was left to stir at room temperature for 3 days. Silica column chromatography with a 5:1 mixture of ethyl acetate and methanol was used to purify the product. Mass of product: 0.5686 g; Percent yield: 83.09%.
1H NMR (CDCl3, 400 MHz) δ 7.635 (m, 1H, CH); 7.142 (m, 2H, CH); 6.327 (m, 2H, CH); 6.223 (m, 2H, CH); 4.922 (m, 4H, CH2); 3.688 (m, 4H, CH2); 1.266 (m, 6H, CH3).
13C{1H} NMR (CDCl3, 100 MHz), δ 156.318 (C=O); 152.945 (C pyridine); 138.705 (CH); 121.274 (CH); 110.728 (CH imidazole); 110.058 (CH imidazole); 48.421 (CH2); 38.537 (CH2); 14.872 (CH3).
ESI-MS (Positive Ion Mode) m/z: 328.1797 (M + H)
ATR-IR (frequency, intensity) 3422.89 cm−1 (br); 3130.93 cm−1 (w); 2977.35 cm−1 (w); 2935.12 cm−1 (w); 1654.86 cm−1 (vs); 1594.88 cm−1 (w); 1576.31 cm−1 (w); 1456.61 cm−1 (s); 1382.13 cm−1 (w); 1351.18 cm−1 (w); 1245.29 cm−1 (s); 1221.60 cm−1 (s); 1163.68 cm−1 (w); 1089.40 cm−1 (w); 1039.83 cm−1 (w); 994.74 cm−1 (w); 950.19 cm−1 (m); 905.13 cm−1 (w); 822.35 cm−1 (m); 758.72 cm−1 (s); 658.42 cm−1 (s); 587.59 cm−1 (w); 546.67 cm−1 (w); 514.40 cm−1 (w).
2.4. Preparation of nickel(II) and cobalt(II) Complexes
2.4.1. Preparation of Bis-(n3-O,O,N)-[2,6-bis(N-isopropyl-N’-methyleneimidazole-2-one)pyridine]nickel(II) Tetrafluoroborate [1]
The ligand precursor with the isopropyl wingtip group (0.156 g, 0.440 mmol) was dissolved in 10 mL of acetonitrile in a 100 mL round-bottomed flask. Nickel(II) tetrafluoroborate hexahydrate (0.150 g, 0.441 mmol) was dissolved in 2 mL of acetonitrile in a separate flask. The nickel(II) solution was combined with the ligand. Upon mixing, the ligand solution changed color from a pale yellow to a spearmint green. Potassium chloride (0.032 g, 0.439 mmol) was added to the solution and left on reflux for eighteen hours. The solvent was then removed under reduced pressure. Mass of Product: 0.349 g Percent Yield: 81.2%
Crystals suitable for X-ray diffraction were grown by dissolving the complex in acetonitrile and allowing diethyl ether vapor to diffuse into the acetonitrile solution containing the complex.
Anal. Calc. for [C38H50N10NiO4][BF4]2·2H2O (979.21): C, 46.61; H, 5.5.6; N, 14.30. Found: C, 46.42; H, 4.97; N, 14.09.
Magnetic susceptibility: 2.59 BM at 294.2 K
UV-Visible data: λ (nm), (ε (M−1cm−1): 264 nm (1860 M−1cm−1)
Cyclic voltammetry solution preparation:
A solution was prepared containing the nickel(II) isopropyl complex (0.0190 g, 0.0247 mmol) and tetrabutylammonium tetrafluoroborate (0.3302 g, 1.003 mmol) dissolved in acetonitrile to make a 10.00 mL solution. The electrodes were glassy carbon (working electrode), silver wire (pseudo-reference), and platinum wire (counter).
2.4.2. Preparation of Bis-(n3-O,O,N)-[2,6-bis(N-ethyl-N’-methyleneimidazole-2-one)pyridine]cobalt(II) Tetrachlorocobaltate [2]
The ligand precursor with the ethyl wingtip group (0.151 g, 0.456 mmol) was dissolved in 10 mL of acetonitrile in a 100 mL round-bottomed flask. Cobalt(II) tetrafluoroborate hexahydrate (0.464 g, 0.430 mmol) was dissolved in 2 mL of acetonitrile in a separate flask. The cobalt(II) solution was combined with the ligand precursor solution, and the solution changed from a pale yellow color to a deep blue. Potassium chloride (0.0342 g, 0.459 mmol) was added to the solution and left on reflux for eighteen hours. The solvent was then removed under reduced pressure. Mass of Product: 0.396 g Percent Yield: 44.8%.
Crystals suitable for X-ray diffraction were grown by dissolving the complex in acetonitrile and allowing diethyl ether vapor to diffuse into the acetonitrile solution containing the complex.
Anal. Calc. for [C34H42CoN10O4][CoCl4]·1CH3CN (955.50): C, 45.25; H, 4.75; N, 16.13. Found: C, 45.50; H, 5.08; N, 15.72.
Magnetic susceptibility results: 1.59 BM at 294.2 K
UV-Visible data: λ (nm), (ε (M−1cm−1): 574 nm (41 M−1cm−1); 594 nm (59 M−1cm−1); 639 nm (63 M−1cm−1)
Cyclic voltammetry solution preparation:
A solution was prepared containing the cobalt(II) complex (0.0198 g, 0.0257 mmol) and tetrabutylammonium tetrafluoroborate (0.3299 g, 1.002 mmol) dissolved in acetonitrile to make a solution (10.00 mL). The electrodes were glassy carbon (working electrode), silver wire (pseudo-reference) and platinum wire (counter).
2.4.3. Preparation of Bis-(n3-O,O,N)-[2,6-bis(nt-butyl-N’-methyleneimidazole-2-one)pyridine]cobalt(II) Tetrafloroborate [3]
The ligand precursor with the t-butyl wingtip group (0.200 g, 0.522 mmol) was dissolved in 10 mL of acetonitrile in a 100 mL round-bottomed flask. Cobalt(II) tetrafluoroborate hexahydrate (0.178 g, 0.518 mmol) was dissolved in 2 mL of acetonitrile in a separate flask. The cobalt(II) solution was combined with the ligand precursor solution, and the solution changed from a pale yellow color to a dark rose-purple. The solution was left on reflux for eighteen hours. The solvent was then removed under reduced pressure. Mass of Product: 0.328 g, Percent Yield: 70.0%.
Crystals suitable for X-ray diffraction were grown by dissolving the complex in acetonitrile and allowing diethyl ether vapor to diffuse into the acetonitrile solution containing the complex. The crystal structure for [3] is available in CSD Communications [35].
Anal. Calc. for [C40H58CoN10O4][BF4]·1H2O (1017.55): C, 49.58; H, 5.94; N, 13.77. Found: C, 49.18; H, 5.55; N, 13.63.
Magnetic susceptibility: 2.59 BM at 294.2 K
UV-Visible data: λ (nm), (ε (M−1cm−1): 596 nM (1290 M−1cm−1); 687 nm (1690 M−1cm−1)
Cyclic voltammetry solution preparation:
A solution was prepared containing the cobalt(II) complex (0.0134 g, 0.0134 mmol) and tetrabutylammonium tetrafluoroborate (0.3329 g, 1.011 mmol) dissolved in acetonitrile to make a 10.00 mL solution. The electrodes were glassy carbon (working electrode), silver wire (pseudo-reference) and platinum wire (counter).
- Characterization:
- Single Crystal X-Ray Crystallography
- Nickel(II) complex [1], cobalt(II) complex [2], cobalt(II) complex [3]
- Experimental
Low-temperature diffraction data (ω-scans) were collected on a Rigaku MicroMax-007HF diffractometer coupled to a Saturn994+ CCD detector with Cu Kα (λ = 1.54178 Å) for the structure of [1]. Low-temperature diffraction data (ω-scans) were collected on a Rigaku MicroMax-007HF diffractometer coupled to a Dectris Pilatus3R detector with Mo Kα (λ = 0.71073 Å) for the structure of [2]. Low-temperature diffraction data (ω-scans) were collected on a Rigaku Synergy-S diffractometer coupled to a HyPix-Arc 100 detector with Cu Kα (λ = 1.54178 Å) for the structure of [3]. The structure of [3] was previously published at CSD Communication [35]. The diffraction images were processed and scaled using Rigaku Oxford Diffraction software (CrysAlisPro; Rigaku OD: The Woodlands, TX, USA, 2015). The structure was solved with SHELXT and was refined against F2 on all data by full-matrix least squares with SHELXL [37]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included in the model at geometrically calculated positions and refined using a riding model.
For complex 1, the isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms to which they are linked (1.5 times for methyl groups). These data were refined as a 2-component twin. The fractional volume contribution of the minor twin component was freely refined to a converged value of 0.53(10). The solution was initially solved in C2/m. The refinement residuals for model agreement were found to be high (>9%). The reflection data was carefully inspected and found to solve best when the metric symmetry was lowered to C2, with the model refined as a racemic twin. This solution shows clear signs of disorder. The atomic positions were split for atoms with large thermal ellipsoids, and their thermal parameters were restrained with rigid bonds and similar restraints. The site occupancy factors were constrained to not exceed a value of 1.0 for any set of two chemically identical disordered atoms. A solvent mask was calculated, and 114 electrons were found in a volume of 580 Å3 in 2 voids per unit cell. This is consistent with the presence of 2[H2O], 1.5[H3C2N] per formula unit which account for 106 electrons per unit cell [38]. The full numbering scheme of compound 1, 007a-22125, can be found in the full details of the X-ray structure determination (CIF), which is included as Supporting Information. CCDC number 2402484 (1) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif (accessed on 9 February 2025).
For complex 2, the isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms to which they are linked (1.5 times for methyl groups). One ethyl arm was disordered. Chemically equivalent C-C distances that were disordered were restrained to be similar. The thermal parameters of atoms that were disordered or near the disorder (N15, C27, C51, and C52) were restrained to be similar. The full numbering scheme of compound 2, 007c-22060, can be found in the full details of the X-ray structure determination (CIF), which is included as Supporting Information. CCDC number 2402485 [2] contains the supplementary crystallographic data for this paper.
For complex 3, the isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms to which they are linked (1.5 times for methyl groups). The full numbering scheme of compound 3, syn-23135, can be found in the full details of the X-ray structure determination (CIF), which is included as Supporting Information. CCDC number 2402486 (syn-23135) contains the supplementary crystallographic data for this paper. This has previously been published at CSD Communication [35].
Table 1 provides the crystal data and structure refinement for complexes 1–3.

Table 1.
Crystal data and structure refinement for complexes 1–3.
3. Results and Discussion
3.1. Preparation and Characterization of the Ligand Precursors
The ligand precursors were prepared in three steps following Scheme 1. Steps 1 and 3 required the use of dry tetrahydrofuran. The pincer ligand precursors in step 3 were purified using SiO2 column chromatography and the products were eluted using ethyl acetate/methanol (5:1 volume/volume ratio). Various reaction times in step 2 of the ligand precursor synthesis are needed depending on what R-group is utilized.
The yields for step one in the synthesis were excellent and the products were prepared in quantitative yield. The imidazole-2-one products in step 2 were 72% or higher. The products in step 3 were prepared in fair yield (22% for R = isopropyl) after purification. The products for step 3 were prepared in very good to excellent yield for R = t-butyl and ethyl. The products of the reactions were soluble in chloroform, acetone, dichloromethane, methanol, ethyl acetate, and dimethyl sulfoxide. All of the ligand precursors were either white or off-white in color.
NMR spectra were obtained of the ligand precursors in CDCl3. For the urea-like product of the ligand precursor (step 1 in the synthesis), the CH2 resonances in the ethyl substituents are diastereotopic. The step 2 product shows a broad N-H resonance at around δ 10.9 ppm. The step 3 product in the preparation of the ONO ligand precursor shows the absence of the N-H resonance, indicating complete deprotonation. There is a plane of symmetry in this ligand precursor, and thus the two halves of the molecule are symmetrically equivalent.
The ligand precursors were also characterized by high-resolution ESI-MS. For the step 1 products, the [M + Na]+ peak was clearly identified. For the step 2 product, the [M + H]+ peak was clearly apparent. For the step 3 product, the [M + Na]+ peak was apparent when R = iPr and the [M + H]+ peak was apparent when R = t-butyl or ethyl.
3.2. Preparation of the Metal Complexes
The ONO ligand precursors were metallated to prepare cobalt(II) and nickel(II) complexes. The preparation of these complexes was accomplished following the approach in Figure 3, Figure 4 and Figure 5. In complexes 1 and 3, [BF4]− is the counter-anion. For complex 2, [CoCl4]2− is the counter-anion. It is unclear why tetrachlorocobaltate is the counter-anion. Potassium chloride was used in the synthesis because this was utilized as a reagent in previous metallations in the preparation of SNS pincer metal complexes [6,7,14,15,16].
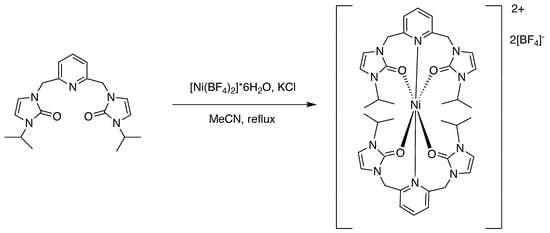
Figure 3.
Preparation of the nickel(II) complex [1] with isopropyl wingtips.
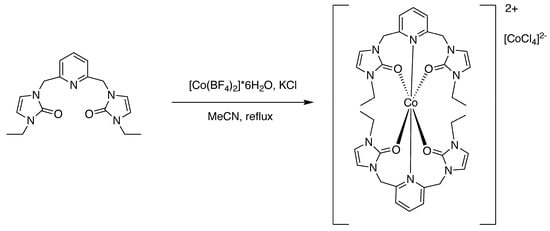
Figure 4.
Preparation of the cobalt(II) complex [2] with ethyl wingtips.
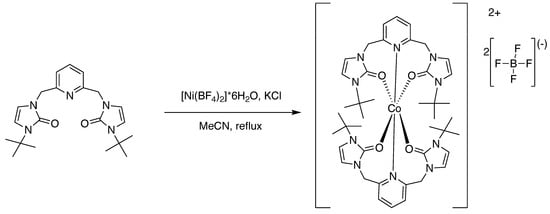
Figure 5.
Preparation of the cobalt(II) complex [3] with t-butyl wingtips.
There were color changes upon adding the metal precursors to a solution containing the ligand precursor. In the preparation of the nickel(II) complex, 1, the solution turned from colorless to a yellow-green color upon adding the metal precursor salt. In the preparation of the cobalt(II) complex, 2, (R = ethyl), the solution turned from colorless to blue in color upon adding the metal precursor salt. In the preparation of the cobalt(II) complex (R = t-butyl), 3, the solution turned from colorless to violet purple in color upon adding the metal precursor salt. The complexes were prepared in fair to good yield. Single crystals suitable for X-ray diffraction were grown by dissolving the complexes in acetonitrile and allowing diethyl ether vapor to slowly diffuse into the solution containing the complex. The elemental analyses results obtained for the metal complexes were consistent with having two tridentate ligands coordinated per metal center. The geometry about the metal center was pseudo-octahedral in each case.
The crystal structure of the nickel(II) complex with isopropyl wingtip groups was determined via single crystal X-ray diffraction, as shown in Figure 6. In this structure, two ligands are coordinated to the metal in a pseudo-octahedral geometry. The approximate symmetry of this structure is a C2h point group. Previous SNS pincer complexes did not afford two ligand precursors per metal center. The unit cell was monoclinic with a C121 space group. The isopropyl wingtip groups are directed away from the nickel(II) center.
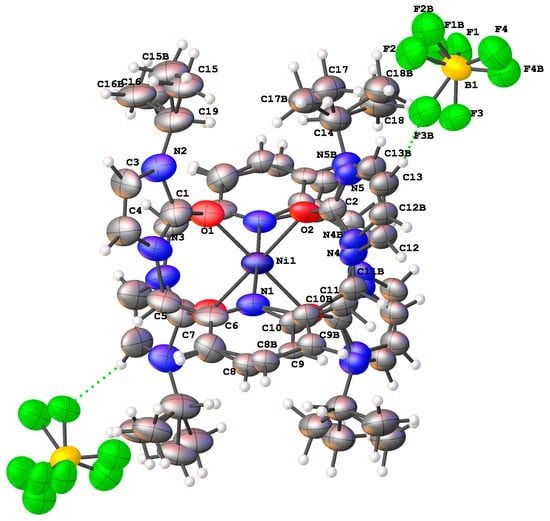
Figure 6.
The single crystal structure of a nickel(II) complex, [1], with isopropyl wingtip groups.
Select bond distances and bond angles for the nickel(II) complex are provided in Table 2 and Table 3. The nickel(II) oxygen bond distances of 2.028(7) Å are consistent with nickel(II)–oxygen bond distances of a pseudo-octahedral metal complex [39]. The Nickel(II)–nitrogen (pyridine) bond distance of 2.296(4) Å is longer than a previously reported nickel(II)–nitrogen (pyridine) bond length of 2.145 Å [40].
Table 2.
Select bond distances in the nickel(II) complex, 1, with isopropyl wingtip groups.
Table 3.
Select bond angles about the nickel atom in the nickel(II) complex, 1, with isopropyl wingtip groups.
The carbon–oxygen bond distances of 1.240(10) Å and 1.290(10) Å are consistent with a carbon–oxygen double bond [41].
The crystal structure of the cobalt(II) complex with ethyl wingtip groups, 2, was determined via single crystal X-ray diffraction, as shown in Figure 7. In this structure, two ligands are coordinated to the metal in a pseudo-octahedral geometry. The counter-anion for this complex is tetrachlorocobaltate. The approximate symmetry of this structure is a C2h point group. Previous SNS pincer complexes did not afford two ligand precursors per metal center [6,7,9,10,11,12,13,14,15,16]. The unit cell is monoclinic with a P21/n space group. Like the nickel(II) complex, the ethyl wingtip groups are directed away from the cobalt(II) center.
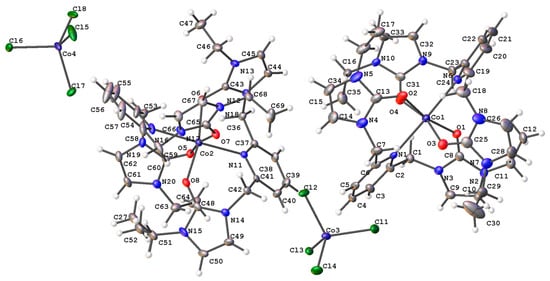
Figure 7.
The single crystal structure of a cobalt(II) complex, 2, with isopropyl wingtip groups.
Select bond distances and bond angles for the cobalt(II) complex are provided in Table 4 and Table 5. The Co-O bond distances are in good agreement with cobalt(II)–oxygen bond distances in the literature for six coordinate, pseudo-octahedral metal complexes. The cobalt(II)–nitrogen bond distance is longer than previously reported cobalt(II)–nitrogen bond distances where the coordinating nitrogen atom is from a pyridine substituent [42]. A previously reported NNN pincer complex where the nitrogen donor atoms came from triazole and pyridine had Co(II)–nitrogen (pyridine) bond distances of 2.148(6) and 2.149(6) Å [43,44]. In this cobalt(II) complex, the cobalt(II)–nitrogen (pyridine) bond distances were 2.346(5) and 2.356(5) Å.
Table 4.
Selected bond distances in the cobalt(II) complex, 2, with ethyl wingtip groups.
Table 5.
Selected bond angles about the cobalt atom in the cobalt(II) complex, 2, with ethyl wingtip groups.
The carbon–oxygen bond distances of 1.230(7), 1.237(8), 1.230(8), and 1.221(7) Å are consistent with a carbon–oxygen double bond [41].
The crystal structure of the cobalt(II) complex with t-butyl wingtip groups, 3, was determined via single crystal X-ray diffraction, shown in Figure 8 [35]. In this structure, two ligands are coordinated to the metal in a pseudo-octahedral geometry. The approximate symmetry of this structure is a C2 point group. Previous SNS pincer complexes did not afford two ligand precursors per metal center [6,7,9,10,11,12,13,14,15,16]. The unit cell is triclinic with a P-1 space group. Like complexes 1 and 2, complex 3 has its wingtip groups directed away from the metal center.
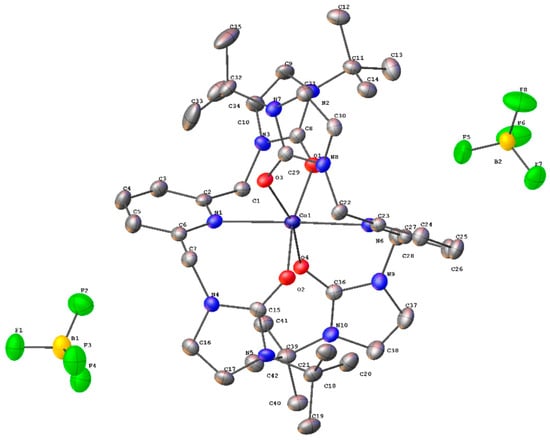
Figure 8.
The single crystal structure of a cobalt(II) complex, 3, with t-butyl wingtip groups [35].
Select bond distances and bond angles for the cobalt(II) complex are provided in Table 6 and Table 7 [35]. The Co–O bond distances are in good agreement with cobalt(II)–oxygen bond distances in the literature for six coordinate, pseudo-octahedral metal complexes. The cobalt(II)–nitrogen bond distance is longer than previously reported cobalt(II)–nitrogen bond distances where the coordinating nitrogen atom is from a pyridine substituent [42]. A previously reported NNN pincer complex where the nitrogen donor atoms came from triazole and pyridine had Co(II)–nitrogen (pyridine) bond distances of 2.148(6) and 2.149(6) Å [43,44]. In this cobalt(II) complex, the cobalt(II)–nitrogen (pyridine) bond distances were 2.379(3) and 2.385(3) Å.
Table 6.
Selected bond distances in the cobalt(II) complex, 3, with t-butyl wingtip groups [35].
Table 7.
Selected bond angles about the cobalt atom in the cobalt(II) complex, 3, with t-butyl wingtip groups [35].
The carbon–oxygen bond distances of 1.258(4), 1.255(4), 1.252(4), and 1.266(4) Å are consistent with a carbon–oxygen double bond. The carbon–oxygen bond distances in the cobalt(II) complex with t-butyl wingtip groups are about 0.02 Å longer than the carbon–oxygen bond distances in the cobalt(II) complex with ethyl wingtip groups [41].
3.3. Magnetic Susceptibility of the Metal Complexes
The magnetic susceptibility of the metal complexes, 1–3, were characterized using the Evans Method [16]. Complex 1 had a magnetic moment of 2.59 BM. This is consistent with having two unpaired electrons in the complex. Complex 2 had a magnetic moment of 1.59 BM. There may be some contribution of the [CoCl4]2− counter-anion for the magnetic moment. Complex 3 had a magnetic moment of 6.67 BM. Perhaps, there may be some ferromagnetic coupling in this complex. Future experiments involve magnetic susceptibility of complex 3 at variable temperatures to further understand if ferromagnetic coupling is indeed occurring in this complex.
3.4. Cyclic Voltammetry
The imidazole-2-one ligand precursor, the ONO pincer ligand precursor, and the metal complexes were characterized using cyclic voltammetry. The experiments were carried out to establish the potentials at which oxidations and reductions occur and whether any of the oxidation and reduction waves are related to each other. The ligand precursors and the metal complexes are non-innocent and therefore redox-active. The cyclic voltammograms were measured in acetonitrile. For the cyclic voltammetry experiments, the supporting electrolyte was tetrabutylammonium tetrafluoroborate.
The imidazole-2-one ligand precursor showed an oxidation wave at 0.96 V that was irreversible. The cyclic voltammogram is provided in Figure 9.
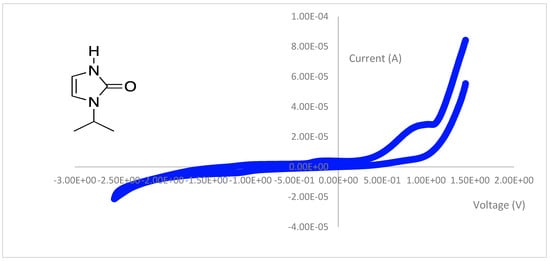
Figure 9.
The cyclic voltammogram of the Step 2 imidazole-2-one product with isopropyl wingtip groups from 2.0 to −2.5 V at 100 mV/s.
The ONO ligand precursor with isopropyl wingtips showed oxidation and reduction waves as shown in Figure 10. The ONO ligand precursor contains two oxidation waves at 1.10 and 1.50 volts, as well as two reduction waves at −2.21 and 1.20 volts. This indicates that the ligand precursor with isopropyl wingtip groups is redox-active, and these processes were irreversible.
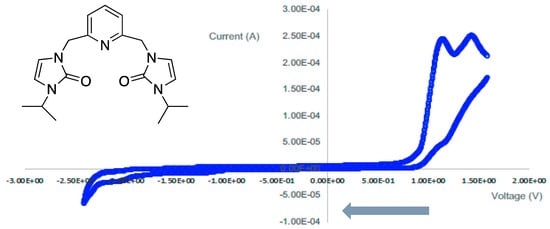
Figure 10.
The cyclic voltammogram of the ligand precursor with isopropyl wingtip groups from 1.8 to −2.5 V at 100 mV/s.
The cyclic voltammograms for the nickel(II) complex, 1, was also acquired.
As shown in Figure 11, the cyclic voltammogram of the nickel(II) complex with isopropyl wingtip groups, 1, contained five oxidation waves (1.63 V, 1.26 V, 1.03 V, 0.12 V, and −1.89 V) and three reduction waves (1.17 V, −1.05 V, and −1.52 V). These oxidation and reduction waves are all irreversible. These results are summarized in Table 8.
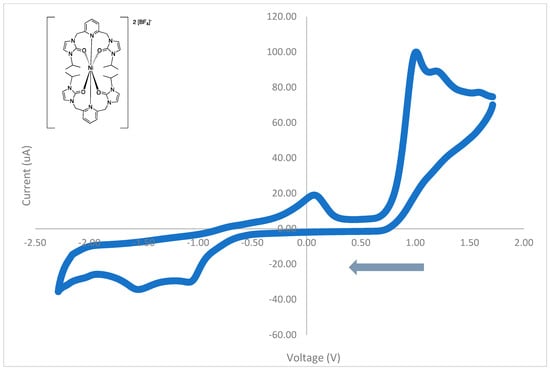
Figure 11.
The cyclic voltammogram of the nickel(II) complex, 1, with isopropyl wingtip groups from 1.8 to −2.2 V at 100 mV/s.
Table 8.
Assignments of peak potentials in cyclic voltammogram for nickel(II) complex with isopropyl wingtips, 1.
Cyclic voltammograms of the cobalt(II) complex with ethyl wingtip groups were also acquired. As shown in Figure 12, the cyclic voltammogram of the cobalt(II) complex with ethyl wingtip groups contains three oxidation waves (1.66 V, 1.36 V, and 1.06 V) and three reduction waves (1.52 V, −1.34 V, and −1.46 V). These oxidation and reduction waves are irreversible, and the complex is redox-active. The results are also summarized in Table 9.
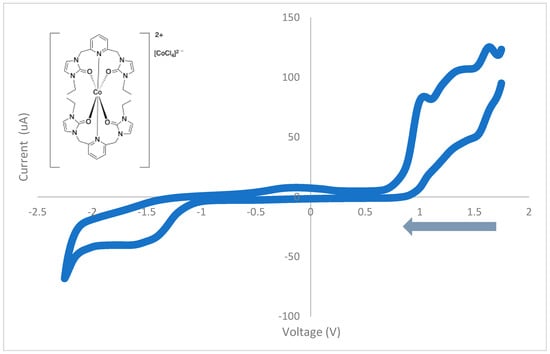
Figure 12.
The cyclic voltammogram of the cobalt(II) complex, 2, with ethyl wingtip groups from 1.8 to −2.2 V at 100 mV/s.
Table 9.
Assignments of peak potentials in cyclic voltammogram for cobalt(II) complex with ethyl wingtips, 2.
Cyclic voltammograms of the cobalt(II) complex with t-butyl wingtip groups were also acquired in acetonitrile. Unlike complex 2, complex 3 has a counter-anion that does not contain a metal-complex anion. As shown in Figure 13, the cyclic voltammogram of the cobalt(II) complex with t-butyl wingtip groups contains three oxidation waves (1.70 V, 1.34 V, and 1.04 V) and three reduction waves (1.56 V, −0.84 V, and −1.57 V). These oxidation and reduction waves are irreversible, and the complex is redox active. The results are also summarized in Table 10.
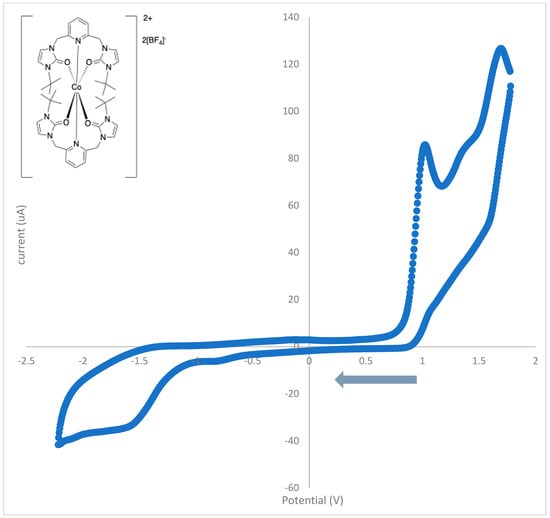
Figure 13.
The cyclic voltammogram of the cobalt(II) complex, 3, with t-butyl wingtip groups from 1.78 to −2.21 V at 100 mV/s.
Table 10.
Assignments of peak potentials in cyclic voltammogram for cobalt(II) complex with t-butyl wingtips, 3.
We have attempted to perform Gaussian calculations to better understand the cyclic voltammetry results. We were not able to obtain results that match our findings. We believe that Gaussian was not handling the unpaired electrons in the nickel(II) and cobalt(II) metal centers properly in the calculations.
3.5. UV-Visible Spectroscopy
The nickel(II) and cobalt(II) complexes were characterized using UV-Visible Spectroscopy to gain further insight on the electronic environment of the complexes. The UV-visible spectra are included in the Supporting Information. As shown in the Supporting Information and summarized in Table 11, the UV-visible spectrum of the nickel(II) complex [1] is distinguished by one feature at 264 nm (ε = 1860 M−1cm−1). The calculated molar absorptivity is consistent with a charge-transfer transition.
Table 11.
UV-Vis data for the cobalt(II) complex 2 (1.17 mM in acetonitrile).
As shown in the Supporting Information and summarized in Table 11, the UV-Visible spectra for the cobalt(II) complex, 2, with ethyl wingtips presented d to d transitions, based on the calculated molar absorptivity values. In comparison to the data collected for the nickel(II) complex, the cobalt(II) complex with ethyl wingtips presented d to d transitions, whereas the nickel(II) complex did not. The cobalt(II) complex with t-butyl wingtips showed charge transfer transitions.
As shown in the Supporting Information and summarized in Table 12, the UV-Visible spectra for the cobalt(II) complex with t-butyl wingtips, 3, presented more intense d to d transitions based on the molar absorptivity values. There may be some charge transfer transitions happening in this complex.
Table 12.
UV-Vis data for the cobalt(II) complex 3 (0.40 mM in acetonitrile).
4. Conclusions
We have prepared a series of pincer ligand precursors with oxygen, nitrogen, and oxygen donor atoms and one nickel(II) and two cobalt(II) complexes that contain a novel ONO pincer ligand. The complexes had pseudo-octahedral geometries about the metal center. Single crystal structures were obtained for each of the complexes. The complexes were paramagnetic as determined by using the Evans method. The nickel(II) complex had an experimentally determined μeff of 2.59 BM, which is consistent with having two unpaired electrons based on the spin-only formula. The cobalt(II) complex with ethyl wingtips had a μeff of 1.59 BM. There will be a contribution from the [CoCl4]2− anion in this magnetic moment calculation. The cobalt(II) complex with t-butyl wingtips had a μeff of 6.67 BM. There may be a ferromagnetic contribution for this complex. UV-Visible spectra were collected for complexes 1–3. For these complexes, charge transfer and d to d bands were noted. Cyclic voltammetry was obtained for complexes 1 and 2. Both complexes were redox-active, and the redox waves that were observed were irreversible. Future experiments will focus on the electrocatalytic activity of these complexes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst15030227/s1.
Author Contributions
Conceptualization, J.R.M. and O.J.C.N.; methodology, J.R.M. and O.J.C.N.; software, J.R.M. and B.Q.M.; validation, J.R.M., O.J.C.N. and B.Q.M.; formal analysis, B.Q.M., A.J.A., S.L.E., J.P.T., N.R.B., M.J.C. and C.J.P.; investigation, J.R.M., O.J.C.N., B.Q.M., A.J.A., N.R.B., S.L.E., J.P.T., M.J.C., C.J.P., S.L.C., A.J.W. and I.P.O.; resources, J.R.M., O.J.C.N. and B.Q.M.; data curation, J.R.M., O.J.C.N., B.Q.M., A.J.A., N.R.B., S.L.E., J.P.T., M.J.C., C.J.P., S.L.C., A.J.W. and I.P.O.; writing—original draft preparation, J.R.M., A.J.A., N.R.B., S.L.E. and J.P.T.; writing—review and editing, J.R.M.; visualization, J.R.M., B.Q.M., A.J.A., N.R.B., S.L.E., J.P.T. and C.J.P.; supervision, J.R.M. and O.J.C.N.; project administration, J.R.M.; funding acquisition, J.R.M., A.J.A., N.R.B., S.L.E., J.P.T., M.J.C., C.J.P., S.L.C. and A.J.W. All authors have read and agreed to the published version of the manuscript.
Funding
J.R.M. acknowledges the National Science Foundation Major Research Instrumentation Program for the purchase of a 400 MHz N.M.R. Spectrometer (CHE-1827854). J.R.M. is grateful for receiving generous support from the NASA CT Space Grant (Award number P-2173) in support of this work. N.R.B. received the Jean Dreyfus Lectureship Summer Research Student Award to work on this project. A.J.A. and C.J.P. received Hardiman Research Scholarships. NRB and MJC received Mancini Fund Scholarships. A.J.A., C.J.P., J.P.T., S.L.E. and S.L.C. received support from the Lawrence Family Faculty Student Research Mentorship Award when working on this project at Fairfield University. This research made use of the West Campus Analytical Core and the Chemical and Biophysical Instrumentation Center (CBIC) at Yale University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was not acquired for these studies since human subjects were not involved.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Peris, E.; Crabtree, R.H. Key Factors in Ligand Pincer Design. Chem. Soc. Rev. 2018, 47, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Gunanathan, C.; Milstein, D. Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev. 2014, 114, 12024–12087. [Google Scholar] [CrossRef] [PubMed]
- Szabo, K.J.; Wendt, O.F. (Eds.) Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis; Wiley VCH: Weinheim, Germany, 2014. [Google Scholar]
- Van Koten, G.; Milstein, D. (Eds.) Organometallic Pincer Complexes; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Van Koten, G.; Gossage, R.A. The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Miecznikowski, J.R.; Lo, W.; Lynn, M.A.; O’Loughlin, B.E.; DiMarzio, A.P.; Martinez, A.M.; Lampe, L.; Foley, K.M.; Keilich, L.C.; Lisi, G.P.; et al. Syntheses, Characterization, Density Functional Theory Calculations, and Activity of Tridentate SNS Zinc Pincer Complexes. Inorg. Chim. Acta 2011, 376, 515–524. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Lo, W.; Lynn, M.A.; Jain, S.; Keilich, L.C.; Kloczko, N.F.; O’Loughlin, B.E.; DiMarzio, A.P.; Foley, K.M.; Lisi, G.P.; et al. Syntheses, Characterization, Density Functional Theory Calculations, and Activity of Tridentate SNS Zinc Pincer Complexes Based on Bis-Imidazole or Bis-Triazole Precursors. Inorg. Chim. Acta 2012, 387, 25–36. [Google Scholar] [CrossRef]
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996, 96, 2239–2314. [Google Scholar] [CrossRef]
- Sunderland, J.R.; Tao, X.; Butrick, E.E.; Keilich, L.C.; Villa, C.E.; Miecznikowski, J.R.; Jain, S. Investigation of liver alcohol dehydrogenase catalysis using an NADH biomimetic and comparison with a synthetic zinc model complex. Polyhedron 2016, 114, 145–151. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Lynn, M.A.; Jasinski, J.P.; Lo, W.; Bak, D.; Pati, M.; Butrick, E.E.; Drozdoski, A.E.R.; Archer, K.A.; Villa, C.E.; et al. Synthesis and characterization of three-and five-coordinate copper(II) complexes based on SNS pincer ligand precursors. Polyhedron 2014, 80, 157–165. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Lynn, M.A.; Jasinski, J.P.; Reinheimer, E.; Bak, D.; Pati, M.; Butrick, E.E.; Drozdoski, A.E.R.; Archer, K.A.; Villa, C.E.; et al. Synthesis, Characterization, and Computational Study of Three-Coordinate SNS Copper(I) Complexes Based on Bis-Thione Ligand Precursors. J. Coord. Chem. 2014, 67, 29–44. [Google Scholar] [CrossRef]
- Lynn, M.A.; Miecznikowski, J.R.; Jasinski, J.P.; Kaur, M.; Mercado, B.Q.; Reinheimer, E.; Almanza, E.; Kharbouch, R.M.; Smith, M.R.; Zygmont, S.E.; et al. Copper(I) SNS Pincer Complexes: Impact of Ligand Design and Solvent Coordination on Conformer Interconversion from Spectroscopic and Computational Studies. Inorg. Chim. Acta 2019, 495, 118996. [Google Scholar] [CrossRef]
- Mast, Z.; Huntzinger, C.G.; Stinson, T.A.; Myren, T.H.T.; Kharbouch, R.M.; Almanza, E.M.; Zygmont, S.E.; Miecznikowski, J.R.; Luca, O.R. Cu(I) SNS triazole and imidazole pincers as electrocatalyst precursors for solar fuel production. Inorg. Chem. Front. 2020, 7, 1012–1015. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Zygmont, S.E.; Jasinski, J.P.; Kaur, M.; Almanza, E.; Kharbouch, R.M.; Bonitatibus, S.C.; Mircovich, E.E.; Le Magueres, P.; Reinheimer, E.; et al. Synthesis, Characterization, and Electrochemistry of SNS Cobalt(II) Tridentate Complexes. Transit. Met. Chem. 2022, 47, 127–137. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Mircovich, E.E.; Bertolotti, N.R.; Corbett, M.J.; Jasinski, J.P.; Reinheimer, E. Synthesis, single crystal structure and spectroscopic characterization of a nickel(II) complex that contains a SNS tridentate ligand. J. Chem. Crystallogr. 2022, 52, 287–296. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Jasinski, J.P.; Bonitatibus, S.C.; Almanza, E.; Kharbouch, R.M.; Zygmont, S.E.; Landy, K.R. Preparation of SNS cobalt(II) pincer model complexes of liver alcohol dehydrogenase. J. Vis. Exp. 2020, 157, e60668. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Veige, A.S. Trianionic pincer and pincer-type metal complexes and catalysts. Chem. Soc. Rev. 2014, 43, 6325–6369. [Google Scholar] [CrossRef]
- Elamathi, C.; Butcher, R.; Prabhakaran, R. Preparation, characterizations and biological evaluations of new copper(II) complexes containing ONO pincer type ligands. Appl. Organomet. Chem. 2018, 32, e4364. [Google Scholar] [CrossRef]
- Masood, S.; Jamshaid, M.; Zafar, M.N.; Mughal, E.U.; Ashfaq, M.; Tahir, M.N. ONO-pincer Zn(II) & Cd(II) complexes: Synthesis, structural characterization, Hirshfeld surface analysis and CT-DNA interactions. J. Mol. Struct. 2024, 1295, 136571. [Google Scholar]
- Szigethy, G.; Heyduk, A.F. Aluminum complexes of the redox active [ONO] pincer ligand. Dalton Trans. 2012, 41, 8144–8152. [Google Scholar] [CrossRef]
- Wong, J.L.; Higgins, R.F.; Bhowmick, I.; Cao, D.X.; Szigethy, G.; Ziller, J.W.; Shores, M.P.; Heyduk, A.F. Bimetallic iron-iron and iron-zinc complexes of the redox active ONO pincer ligand. Chem. Sci. 2016, 7, 1594–1599. [Google Scholar] [CrossRef]
- Hananouchi, S.; Krull, B.T.; Ziller, J.W.; Furche, F.; Heyduk, A.F. Metal effects on ligand non-innocence in Group 5 complexes of the redox-active ONO pincer ligand. Dalton Trans. 2014, 43, 17991–18000. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Ghiviriga, I.; Abboud, K.A.; Veige, A.S. A New ONO3− Trianionic Pincer-Type Ligand for Generating Highly Nucleophilic Metal–Carbon Multiple Bonds. J. Am. Chem. Soc. 2012, 134, 11185–11195. [Google Scholar] [CrossRef]
- Pascaulini, M.E.; DiRusso, N.V.; Quintero, P.A.; Thuijs, A.E.; Pinkowicz, D.; Abboud, K.A.; Dunbar, K.R.; Christou, G.; Meisel, M.W.; Veige, A.S. Synthesis, Characterization, and Reactivity of Iron(III) Complexes Supported by a Trianionic ONO3− Pincer Ligand. Inorg. Chem. 2014, 53, 13078–13088. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Greger, I.; Perez-Torrente, J.J.; Jimenez, M.V.; Modrego, F.J.; Lahoz, F.J.; Oro, L.A. ONO Dianionic Pincer-Type Ligand Precursors for the Synthesis of σ,π-Cyclooctenyl Iridium(III) Complexes: Formation Mechanism and Coordination Chemistry. Organometallics 2013, 32, 6903–6917. [Google Scholar] [CrossRef]
- Delgado-Rangel, L.H.; Reyes-Marquez, V.; Moreno-Narvaez, M.E.; Aragon-Muriel, A.; Parra-Unda, J.R.; Cruz-Navarro, J.A.; Martinez-Torres, M.A.; Valdes, H.; Morales-Morales, D. Biological Activity of Vanadium Pincer Complexes. New J. Chem. 2025. Advance Article. [Google Scholar] [CrossRef]
- Dhibar, P.; Chandra, A.; Paul, P.; Samaresh, B. Ancillary ligand induced variation in electronic spectral and catalytic properties of heteroleptic ONO-pincer complexes of ruthenium. New J. Chem. 2025, 49, 2674–2684. [Google Scholar] [CrossRef]
- Pennamuthiriyan, A.; Rengan, R.; Malecki, J.G. Sustainable Synthesis of Substituted 1,3,5-Triazines by [ONO]-Pincer-Supported Nickel(II) Complexes via an Acceptorless Dehydrogenative Coupling Strategy. J. Org. Chem. 2025, 90, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Garcia-Bosch, I. Metal Complexes Bearing Redox-Active Supporting Ligands that promote Chemical Transformations involving protons and electrons. In Redox-Active Ligands: Concepts and Catalysis; Desage-El Murr, M., Ed.; Wiley: Hoboken, NJ, USA, 2024. [Google Scholar] [CrossRef]
- Awe, B.; Swart, G.; Erasmus, E.; Malan, F.P. Synthesis, characterization, and catalytic evaluation of Ru-ONO complexes featuring N- and P-based ligands. Eur. J. Inorg. Chem. 2024, 28, e202400619. [Google Scholar] [CrossRef]
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT-Integrated space group and crystal structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Nicaise, O.J.C.N.; Mercado, B.Q.; Araujo, A.J.; Bertolotti, N.R.; Erickson, S.L.; Trucchio, J.P.; Corbett, M.J.; Padover, C.J.; Coulombe, S.L.; et al. CCDC 2402486: Experimental Crystal Structure Determination. CSD Commun. 2025. [Google Scholar] [CrossRef]
- Wong, O.; Tsuzuki, N.; Richardson, M.; Rytting, H.; Konishi, R.; Higuchi, T. An Improved Synthesis of 1-alykyl-4-imidazoline-2-ones. Heterocycles 1987, 26, 3153–3158. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, C71, 9–18. [Google Scholar] [CrossRef]
- Gonzalez Nieves, K.; Pinero Cruz, D.M. Crystal structure of a nickel compound comprising two nickel(II) complexes with different ligand environments: [Ni(tren)(H2O)2][Ni(H2O)6](SO4)2. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 314–317. [Google Scholar] [CrossRef]
- Baldwin, M.J.; Krause Bauer, J.A. cis-Bis(ethylenediamine)bis(pyridine)nickel(II) dintrate. Cryst. Struct. Commun. 1999, C55, 1772–1773. [Google Scholar] [CrossRef]
- Glocker, G. Carbon-oxygen bond energies and bond distances. J. Phys. Chem. 1958, 62, 1049–1054. [Google Scholar] [CrossRef]
- Ribeiro, M.A.; Lanznaster, M.; Silva, M.M.P.; Resende, J.A.L.C.; Pinheiro, M.V.B.; Krambrock, K.; Stumpf, H.O.; Pinheiro, C.B. Cobalt lawsone complexes: Searching for new valence tautomers. Dalton Trans. 2013, 42, 5462. [Google Scholar] [CrossRef]
- Miecznikowski, J.R.; Jasinski, J.P.; Ostrowski, T.J.; Landy, K.R.; Bonitatibus, S.C.; Smolinsky, A.N.; Bertolotti, N.R. Crystal Structure and spectroscopic properties of aqua-dichloro{1,1′-[(pyridine-2,6-diyl-κN)(bis-(methylene)]bis(4-butyl-4,5-dihydro-1H-1,2,4-triazole-5-thione-κN2)}cobalt(II). Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, E76, 1757–1761. [Google Scholar] [CrossRef]
- Fang, Z.-Y.; Zhang, L.; Ma, J.-P.; Zhao, L.; Wang, S.-L.; Xie, N.-H.; Liu, Y.-Q.; Guo, X.-Y.; Qin, J. Dinuclear cobalt and nickel complexes of a mercaptoacetic acid substituted 1,2,4 triazole ligand: Syntheses, structures, and urease inhibitory studies. Acta Crystallogr. Sect. C Struct. Chem. 2019, C75, 1658–1665. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).