



Crystal Structure of New Zinc-Hydroxy-Sulfate-Hydrate Zn4(OH)6SO4·2–2.25H2O

Abstract
1. Introduction
2. Materials and Methods
2.1. Obtaining a New Phase
2.2. Methods
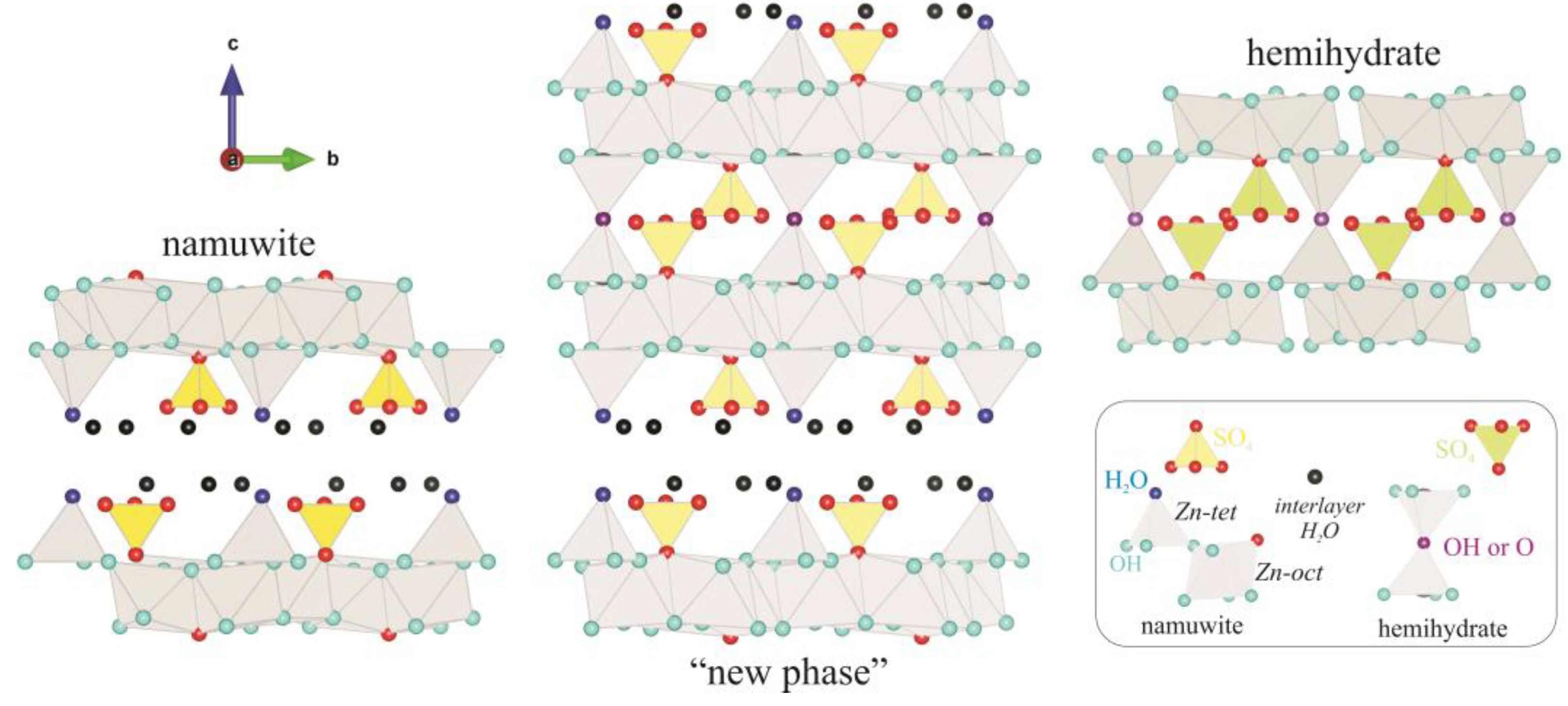
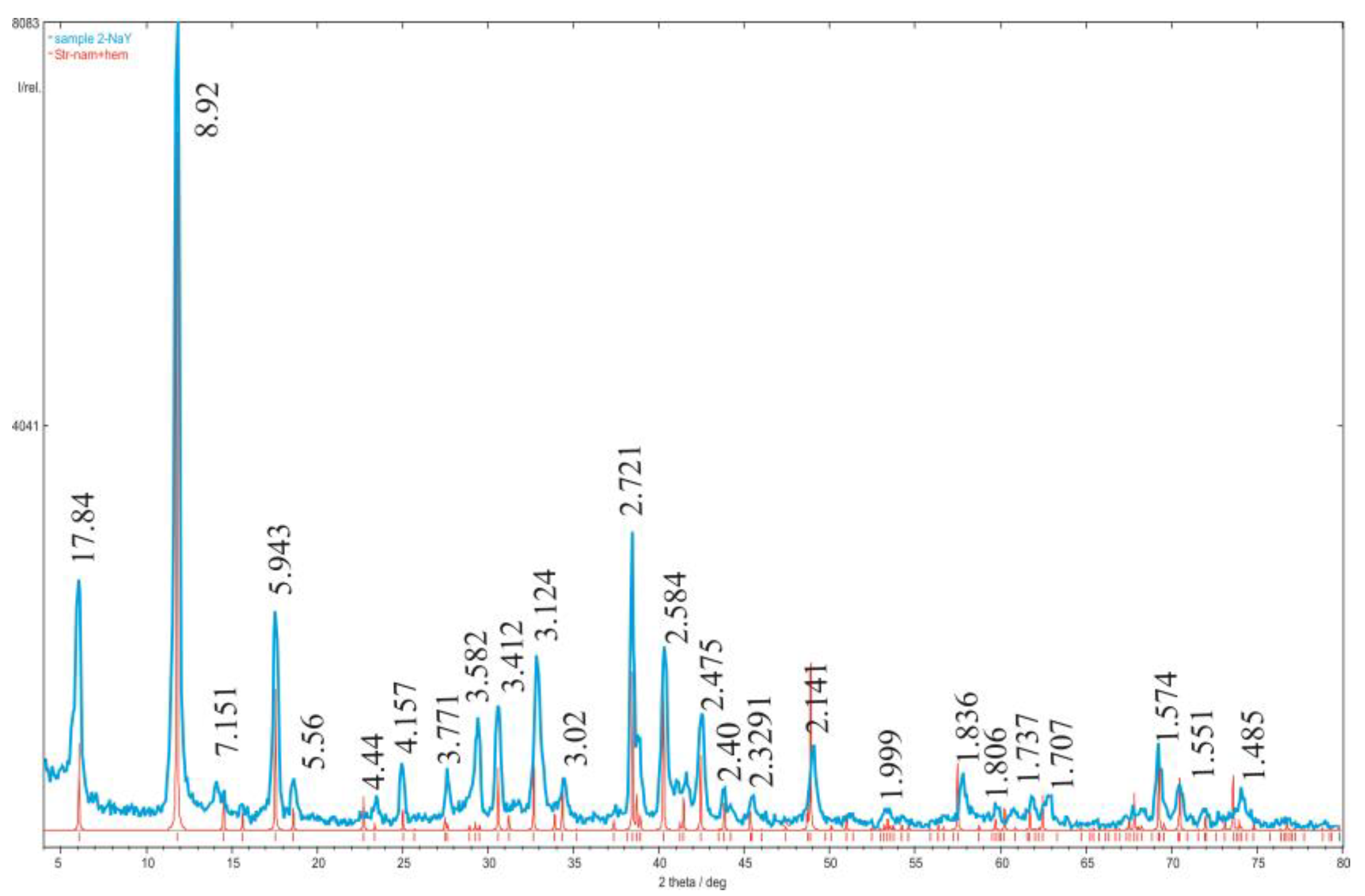
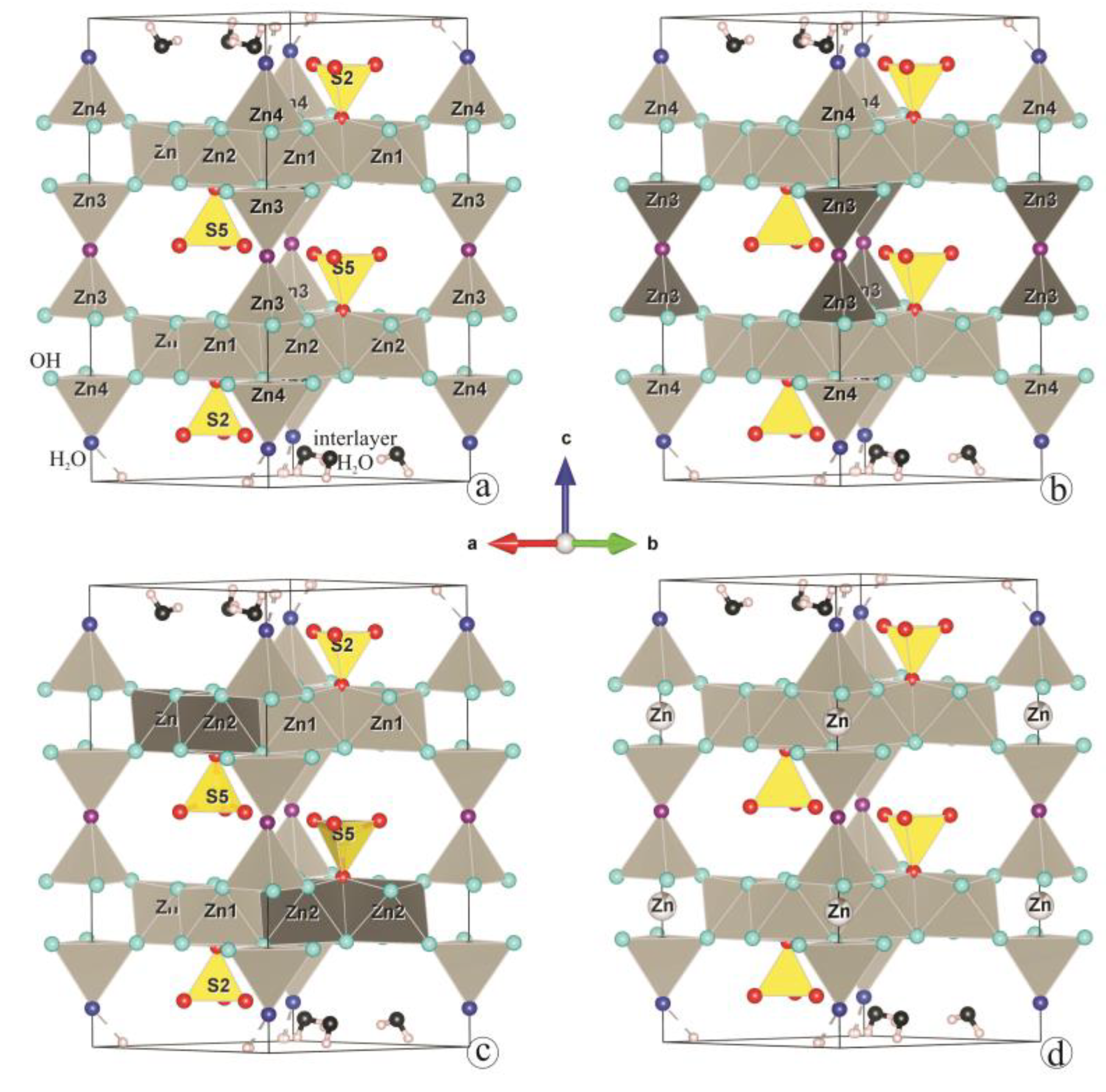
3. Results and Discussion
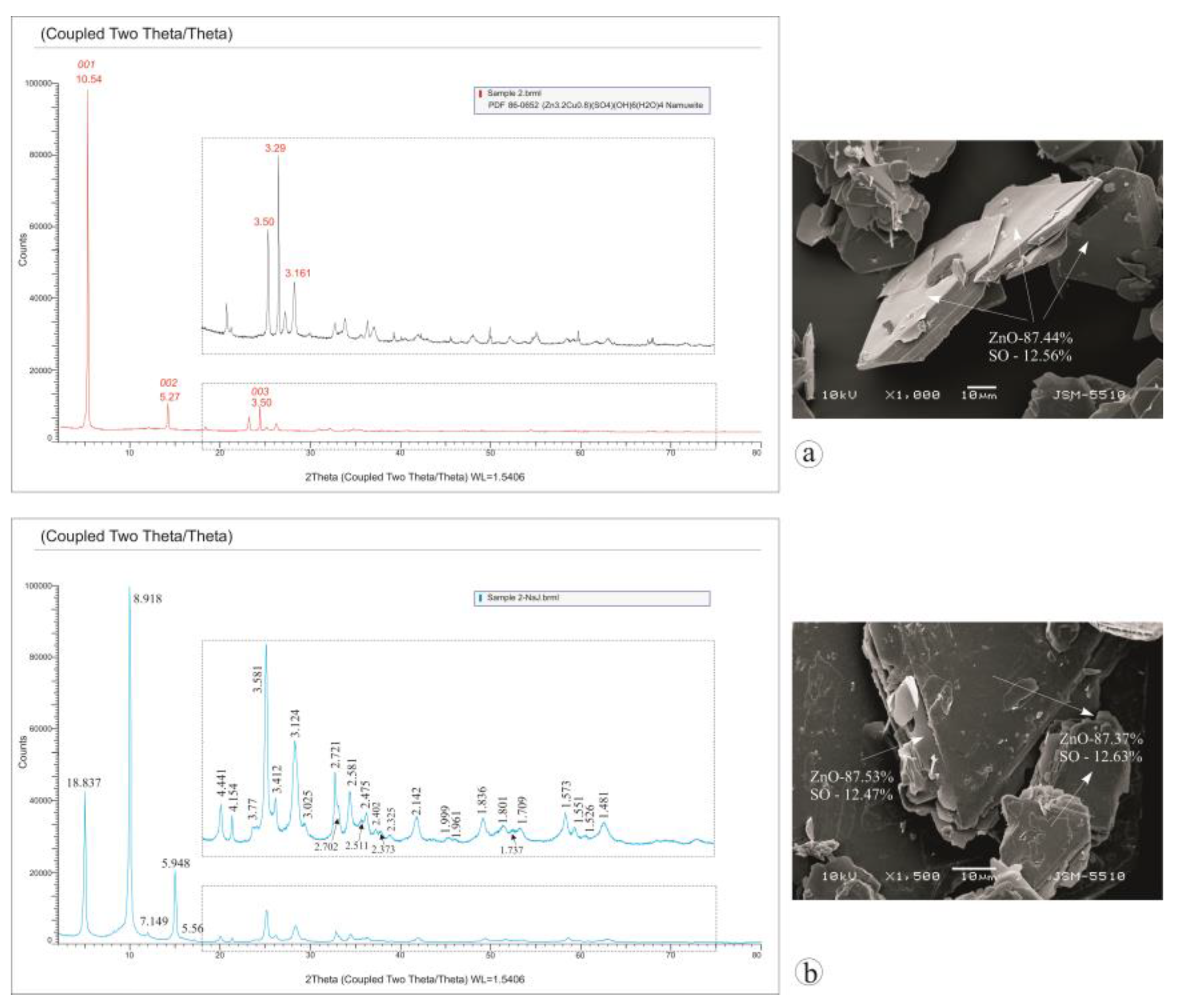
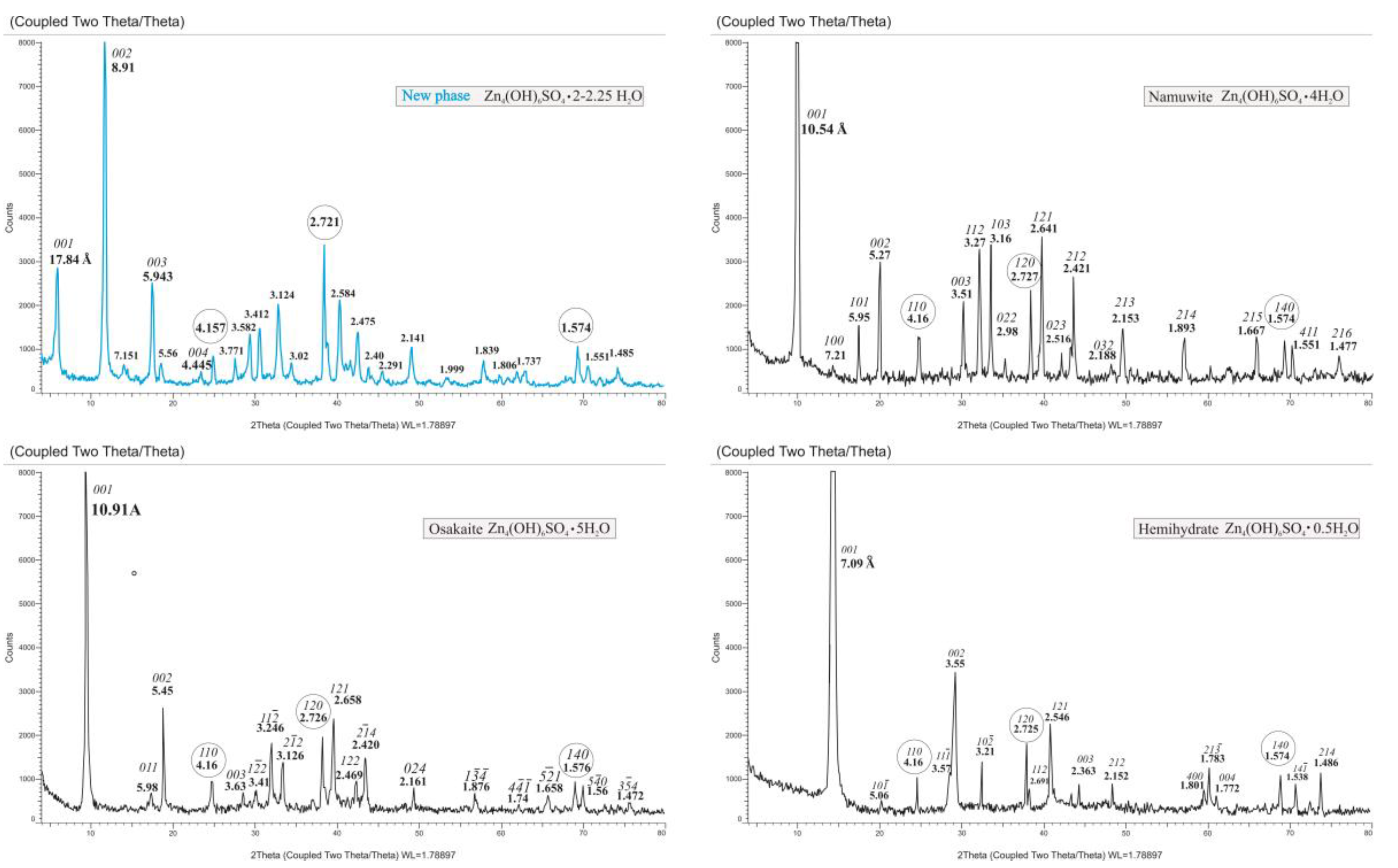
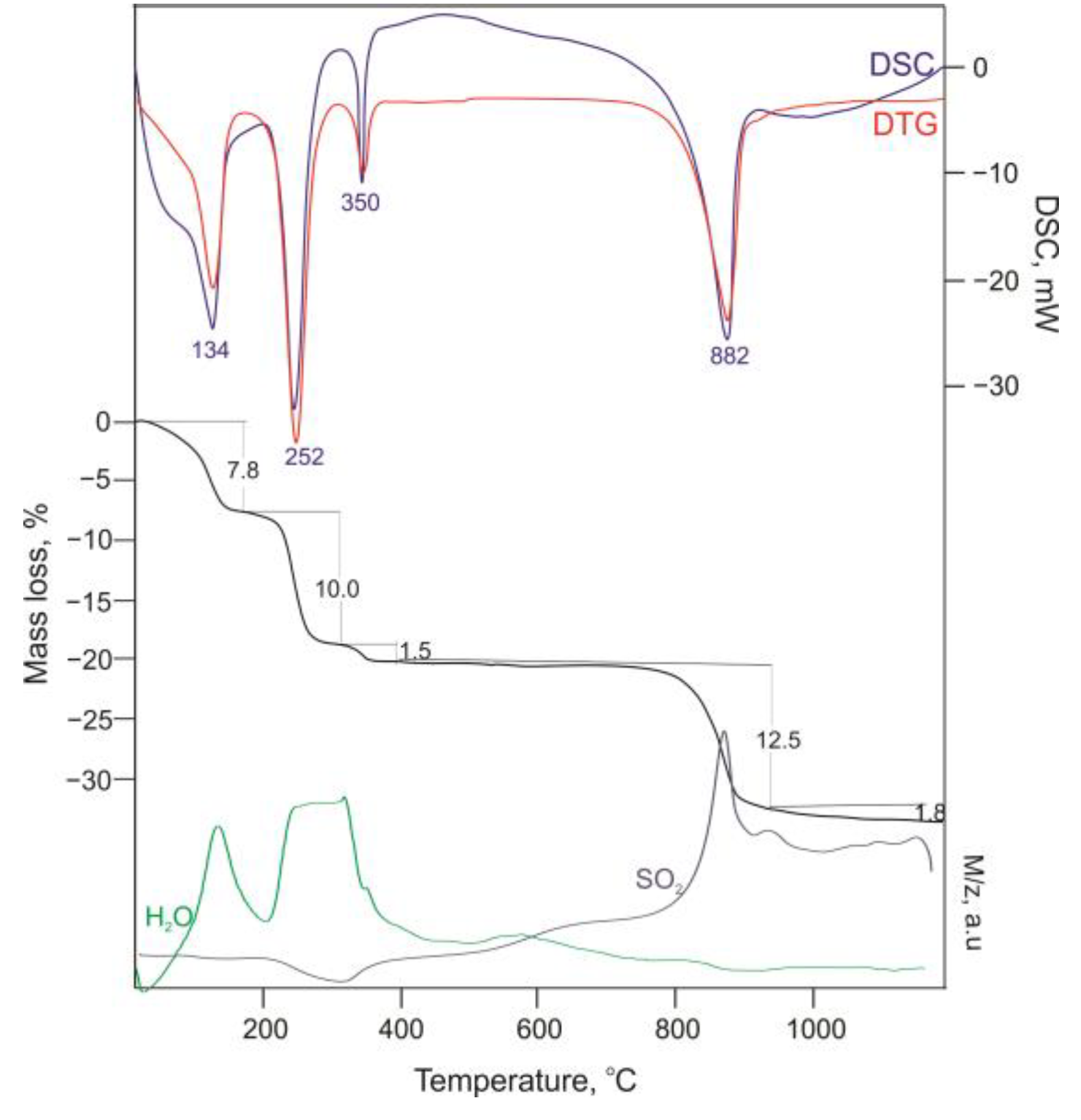
3.1. Characterization of the Starting Materials and the Choice of the Structural Model
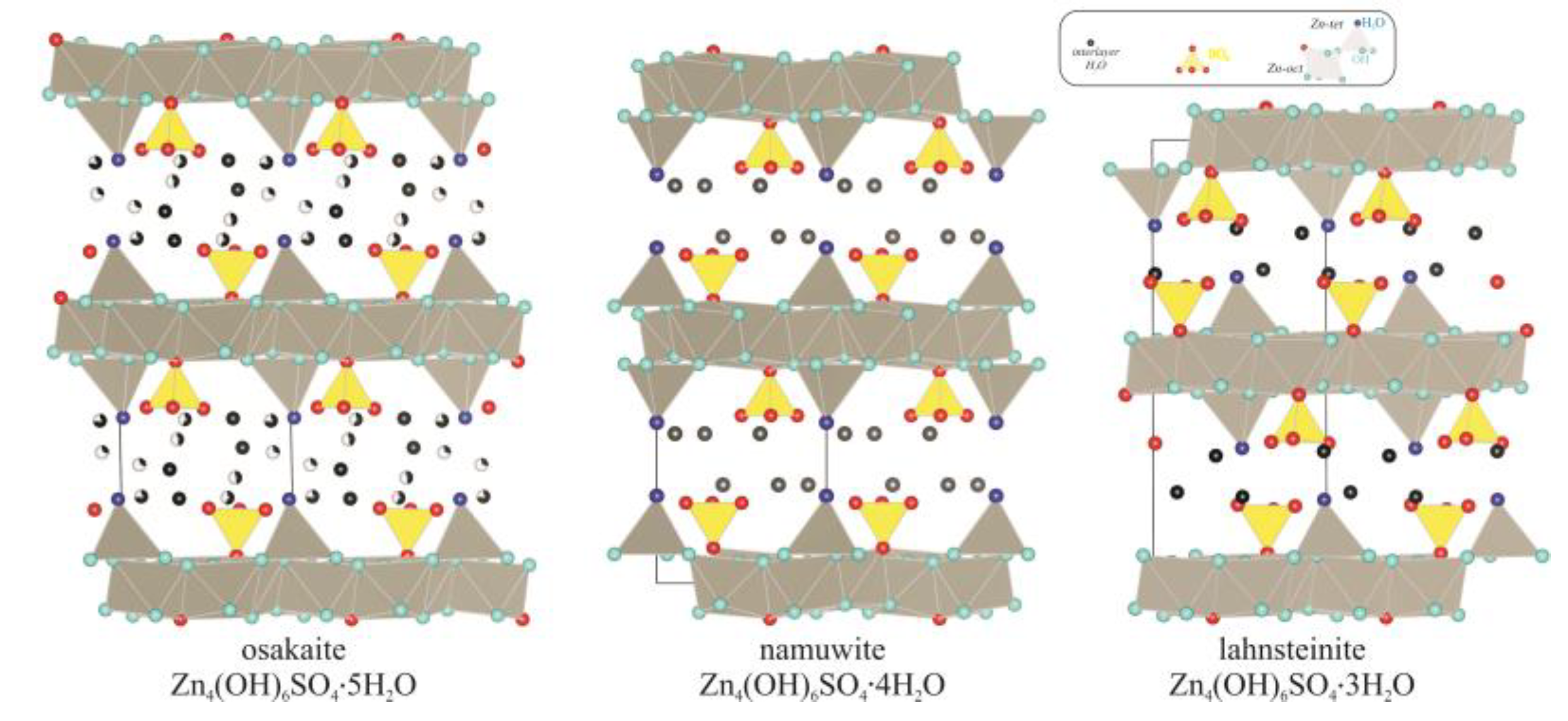
3.2. Structure Refinement and Description
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawthorne, F.C.; Schindler, M. Understanding the weakly bonded constituents in oxysalt minerals. Z. Krist. 2008, 223, 41–68. [Google Scholar] [CrossRef]
- Stanimirova, T. Crystal chemical preconditions for the species diversity of minerals-hydroxy-salts. Geoscience 2017, 78, 35–36. [Google Scholar]
- Carbajal Arizaga, G.G.; Satyanarayana, K.G.; Wypych, F. Layered hydroxide salts: Synthesis, properties and potential applications. Solid State Ion. 2007, 178, 1143–1162. [Google Scholar] [CrossRef]
- Maruyama, S.A.; Krause, F.; Tavares Filho, S.R.; Leitão, A.A.; Wypych, F. Synthesis, cation exchange and dehydration/rehydration of sodium gordaite: NaZn4(OH)6(SO4)Cl·6H2O. Appl. Clay Sci. 2017, 146, 100–105. [Google Scholar] [CrossRef]
- Stanimirova, T. Exchange reactions of zinc hydroxide-sulfate minerals in halide solutions. Appl. Clay Sci. 2019, 168, 396–408. [Google Scholar] [CrossRef]
- Leal, D.A.; Silva, G.M.; Tedim, J.; Wypych, F.; Marino, C.E.B. Synthesis and characterization of gordaite, osakaite and simonkolleite by different methods: Comparison, phase interconversion, and potential corrosion protection applications. J. Solid State Chem. 2020, 291, 121595. [Google Scholar] [CrossRef]
- Nakagaki, S.; Machado, G.S.; Stival, J.F.; dos Santos, E.H.; Silva, G.M.; Wypych, F. Natural and synthetic layered hydroxide salts (LHS): Recent advances and application perspectives emphasizing catalysis. Prog. Solid State Chem. 2021, 64, 100335. [Google Scholar] [CrossRef]
- Silva, G.M.; Wypych, F. A novel and facile synthesis route for obtaining highly crystalline impurity free layered hydroxide sulfates: Gordaite and osakaite. Inorg. Chem. Commun. 2022, 143, 109723. [Google Scholar] [CrossRef]
- Stanimirova, T.; Delcheva, Z.; Petrova, N. New phase obtained at mutual transformations of zinc hydroxy-salts. Bulg. Chem. Commun. 2018, 50, 63–72. [Google Scholar]
- Maruyama, S.A.; Molgero Westrup, K.C.; Nakagaki, S.; Wypych, F. Immobilization of a cationic manganese (III) porphyrin on lithium gordaite (LiZn4(OH)6(SO4)Cl·6H2O), a layered hydroxide salt with cation exchange capacity. Appl. Clay Sci. 2017, 139, 108–111. [Google Scholar] [CrossRef]
- Bear, I.J.; Gray, I.E.; Madsen, I.C.; Newnham, I.E.; Rogers, L.J. Structures of basic zinc sulfates 3Zn(OH)2·ZnSO4·mH2O, m = 3 and 5. Acta Crystallogr. 1986, B42, 32–39. [Google Scholar] [CrossRef]
- Bear, I.J.; Gray, I.E.; Madsen, I.C.; Newnham, I.E.; Rogers, L.J. The ZnSO4·3Zn(OH)2·H2O system. I. Phase formation. Aust. J. Chem. 1987, 40, 539–556. [Google Scholar] [CrossRef]
- Moezzi, A.; Cortie, M.B.; McDonagh, A.M. Zinc hydroxide sulfate and its transformation to crystalline zinc oxide. Dalton Trans. 2013, 42, 14432–14437. [Google Scholar] [CrossRef]
- Stanimirova, T.; Kerestedjian, T.; Kirov, G. Dehydration and rehydration of Zn-hydroxy sulfate minerals with interrupted decorated hydroxide sheets. Appl. Clay Sci. 2017, 135, 16–26. [Google Scholar] [CrossRef]
- Groat, L.A. The crystal structure of namuwite, a mineral with Zn in tetrahedral and octahedral coordination, and its relationship to synthetic basic zinc sulfates. Am. Mineral. 1996, 81, 238–243. [Google Scholar] [CrossRef]
- Adiwidjaja, G.; Frise, K.; Klaska, K.-H.; Schlüter, J. The crystal structure of gordaite. NaZn4(SO4) (OH)6Cl·6H2O. Z. Krist 1997, 212, 704–707. [Google Scholar]
- Burns, P.C.; Roberts, A.C.; Nikischer, A.J. The crystal structure of Ca[Zn8(SO4)2(OH)12Cl2](H2O)9, a new phase from slag dumps at Val Varena, Italy. Eur. J. Mineral. 1998, 10, 923–930. [Google Scholar] [CrossRef]
- Ohnishi, M.; Kusachi, I.; Kobayashi, S. Osakaite, Zn4SO4(OH)6·5H2O, a new mineral fromthe Hiraomine, Osaka, Japan. Can. Mineral. 2007, 45, 1511–1517. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Rastsvetaeva, R.K.; Aksenov, S.M.; Pekov, I.V.; Belakovskiy, D.I.; Blass, G.; Möhn, G. Lahnsteinite, Zn4(SO4)(OH)6·3H2O, a new mineral from the Friedrichssegen Mine, Germany. Geol. Ore Depos. 2013, 55, 663–668. [Google Scholar] [CrossRef]
- Bevins, R.E.; Turgoose, S.; Williams, P.A. Namuwite, (Zn,Cu)4SO4(OH)6·4H2O, a new mineral from Wels. Mineral. Mag. 1982, 46, 51–54. [Google Scholar] [CrossRef]
- Drits, V.A.; Sokolova, T.N.; Sokolova, G.V.; Cherkashin, V.I. New Members of the Hydrotalcite-Manasseite Group. Clays Clay Miner. 1987, 35, 401–417. [Google Scholar] [CrossRef]
- Giester, G.; Rieck, B. Bechererite, (Zn,Cu)6Zn2(OH)13[(S,Si)(O,OH)4]2, a novel mineral species from the Tonopah-Belmont mine, Arizonia. Am. Mineral. 1996, 81, 244–248. [Google Scholar] [CrossRef]
- Hoffmann, C.; Armbruster, T.; Giester, G. The acentric structure (P3) of bechererite, Zn7Cu(OH)13[SiO(OH)3SO4]. Am. Mineral. 1997, 82, 1014–1018. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Kraus, W.; Nolze, C. POWDER CELL—A program for the representation and manipulation of crystal structure and calculation of the resulting X-ray powder pattern. J. Appl. Crystallogr. 1996, 29, 301–303. [Google Scholar] [CrossRef]
- CrysAlis Pro. Agilent Technologies; UK Ltd.: Yarnton, UK, 2011. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Baur, W.H. The geometry of polyhedral distortions. Predictive relationships for the phosphate group. Acta Crystallogr. B 1974, 30, 1195–1215. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral/Phase | Formula | d001, Å * |
|---|---|---|
| osakaite | Zn4(OH)6SO4·5H2O | 10.90 |
| namuwite | Zn4(OH)6SO4·4H2O | 10.54 |
| lahnsteinite | Zn4(OH)6SO4·3H2O | 9.30 |
| dihydrate | Zn4(OH)6SO4·2H2O | 8–7.4 |
| monohydrate | Zn4(OH)6SO4·1H2O | 7.23 |
| hemihydrate | Zn4(OH)6SO4·0.5H2O | 7.09 |
| Sample | New Phase |
|---|---|
| Empirical formula/Z | H15.75 O48.81 S4 Zn15.87/1 |
| Formula weight | 1962.5 |
| Temperature/K | 290(2) |
| Crystal system | trigonal |
| Space group | P-3 |
| a/Å | 8.3418(15) |
| b/Å | 8.3418(15) |
| c/Å | 17.595(7) |
| α/° | 90 |
| β/° | 90 |
| γ/° | 120 |
| Volume/Å3 | 1060.3(6) |
| Z | 2 |
| ρcalc g/cm3 | 3.073 |
| F(000) | 946 |
| Crystal size/mm3 | 0.02 × 0.02 × 0.01 |
| Radiation | MoKα (λ = 0.71073) |
| Θ range for the data collection/° | 3.048 to 25.242 |
| Index ranges | −10 ≤ h ≤ 9, −9 ≤ k ≤ 10, −17 ≤ l ≤ 21 |
| Reflections collected | 3238 |
| Independent reflections | 1482 (Rint = 0.0879, Rsigma = 0.1096) |
| Data/restraints/parameters | 1482/0/92 |
| Goodness-of-fit on F2 | 1.604 |
| Final R indexes (I >= 2σ (I)) | R1 = 0.1918, wR2 = 0.4808 |
| Final R indexes (all data) | R1 = 0.2441, wR2 = 0.5137 |
| Largest diff. peak/hole/e Å−3 | 4.742/−5.521 |
| Atom | X | Y | Z | Occupancy | U(eq) |
|---|---|---|---|---|---|
| O1 | 0.525(2) | 0.902(2) | 0.234(1) | 1 | 23(4) |
| O2 | 0.752(2) | 0.944(2) | 0.3569(13) | 1 | 34(5) |
| O3 | 0.333333 | 0.666667 | 0.367(3) | 1 | 60(11) |
| O4 | 0.476(2) | 1.101(2) | 0.3462(12) | 1 | 33(5) |
| O5 | 0.945(2) | 1.192(2) | 0.2271(10) | 1 | 23(4) |
| O6 | 0.311(3) | 0.481(3) | 0.4887(14) | 1 | 64(7) |
| O7 | 0.666667 | 1.333333 | 0.217(2) | 1 | 32(8) |
| O8 | 0.825(4) | 1.321(5) | 0.102(2) | 1 | 111(12) |
| O9 | 1.000 | 1.000 | 0.500 | 0.85 | 230(80) |
| O10 | 1.000 | 1.000 | 0.084(2) | 0.85 | 89(19) |
| OW1 | 0.1049(4) | 0.7070(3) | 0.0570(2) | 1 | 260(40) |
| S2 | 0.666667 | 1.333333 | 0.1311(8) | 1 | 41(4) |
| S5 | 0.333333 | 0.666667 | 0.4594(11) | 1 | 61(5) |
| Zn1 | 0.7145(3) | 1.1319(4) | 0.2989(2) | 1 | 33(16) |
| Zn2 | 0.5801(4) | 0.7135(3) | 0.2930(2) | 1 | 34(17) |
| Zn3 | 1.000 | 0.3954(4) | 0.024(2) | 0.85 | 26(2) |
| Zn4 | 1.000 | 0.1976(4) | 0.031(2) | 0.85 | 34(2) |
| Zn | 1.000 | 0.294 | 0.069(18) | 0.07 | 15(20) |
| Crystal Phase | Formula | SG | Unit Cell Parameters | |||||
|---|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | |||
| Namuwite | Zn4(OH)6SO4.4H2O (1) | P-3 | 8.330 | 8.330 | 10.540 | 90 | 90 | 120 |
| Hemihydrate | Zn4(OH)6SO4.0.5H2O (2) | P-3 | 8.356 | 8.357 | 7.084 | 90 | 90 | 120 |
| New phase | Zn4(OH)6SO4·2–2.25H2O | P-3 | 8.3418 | 8.3418 | 17.595 | 90 | 90 | 120 |
| Bond | Zn tetrahedron | DI | Bond | Zn octahedron | DI | Bond | SO4 | DI | |
|---|---|---|---|---|---|---|---|---|---|
| New phase | Zn3–O2 Zn3–O9 | 3 × 1.998 1.843 | 0.0301 | Zn2–O4 Zn2–O2 Zn2–O5 Zn2–O1 Zn2–O1 Zn2–O3 | 2.027 2.064 2.107 2.120 2.128 2.301 | 0.0282 | S5–O6 S5–O3 | 3 × 1.552 1.631 | 0.0175 |
| Zn4–O5 Zn4–O10 | 3 × 1.941 2.001 | 0.0105 | Zn1–O4 Zn1–O2 Zn1–O1 Zn1–O4 Zn1–O5 Zn1–O7 | 2.011 2.024 2.107 2.052 2.141 2.392 | 0.0459 | S2–O8 S2–O7 | 3 × 1.471 1.511 | 0.0112 | |
| Namuwite * | Zn1–OH2 Zn1–Wat1 | 3 × 1.954 1.947 | 0.0014 | Zn2–OH1 Zn2–OH1 Zn2–OH1 Zn2–OH2 Zn2–OH2 Zn2–O1 | 2.018 2.052 2.055 2.097 2.159 2.431 | 0.0499 | S–O2 S–O1 | 3 × 1.458 1.411 | 0.0120 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanimirova, T.; Nikolova, R.; Petrova, N. Crystal Structure of New Zinc-Hydroxy-Sulfate-Hydrate Zn4(OH)6SO4·2–2.25H2O. Crystals 2024, 14, 183. https://doi.org/10.3390/cryst14020183
Stanimirova T, Nikolova R, Petrova N. Crystal Structure of New Zinc-Hydroxy-Sulfate-Hydrate Zn4(OH)6SO4·2–2.25H2O. Crystals. 2024; 14(2):183. https://doi.org/10.3390/cryst14020183
Chicago/Turabian StyleStanimirova, Tsveta, Rositsa Nikolova, and Nadia Petrova. 2024. "Crystal Structure of New Zinc-Hydroxy-Sulfate-Hydrate Zn4(OH)6SO4·2–2.25H2O" Crystals 14, no. 2: 183. https://doi.org/10.3390/cryst14020183
APA StyleStanimirova, T., Nikolova, R., & Petrova, N. (2024). Crystal Structure of New Zinc-Hydroxy-Sulfate-Hydrate Zn4(OH)6SO4·2–2.25H2O. Crystals, 14(2), 183. https://doi.org/10.3390/cryst14020183