

Dissolution and Pharmacokinetic Studies of Paracetamol-4,4′-Bipyridine Cocrystals Obtained Using Four Methods

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment
2.2.1. Polarized Light Microscopy (PLM)
2.2.2. Single-Crystal X-Ray Diffraction (SXRD)
2.2.3. Powder X-Ray Diffraction (PXRD)
2.2.4. Thermal Gravity Analysis (TGA)
2.2.5. Elemental Analysis (EA)
2.2.6. Infrared Spectral Analysis (IR)
2.2.7. Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS)
2.2.8. Pharmacokinetic Study (PK)
2.3. Synthesis
2.3.1. Ultrasonic Method
2.3.2. Solution Method
2.3.3. Condensation Reflux Method
2.3.4. Grinding Method
3. Results and Discussion
3.1. PLM Photograph
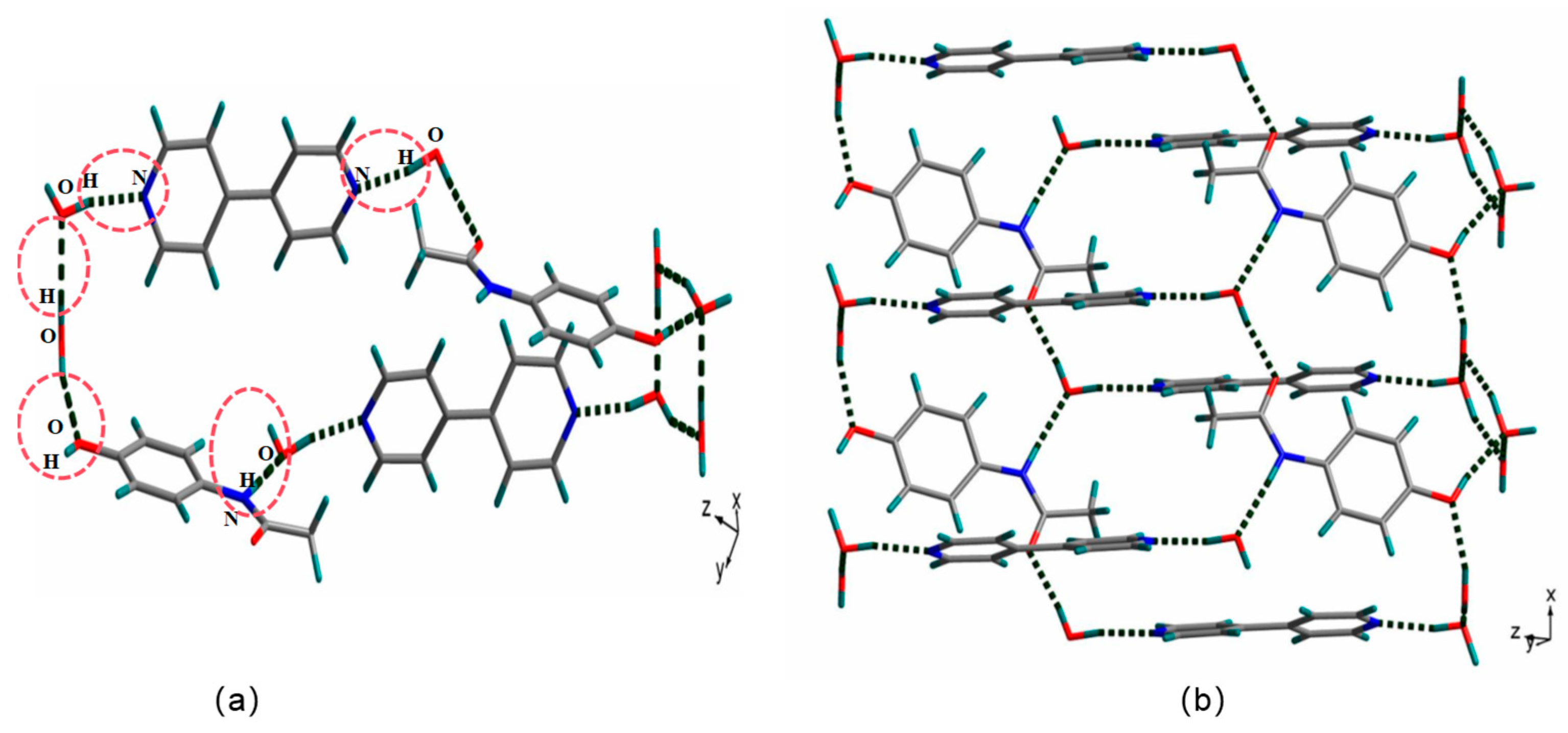
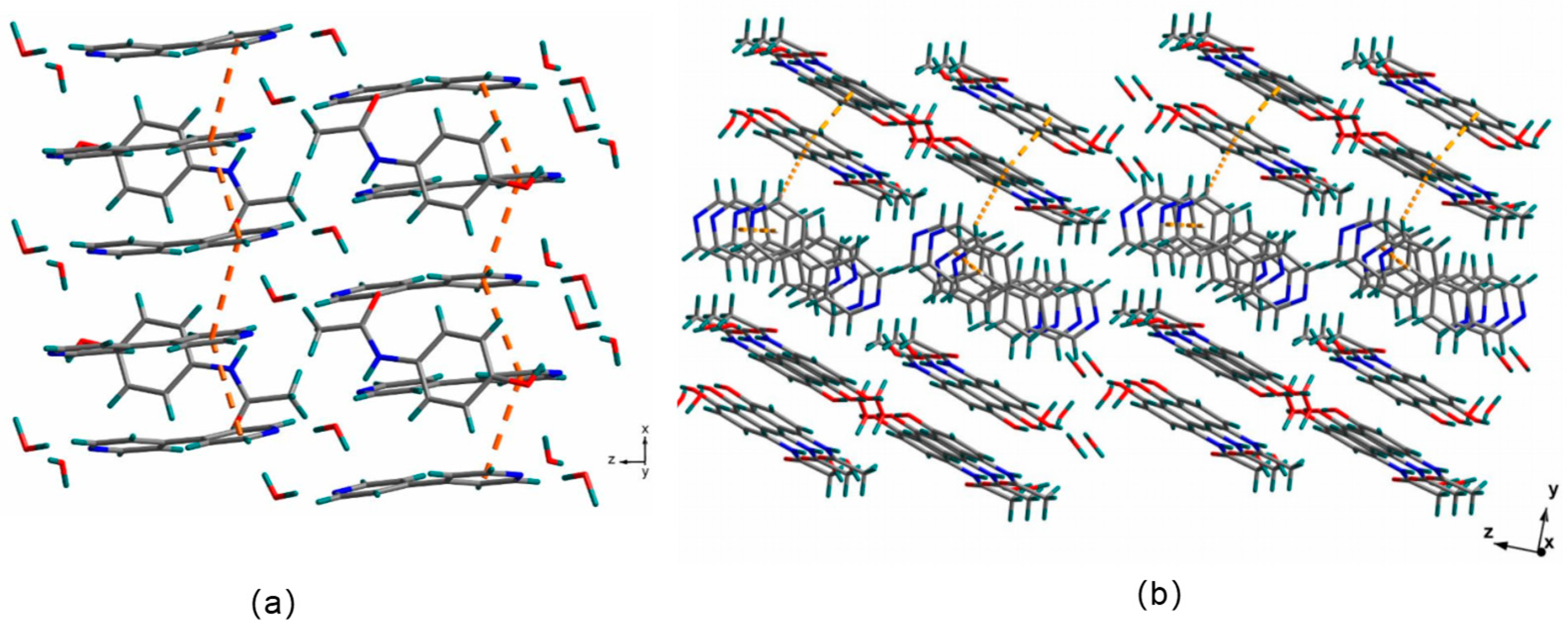
3.2. Crystal Structures
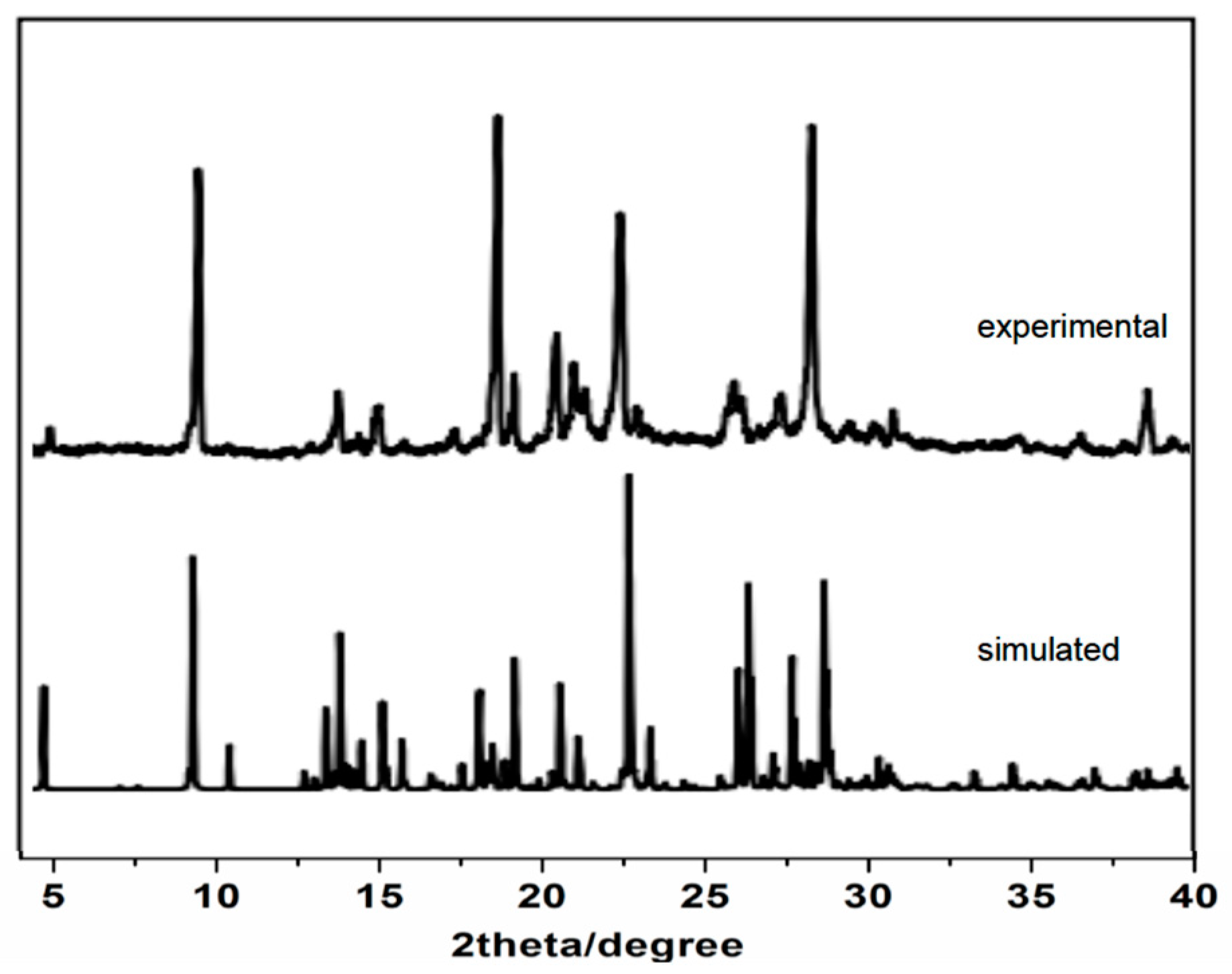
3.3. PXRD Analysis
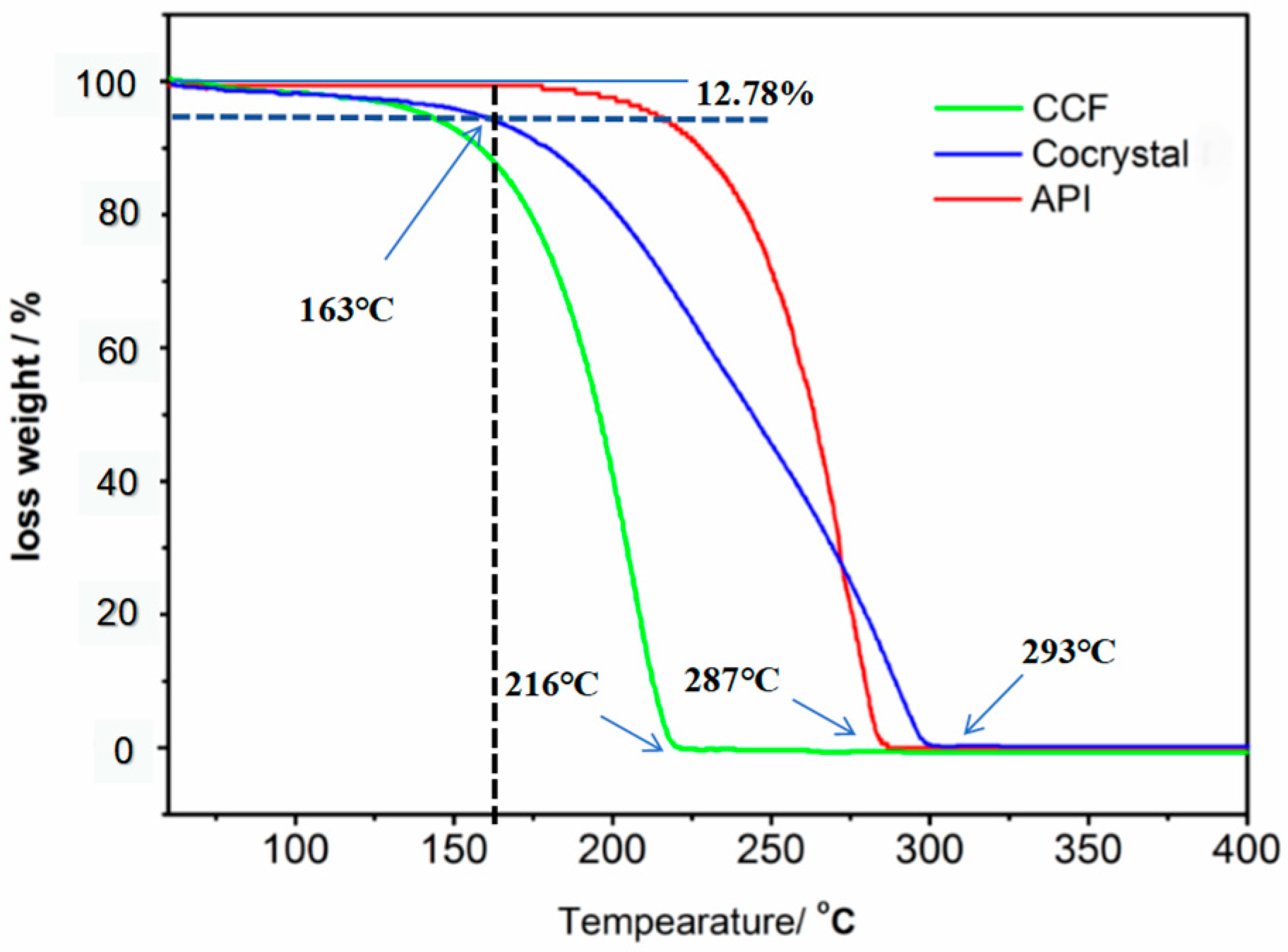
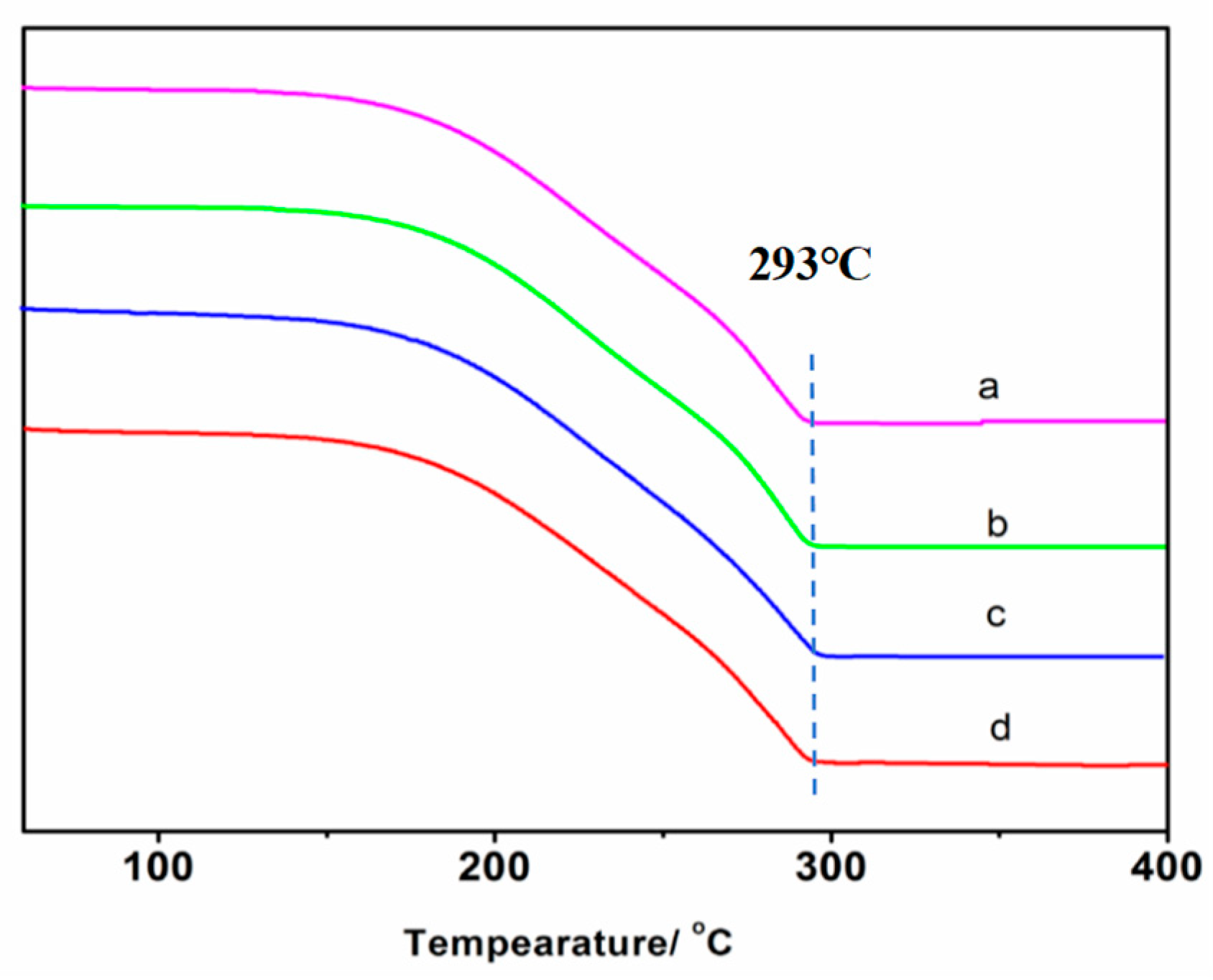
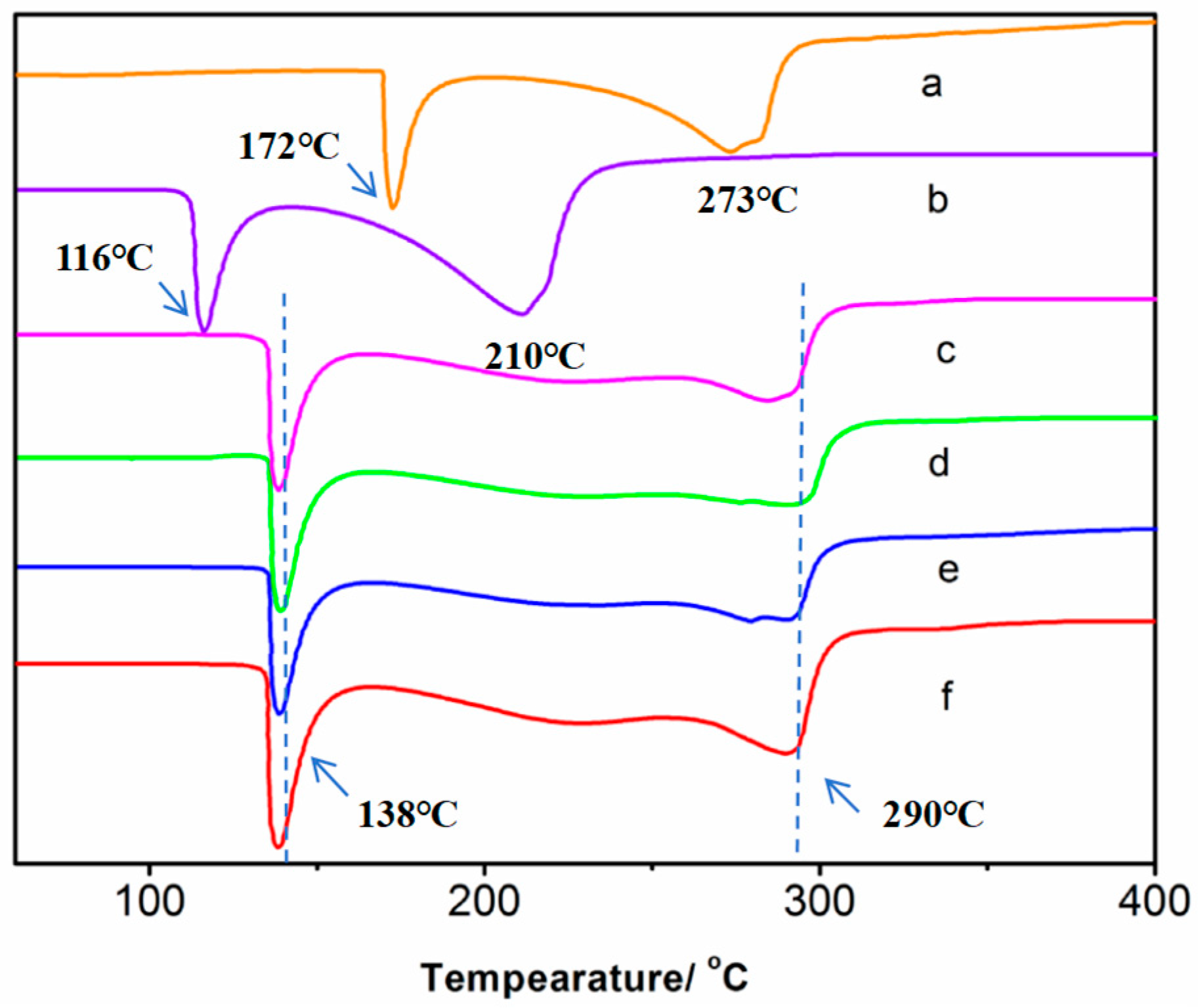
3.4. Thermal Analysis
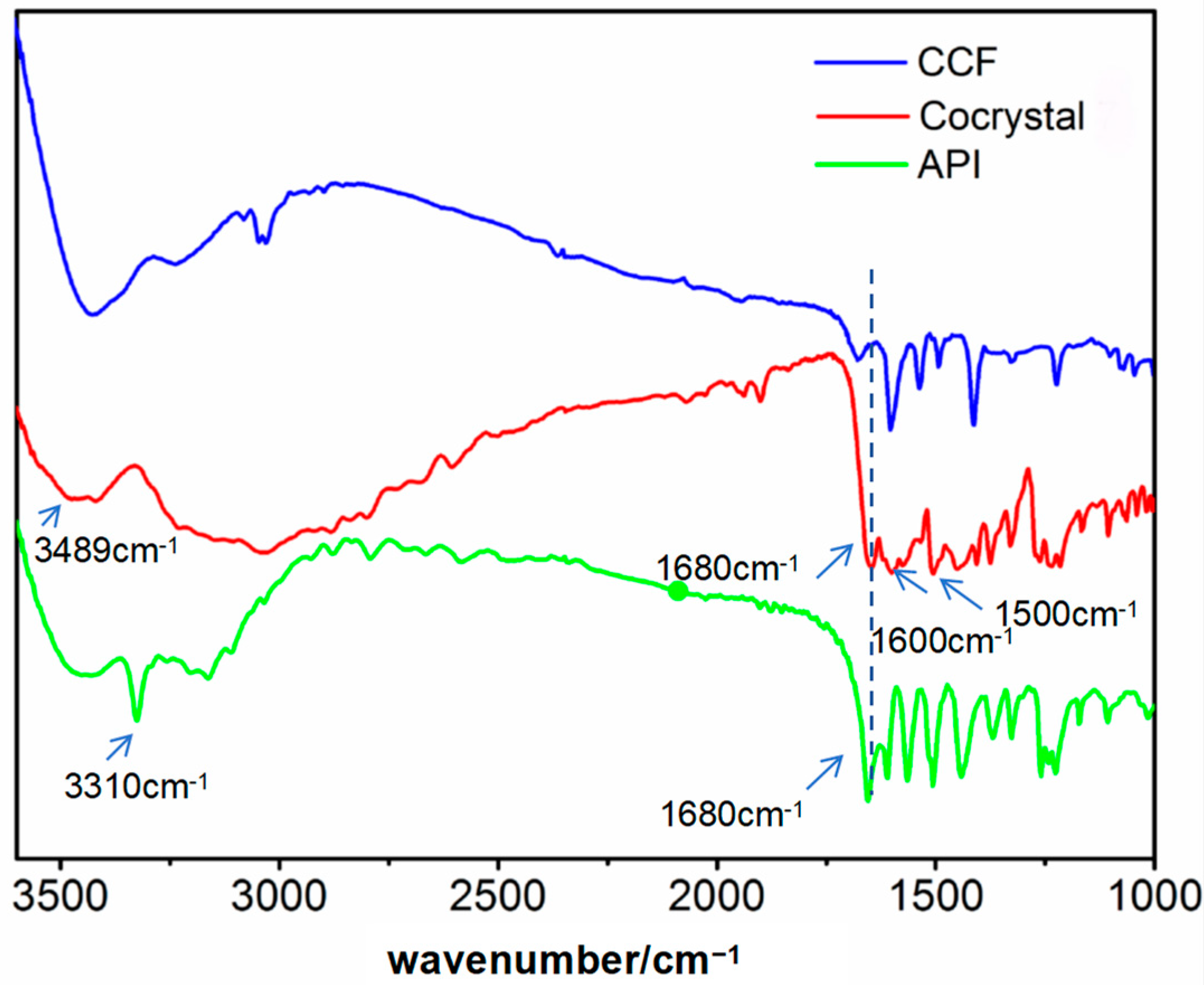
3.5. IR
3.6. EA
3.7. Dissolution Analysis
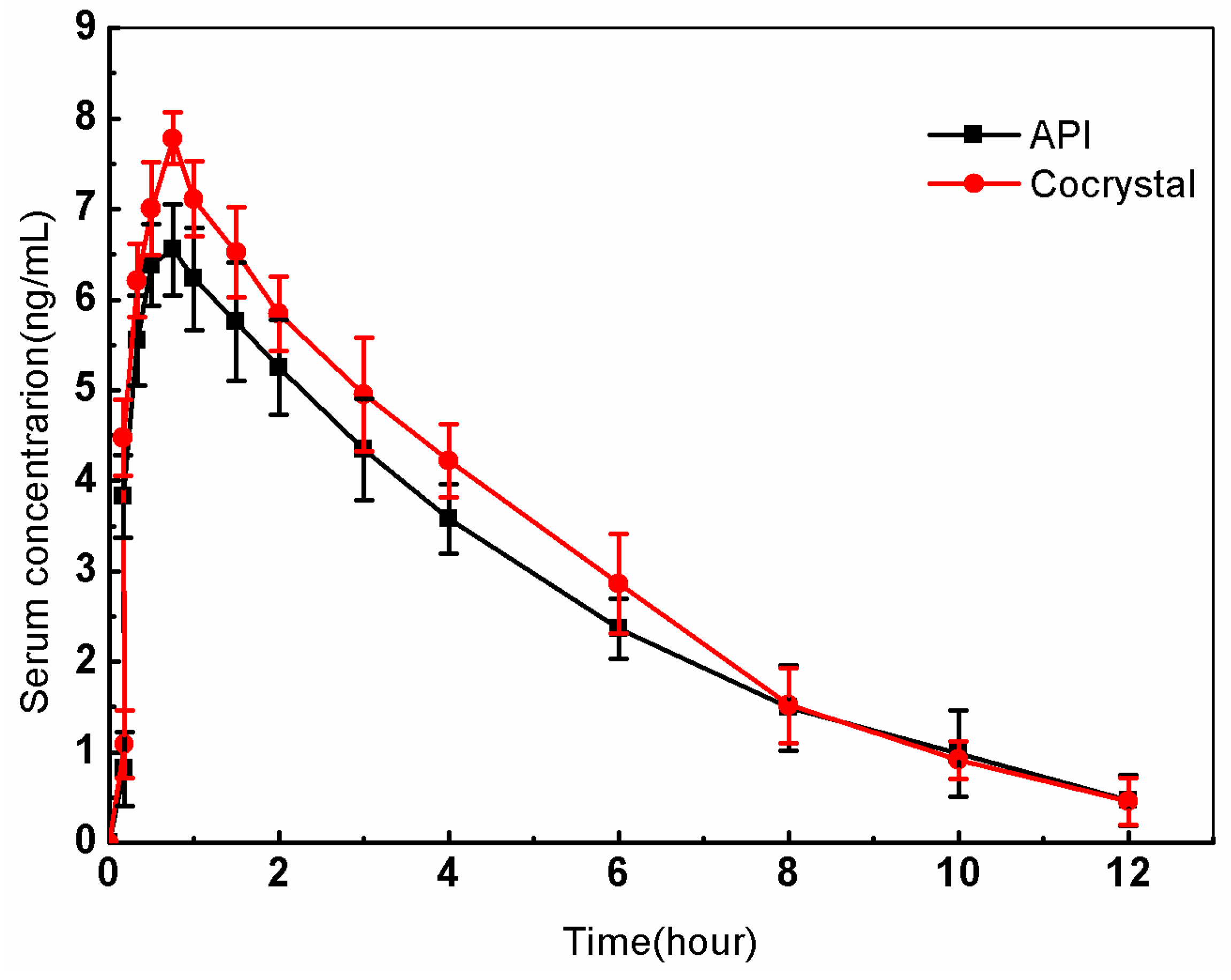
3.8. Pharmacokinetics (PK)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Li, M.; Deng, S.; Yu, T.; Ma, Y.; Yang, H.; Ma, L. A network pharmacology-integrated metabolomics strategy for clarifying the action mechanisms of Schisandrae Chinensis Fructus for treating drug-induced liver injury by acetaminophen. Bioorg. Med. Chem. 2021, 31, 115992. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.F. Paracetamol: Past, present, and future. Am. J. Ther. 2000, 7, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Freo, U.; Ruocco, C.; Valerio, A.; Scagnol, I.; Nisoli, E. Paracetamol: A review of guideline recommendations. J. Clin. Med. 2021, 10, 3420. [Google Scholar] [CrossRef]
- Cao, Z.; Han, K.; Lu, H.; Illangamudalige, S.; Shaheed, C.A.; Chen, L.; Mathieson, S. Paracetamol Combination Therapy for Back Pain and Osteoarthritis: A Systematic Review and Meta-Analyses. Drugs 2024, 84, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Abdelbary, G.; Prinderre, P.; Eouani, C.; Joachim, J.; Reynier, J.P.; Piccerelle, P.H. The preparation of orally disintegrating tablets using a hydrophilic waxy binder. Int. J. Pharm. 2004, 278, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Seager, H. Drug-delivery products and the Zydis fast-dissolving dosage form. J. Pharm. Pharmacol. 1998, 50, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.F. Paracetamol (acetaminophen) poisoning: The early years. Br. J. Clin. Pharmacol. 2024, 90, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Xu, Z.; Qiao, Z.; Wang, X.; Yang, C. Flaxseed lignan alleviates the paracetamol-induced hepatotoxicity associated with regulation of gut microbiota and serum metabolome. Nutrients 2024, 16, 295. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Singh, T.; Singh, V.; Singh, B.; Kaur, S.; Ahmad, S.F.; Singh, B. Ehretia laevis mitigates paracetamol-induced hepatotoxicity by attenuating oxidative stress and inflammation in rats. Int. Immunopharmacol. 2024, 143, 113565. [Google Scholar] [CrossRef]
- Cousin, G.; Bruna, E.; Gendrot, E. Rapidly Disintegratable Multiparticular Tablet. U.S. Patent 5,464,632, 7 November 1995. [Google Scholar]
- Mallet, C.; Desmeules, J.; Pegahi, R.; Eschalier, A. An updated review on the metabolite (AM404)-mediated central mechanism of action of paracetamol (acetaminophen): Experimental evidence and potential clinical impact. J. Pain Res. 2023, 16, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Chidiac, A.S.; Buckley, N.A.; Noghrehchi, F.; Cairns, R. Paracetamol (acetaminophen) overdose and hepatotoxicity: Mechanism, treatment, prevention measures, and estimates of burden of disease. Expert Opin. Drug Metab. Toxicol. 2023, 19, 297–317. [Google Scholar] [CrossRef]
- Majumdar, S.; Sloan, K.B. Synthesis and topical delivery of N-alkyl-N-alkyloxycarbonylaminomethyl prodrugs of a model phenolic drug: Acetaminophen. Int. J. Pharm. 2007, 337, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Devarajan-Ketha, H.; Sloan, K.B. N,N′-Dialkylaminoalkylcarbonyl (DAAC) prodrugs and aminoalkylcarbonyl (AAC) prodrugs of 4-hydroxyacetanilide and naltrexone with improved skin permeation properties. Bioorg. Med. Chem. Lett. 2011, 21, 4078–4082. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, P.; Kolekar, K.; Khot, C.; Dabhole, S.; Salawi, A.; Sabei, F.Y.; Patravale, V. Co-crystal nanoarchitectonics as an emerging strategy in attenuating cancer: Fundamentals and applications. J. Control. Release 2023, 353, 1150–1170. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tang, Y.; Wu, Q.; Li, W.; Zhou, L.; Wang, M.; Zou, F. A drug–drug cocrystal strategy to regulate stability and solubility: A case study of temozolomide/caffeic acid. J. Mol. Struct. 2024, 1312, 138577. [Google Scholar] [CrossRef]
- Alvani, A.; Shayanfar, A. Cocrystallization for Improving Anticancer Activity of Drugs. Cryst. Res. Technol. 2024, 59, 2300253. [Google Scholar] [CrossRef]
- Sahoo, R.N.; Patel, A.; Satapathy, B.S.; Mallick, S. Formulation development and characterization of lamotrigine-salicylic acid crystalline product: A strategy to improve oral release of drug for better management of epilepsy. Indian J. Chem. Technol. 2021, 28, 356–362. [Google Scholar]
- Wang, X.; Wang, L.; Yao, C.; Xie, G.; Song, S.; Li, H.; Tao, X. Novel formulations of the antiviral drug favipiravir: Improving permeability and tabletability. Cryst. Growth Des. 2021, 21, 3807–3817. [Google Scholar] [CrossRef]
- Liang, X.; Liu, S.; Li, Z.; Deng, Y.; Jiang, Y.; Yang, H. Efficient cocrystal coformer screening based on a Machine learning Strategy: A case study for the preparation of imatinib cocrystal with enhanced physicochemical properties. Eur. J. Pharm. Biopharm. 2024, 196, 114201. [Google Scholar] [CrossRef] [PubMed]
- Mezaal, E.N.; Sadiq, K.A.; Jabbar, M.M.; Al-Noor, T.H.; Azooz, E.A.; Al-Mulla, E.A.J. Green methods for determination of paracetamol in drug samples: A comparative study. Green Anal. Chem. 2024, 10, 100123. [Google Scholar] [CrossRef]
- Chettri, A.; Subba, A.; Singh, G.P.; Bag, P.P. Pharmaceutical co-crystals: A green way to enhance drug stability and solubility for improved therapeutic efficacy. J. Pharm. Pharmacol. 2024, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, X.R. Synthesis of two novel azilsartan cocrystals: Preparation, physicochemical characterization and solubility studies. Crystals 2020, 10, 739. [Google Scholar] [CrossRef]
- Fala, L. Entresto: First-in-class angiotensin receptor neprilysin inhibitor FDA approved for patients with heart failure. Am. Health Drug Benefits 2015, 8, 330. [Google Scholar] [PubMed]
- Berg, D.D.; Braunwald, E.; DeVore, A.D.; Lala, A.; Pinney, S.P.; Duffy, C.I.; Morrow, D.A. Efficacy and safety of sacubitril/valsartan by dose level achieved in the PIONEER-HF trial. Heart Fail. 2020, 8, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Gascom, N.; Almansa, C.; Merlos, M.; Miguel Vela, J.; Encina, G.; Morte, A.; Smith, K.; Plata-Salaman, C. Co-crystals of tramadol-celecoxib: Preclinical and clinical evaluation of a novel analgesic. Expert Opin. Investig. Drugs 2019, 28, 399–409. [Google Scholar] [CrossRef]
- Yousef, M.A.; Vangala, V.R. Pharmaceutical cocrystals: Molecules, crystals, formulations, medicines. Cryst. Growth Des. 2019, 19, 7420–7438. [Google Scholar] [CrossRef]
- Karki, S.; Friščić, T.; Fabián, L.; Laity, P.R.; Day, G.M.; Jones, W. Improving Mechanical Properties of Crystalline Solids by Cocrystal Formation: New Compressible Forms of Paracetamol. Adv. Mater. 2009, 21, 3905–3909. [Google Scholar] [CrossRef]
- Wang, X.; Du, S.; Zhang, R.; Jia, X.; Yang, T.; Zhang, X. Drug-drug cocrystals: Opportunities and challenges. Asian J. Pharm. Sci. 2021, 16, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Liu, J.; Ran, L. A review of pharmaceutical nano-cocrystals: A novel strategy to improve the chemical and physical properties for poorly soluble drugs. Crystals 2021, 11, 463. [Google Scholar] [CrossRef]
- Mirocki, A.; Conterosito, E.; Palin, L.; Sikorski, A.; Milanesio, M.; Lopresti, M. Crystal structure of a new 1: 1 acridine-diclofenac salt, obtained with high yield by a mechanochemical approach. Crystals 2022, 12, 1573. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Ye, X.; Yao, C.; Song, S.; Qu, Y.; Tao, X. Efficient Screening of Pharmaceutical Cocrystals by Microspacing In-Air Sublimation. J. Am. Chem. Soc. 2024, 146, 11592–11598. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, A.J.; Sugden, P.; Wilson, C.C.; Castro-Dominguez, B. Elusive Seed Formation via Electrical Confinement: Control of a Novel Cocrystal in Cooling Crystallization. Cryst. Growth Des. 2021, 21, 3310–3315. [Google Scholar] [CrossRef]
- Ismail, K.M.; Hassan, S.S.; Medany, S.S.; Hefnawy, M.A. A facile sonochemical synthesis of the Zn-based metal–organic framework for electrochemical sensing of paracetamol. Mater. Adv. 2024, 5, 5870–5884. [Google Scholar] [CrossRef]
- Acebedo-Martínez, F.J.; Alarcón-Payer, C.; Barrales-Ruiz, H.M.; Niclós-Gutiérrez, J.; Domínguez-Martín, A.; Choquesillo-Lazarte, D. Towards the Development of Novel Diclofenac Multicomponent Pharmaceutical Solids. Crystals 2022, 12, 1038. [Google Scholar] [CrossRef]
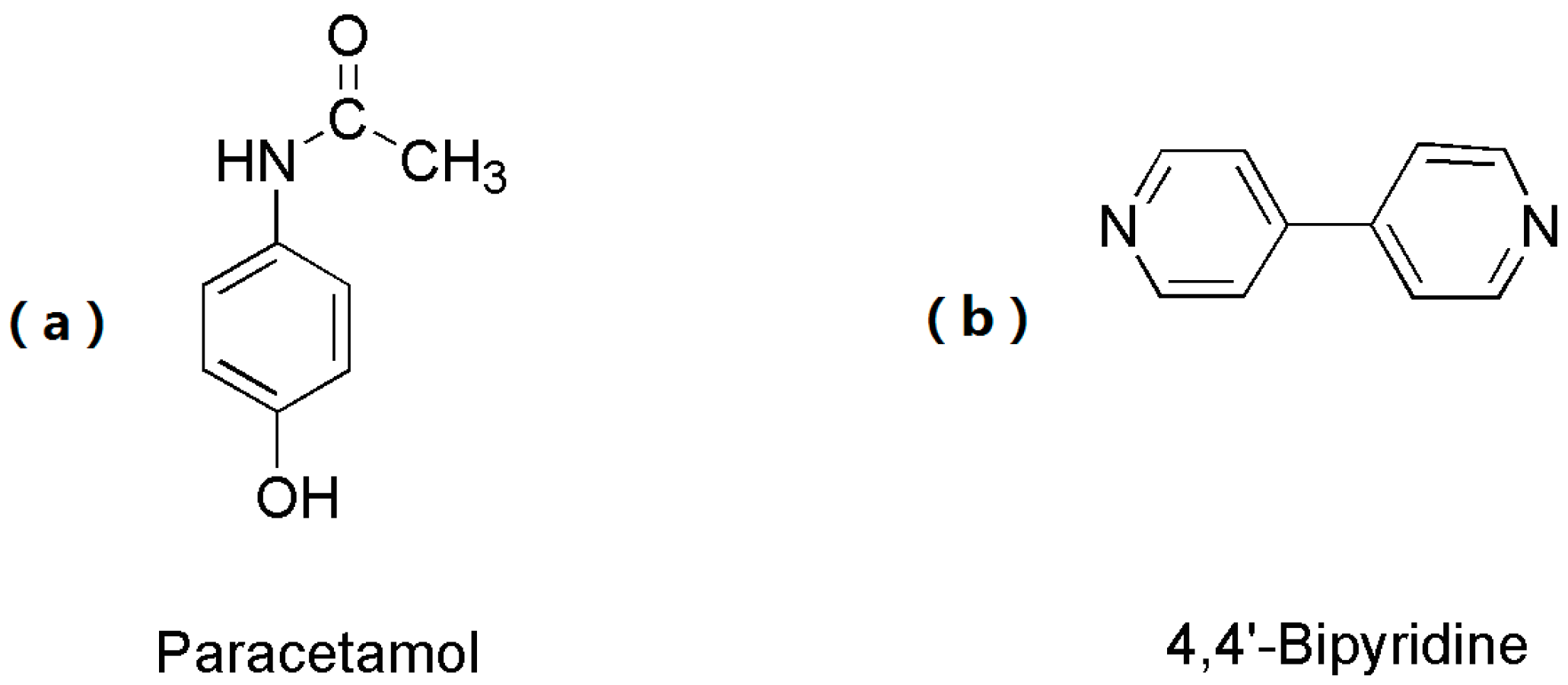




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cocrystal |
|---|---|
| Chemical formula | C36H44N6O9 |
| Original API | paracetamol |
| Cocrystal former | 4,4′-bipyridine |
| Formula weight | 704.77 |
| Crystal system | Triclinic |
| Space group | P-1 |
| a (Å) | 6.9380 (5) |
| b (Å) | 13.007 (6) |
| c (Å) | 18.998 (12) |
| α (°) | 100.443 (2) |
| β (°) | 92.3710 (10) |
| γ (°) | 99.250 (10) |
| vol (Å3) | 1725.47 (8) |
| dcal (g·cm−3) | 1.413 |
| Z | 2 |
| Nref | 751 |
| T (K) | 296 (2) |
| R1 | 0.0590 |
| wR2 | 0.1956 |
| Gof | 1.069 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Huang, Y.; Li, J.; Chen, Y.; Lian, J. Dissolution and Pharmacokinetic Studies of Paracetamol-4,4′-Bipyridine Cocrystals Obtained Using Four Methods. Crystals 2025, 15, 70. https://doi.org/10.3390/cryst15010070
Zhang X, Huang Y, Li J, Chen Y, Lian J. Dissolution and Pharmacokinetic Studies of Paracetamol-4,4′-Bipyridine Cocrystals Obtained Using Four Methods. Crystals. 2025; 15(1):70. https://doi.org/10.3390/cryst15010070
Chicago/Turabian StyleZhang, Xiaoming, Yejia Huang, Jinliang Li, Yiying Chen, and Jialing Lian. 2025. "Dissolution and Pharmacokinetic Studies of Paracetamol-4,4′-Bipyridine Cocrystals Obtained Using Four Methods" Crystals 15, no. 1: 70. https://doi.org/10.3390/cryst15010070
APA StyleZhang, X., Huang, Y., Li, J., Chen, Y., & Lian, J. (2025). Dissolution and Pharmacokinetic Studies of Paracetamol-4,4′-Bipyridine Cocrystals Obtained Using Four Methods. Crystals, 15(1), 70. https://doi.org/10.3390/cryst15010070