Abstract
Recent simulation studies have revealed a wealth of distinct crystal polymorphs encountered in the self-organization of polymer systems driven by entropy or free energy. The present analysis, based on the concept of self-avoiding random walks (SAWs) on crystal lattices, is useful to calculate upper bounds for the entropy difference of the crystals that are formed during polymer crystallization and thus to predict the thermodynamic stability of distinct polymorphs. Here, we compare two pairs of crystals sharing the same coordination number, ncoord: hexagonal close-packed (HCP) and face centered cubic (FCC), both having ncoord = 12 and the same packing density, and the less dense simple hexagonal (HEX) and body centered cubic (BCC) lattices, with ncoord = 8. In both cases, once a critical number of steps is reached, one of the crystals shows a higher number of SAWs compatible with its geometry. We explain the observed trends in terms of the bending and torsion angles as imposed by the geometric constraints of the crystal lattice.
1. Introduction
The term “soft matter” refers to a class of physical systems which includes polymers, colloids, granular media, surfactants, and gels. The common feature of all these diverse materials is that they consist of units whose size is significantly larger than the constituent atoms [1]. One of the main characteristics of soft matter is the existence of thermal fluctuations which are mainly manifested as Brownian motion of atoms, particles, and molecules. Due to this, the molecular shape and size are constantly changing even under conditions of equilibrium. Thus, to describe equilibrium local and global structure, a statistical approach is required. Additionally, soft matter systems are characterized by spontaneous self-assembly, self-organization, and phase transitions. An important tool to aid in the statistical description of mesoscopic structure, but also of phenomena and processes relevant to soft matter, in general, and polymer science, in particular, is the concept of random walk (RW) and its variation in the form of self-avoiding random walk (SAW) [2].
The mathematical concept of a SAW corresponds to a trajectory of fixed step length that grows randomly on a lattice under the condition that it cannot visit the same point twice. In a strict mathematical sense, several salient aspects of SAW are still unknown or only partially resolved [2]. Still, this has not been an obstacle for its successful application in a very wide range of physical systems and topics in chemistry, physics, polymer science, material technology, mathematics, process optimization, computer science, and biology [3,4,5,6,7,8,9,10,11,12]. Over the years, significant advances have been made in the algorithms related to SAW identification and enumeration, further evolving its usage in diverse problems, especially ones related to synthetic and biological macromolecules. The excluded volume effect in polymer melts and solutions is, in fact, intimately related to the condition of self-avoidance in random walks [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29].
Self-avoiding random walks (SAWs) and polygons (SAPs) have been studied extensively on 2D and 3D lattices [30] with specific examples being the honeycomb [31,32,33,34], square [35,36], triangular [37], simple cubic [35,38,39,40], body centered cubic (BCC) [41], and face centered cubic (FCC) [41,42] lattices. Due to their importance and general applicability, significant algorithmic and theoretical advances have been made in the enumeration, characterization, identification, and scaling behavior of SAWs, SAPs, and their variants [19,20,33,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62]. It is interesting to notice that while the simple cubic (SC), BCC, and FCC crystals have been extensively studied in the literature, no such wealth of information exists for the hexagonal close-packed (HCP) one. This could be attributed to a possible assumption that the HCP and FCC crystals are characterized by the same number of SAWs, independent of the step size [63], because both crystals have the same coordination number (ncoord = 12) and packing density (φ = ). As will be demonstrated in the continuation, such a claim is not valid: beyond a certain SAW length (number of steps) the number of SAWs compatible with the HCP crystal becomes marginally higher than the one for the FCC crystal, the difference increasing as the number of SAW steps grows.
In the past, we employed a SAW-based analysis to enumerate the possible configurations of single-chain crystals and thus explain their thermodynamic stability in regular lattices in slits (tubes) [64] and plates [65]. These studies have been motivated by the spontaneous, entropy-driven crystallization of chains of tangent hard spheres as demonstrated in Monte Carlo (MC) simulations of dense packings under various conditions of spatial confinement [66,67,68]. Very recently, it was documented that starting from random (disordered) packings, freely jointed chains of hard spheres in the bulk show a transition to the ordered state following Ostwald’s rule of stages [69]: initially a random hexagonal close-packed (rHCP) morphology is formed of mixed HCP and FCC character [70,71]. Given enough observation time, the rHCP ordered morphology is eventually succeeded by an almost defect-free (perfect) FCC crystal [71]. In parallel modeling efforts, a wealth of distinct crystal polymorphs, including non-compact crystals like the body centered cubic (BCC) and holoedric 6/mmm (simple hexagonal, HEX), has also been encountered in the (free) energy-driven self-assembly of freely jointed chains of tangent monomers interacting with the square well potential under very dilute conditions [72].
With respect to the stability of the HCP and FCC crystals made of athermal polymer chains, semi-analytical calculations were presented in [70], based on the separation of translational and conformational degrees of freedom. Resulting upper-bound estimates demonstrate that the conformational entropy of an HCP crystal of hard-sphere polymers is higher than the one of the FCC crystal by a margin of 0.331 × 10−5 k per monomer, where k is Boltzmann’s constant. However, this minute difference is significantly smaller than the free energy advantage of the FCC crystal in terms of translational entropy. The latter can be assumed to be very similar, if not identical, to the translational entropy of monomers disregarding the constraints imposed by chain connectivity. A widely accepted value for the entropic difference between FCC and HCP crystals for monomeric hard spheres, corresponding to approximately 112 × 10−5 k per monomer, has been established in the literature [73,74,75], depending further on packing density [76]. The semi-analytical predictions of [70] have been supported by extremely long molecular simulations based on Monte Carlo algorithms demonstrating FCC perfection in athermal polymer crystallization of entangled chains [66,77].
In the present contribution, we further support the semi-analytical calculations of [70] by exhaustive enumeration of the self-avoiding random walks (SAWs) compatible with the inter-site geometry as encountered in the FCC and HCP crystals. We also analyze the SAW statistics, including the local geometry (bending and torsion angles) and the global size (distribution and mean of the end-to-end distance). Two additional crystals, the HEX and BCC, both having ncoord = 8, encountered in simulations of chains whose monomers interact with the square well potential [72], are considered in the current work.
The manuscript is organized as follows: Section 2 presents the model, the reference crystals and the method employed for the SAW enumeration. Section 3 hosts the results on the local and global properties of SAWs on the HCP, FCC, HEX, and BCC lattices. The discussion of the results can be found in Section 4. The manuscript is concluded with Section 5 summarizing the main conclusions and listing future extensions.
2. Materials and Methods
As reported in the Introduction, the present work forms part of an ongoing modeling study of (free) energy- [72,78] and entropy-driven [70,71,79,80] self-organization of idealized systems based on polymers interacting with the hard sphere or the square well potential [81]. Four different crystals have been considered in this work: the hexagonal close-packed (HCP), face centered cubic (FCC), body centered cubic (BCC or BC), and holoedric 6/mmm (simple hexagonal, HEX), whose periodic structure and salient characteristics can be found in [77,82]. The HCP and FCC ones have been selected because they compete as emerging and resulting structures in the disorder–order transition of freely jointed chains of hard spheres at sufficiently high packing densities [70,71,79,80]. The HEX and BCC crystals, along with the HCP and FCC crystals and the Frank–Kasper [83,84] phase, are encountered as final stable morphologies in the crystallization of clusters formed from chains interacting with the square well attractive potential [72,78] at dilute conditions. Density-based [85,86] and geometric [72] arguments can accurately explain the dominance of non-compact crystals in specific ranges of the interaction potential in two and three dimensions.
In the present work, a polymer is represented as a linear chain of strictly tangent hard spheres. This chain is grown on the sites of a crystal, so its bonded geometry is defined by the linear architecture of the chain, the tangency condition of the bonded atoms, and the spatial constraints imposed by the crystal sites. As in our past works [64,65], “monomer” refers to each of the hard spheres that constitute the polymer chain, while “site” refers to the each of the sites of the ideal crystal. A single chain is grown on each reference lattice, with a monomer occupying a single site, and bonded monomers lying on adjacent sites, which are thus separated by a bond/step length. With respect to bond geometry, bending (θ) and torsion (ϕ) angles are formed by successive triplets and quartets of monomers along the chain backbone. These angles must respect the connectivity and geometry of each lattice. Accordingly, bending and torsion angles along the polymer chain are different for different crystal types, as can be seen in Table 1 (bending angles) and Table 2 (torsion angles). Chains on the HCP crystal show the largest variety, with 6 and 19 distinct bending and torsion angles, respectively, while SAWs on the BCC crystal have only 3 compatible bending angles.

Table 1.
Bending angles, θ, which are compatible with each reference crystal. Compatibility and incompatibility are indicated by the “+” and “−” symbols, respectively.
Table 2.
Torsion angles, ϕ, which are compatible with each reference crystal. Compatibility and incompatibility are indicated by the “+” and “−” symbols, respectively.
Our homemade algorithm is based on direct enumeration of SAWs, as also implemented in our past works on SAWs under confinement [64,65]. The approach includes the exhaustive identification of all possible sequences of steps which fulfil two essential conditions: (1) the geometry imposed by the neighbor connectivity of the corresponding crystal, and (2) the self-avoidance of the random walk according to which the same crystal site cannot be visited twice by different segments (steps) of the SAW. The step length of the SAW is taken as unity and coincides with the distance between nearest neighbors in the crystal. Since the SAW growth is subjected only to the two conditions mentioned above, the selected bending (between two successive steps) and torsion (between three successive steps) angles adopt discrete values according to the imposed neighbor geometry of the corresponding crystals (as reported in Table 1 and Table 2). Figure 1 hosts segments of the four reference crystals (HCP, FCC, BCC, and HEX) along with a SAW of 9 steps (N = 9) grown on each one of them. Crystal sites are represented as spheres while the SAW steps are shown as orange lines.
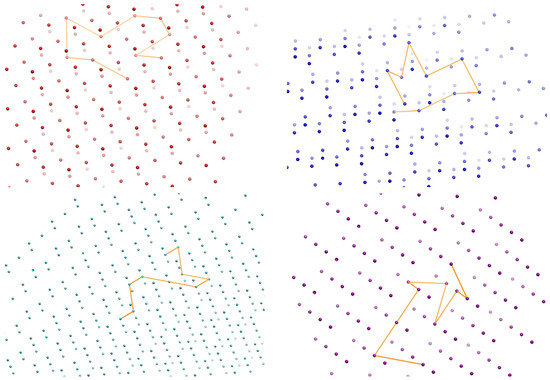
Figure 1.
Snapshots showing a self-avoiding random walk of 9 steps (N = 9) grown on the reference crystals: FCC (top left), HCP (top right), BCC (bottom left), and HEX (bottom right). FCC, HCP, HEX and BCC crystal sites are shown in red, blue, cyan and purple colors, respectively; SAW steps are shown as orange lines. Images created with the VMD visualization software (version 1.6.3) [87].
A SAW of N steps consists of the ordered sequence of sites ωN(0), ωN(1), …, ωN(N − 1), ωN(N) with ωN(0) being the SAW origin. In contrast to SAWs on spatially restricted lattices [64,65], the ones studied here correspond to the unconstrained bulk case. Thus, any site can be selected as the origin of the SAW, ωN(0), due to the maximal symmetry of the bulk lattice. Self-avoidance of the random walk (or equivalently excluded volume of polymer) is fulfilled as ωN(i) ≠ ωN(j) for all i ≠ j. Bond tangency is satisfied as |ωN(i + 1) − ωN(i)| = 1, with i [0, 1, …, N − 1] and |ω| = (ω·ω)(1/2) denoting the Euclidean norm. The size of the SAW is quantified by the square end-to-end distance: |ωN|2 = (ωN(N) − ωN(0)). (ωN(N) − ωN(0)).
For a given number of steps, N, and a reference crystal X (HCP, FCC, HEX, or BCC) our algorithm is based on the direct enumeration of the total number of distinct SAWs, cN(X), and proceeds with the calculation of all bending and torsion angles and of the square end-to-end distance of each identified SAW. Accordingly, for a SAW of N steps, the mean square end-to-end distance, ⟨|ωN|2 ⟩(X), can be readily calculated as:
The scaling of the number of distinct SAWs, cN, and of the average SAW size, ⟨|ωN|2 ⟩, as a function of the number of SAW steps is given by exponential-power-law asymptotic expressions [2,36,38,39,40,60,88]:
where A and D are the critical amplitudes, μ is the connective constant, and γ and ν are the critical exponents. The critical exponents are considered universal, while the values of A, D, and μ are lattice dependent.
Starting from the work of Orr [22], elegant methods and efficient algorithms have been developed over the years to tackle the exponentially difficult SAW enumeration problem [6,9,36,38,39,61,62,89,90], reaching various high-N SAWs in distinct lattices. We should note here that our SAW enumeration algorithm is not as efficient as the state-of-the-art methods described above, and thus our analysis is limited to SAWs of intermediate number of steps. However, this should not be considered as a potential disadvantage as extensive off-lattice simulations, under a wide variety of conditions, have clearly demonstrated that, beyond a moderate value, chain length has practically no effect on the crystallization of athermal packings of fully flexible chains. The phase behavior and the established ordered morphologies are the same, independent of the average length of chains being as low as 12 or as high as 1000 monomers, the former value corresponding to short oligomers while the latter to well-entangled chains, deep in the polymeric regime [67,79,80].
The main objective of the present work is to provide a quantitative basis for the study of the thermodynamic stability of the HCP and FCC crystals made of fully flexible, athermal polymers. As mentioned in the Introduction, by assuming independence of the translational and conformational degrees of freedom, an argument which is analyzed in detail in [70], the total entropy of the crystal can be considered as the summation of two distinct contributions: the conformational one, ΔSconf, dictated by chain connectivity respecting the geometric constraints of the reference crystal, and the translational one, ΔStran, which should be very similar to the one of monomeric systems of hard spheres, which are free of any constraints imposed by chain connectivity. Accordingly, the entropy difference of the HCP and FCC crystals can be written as [70]:
where Ξ is the complete set (partition function) of all multichain configurations compatible with the polymer model (here, freely jointed chains of tangent hard spheres) and the lattice geometry (here, HCP or FCC), and || denote the cardinality of a set [70]. The translational contribution is known from past studies on monomeric analogs [73,74,75]. Thus, the evaluation of Ξ for the multichain configurations is required to arrive at an accurate estimate for . However, such information is not available for any of the crystals. Accordingly, we need to establish an upper-bound, single-chain estimation. The concept of random walks (RWs) would not shed any light given that both the HCP and FCC crystals have the same coordination number and as such the same number of RWs over the whole range of steps. Toward this end, as a more refined criterion, we resort to self-avoiding random walks (SAWs), providing a tighter and more discriminating upper-bound estimate for the difference in the configurational entropy as [70]:
3. Results
Table 3 presents the total number of distinct SAWs, cN, and the mean square end-to-end distance, ⟨|ωN|2⟩, as a function of SAW steps, N, for the FCC and HCP lattices, along with the difference in the number of SAWs, ΔcN (=cN(HCP) − cN(FCC)). The corresponding data for the BCC and HEX crystals are presented in Table 4, which further hosts a column showcasing the difference in the mean square end-to-end distance. No such column exists in Table 3 because within the reported accuracy (four significant digits) there is no difference in the average SAW size between the HCP and the FCC crystals.
Table 3.
Distinct number of SAWS, cN, and mean square end-to-end distance, ⟨|ωN|2⟩, as a function of number of SAW steps, N, for the FCC and HCP crystals. The difference in the number of SAWs ΔcN = cN(HCP) − cN(FCC) is also reported. Within the reported accuracy, there is no difference in the mean square end-to-end distance between the HCP and the FCC crystals.
Table 4.
Distinct number of SAWS, cN, and mean square end-to-end distance, ⟨|ωN|2⟩, as a function of number of SAW steps, N, for the BCC and HEX crystals. The differences ΔcN = cN(BCC) − cN(HEX) and Δ⟨|ωN|2⟩ = ⟨|ωN|2⟩ (BCC) − ⟨|ωN|2⟩ (HEX) are also reported.
The FCC and HCP crystals show the same number of SAWs up to N = 5 steps. However, for N ≥ 6, the number of SAWs on the two lattices starts to deviate with the HCP crystal showing systematically more SAWs than the FCC one. At N = 6, ΔcN = 12, the relative difference is approximately 7.6 × 10−6. For the longest SAW studied here (N = 12) ΔcN = 60,617,100, which corresponds to a relative difference of 3.4 × 10−5, showing increasing trends as the SAW grows in length. In parallel, as can be seen by the comparison of the related rows in Table 3, the average size of the SAWs in the HCP and FCC lattices is the same within a tolerance of 10−5 for all values of N.
Similar trends are observed in the comparison of the BCC and HEX crystals as listed in Table 4. For N = 3, the BCC crystal has 12 more SAWs compared to the HEX crystal with the relative difference being 0.031, significantly higher than the one observed for the HCP-FCC pair at the corresponding deviation point (N = 6). For the longest SAW studied here (N = 14), the absolute and relative differences for the BCC-HEX pair increase to 89,206,013,508 and 0.20, respectively. Accordingly, even if both lattices have the same coordination number (ncoord = 8) the single-chain conformations, as quantified by the number of SAWs, are significantly fewer in HEX compared to BCC. With respect to the average size, the polymer grown on the HEX lattice is systematically longer than the one on the BCC with the relative difference being approximately 0.053 for N = 14.
The left panel of Figure 2 shows the logarithm of the total number of distinct SAWs, cN, as a function of the logarithm of the total number of SAW steps, N, for all lattices studied here. The differences of the two pairs are also shown. On the right panel of Figure 2, we can observe the dependence of the logarithm of the mean square end-to-end distance, ⟨|ωN|2 ⟩(X), as calculated from Equation (1), on the logarithm of the number of SAW steps, N. Furthermore, we have fitted all available data with best linear fits corresponding to the scaling formula of Equation (3). Figure 2 shows one such best linear fit corresponding to the data for the HEX crystal, with the reliability fitting coefficient being practically equal to 1. The complete set of the parameter values, according to Equations (2) and (3), as obtained from best linear fits on current SAW data, are reported in Table 5. Very little variation is observed for both the critical amplitude and exponent between the different lattices, especially when the FCC and HCP crystals are compared. A minimal trend suggests that the exponent increases slightly for the crystals of the lower coordination number, while the opposite behavior is observed for the amplitude. Given the short or intermediate length of the studied SAWs, the universal exponent of 0.588 is nicely matched for the HCP and FCC crystals. Between the HCP and the FCC crystal there is no appreciable difference suggesting that the corresponding SAW sizes will be very similar, but strictly not identical, for the limit of infinitely long chains (N → ∞).
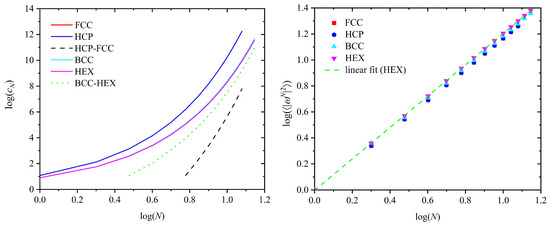
Figure 2.
(Left panel): Logarithm of the total number of distinct SAWs, cN, as a function of the logarithm of the total number of SAW steps, N, for all lattices studied here: HCP, FCC, BCC, and HEX. The corresponding differences between the pairs: ΔcN(HCP-FCC) and ΔcN(BCC-HEX) are also reported. The red curve (FCC) is obscured by the blue (HCP) one and the cyan (BCC) by the magenta (HEX) due to minimal differences. (Right panel): Logarithm of the mean square end-to-end distance, ⟨|ωN|2⟩, as a function of the logarithm of the total number of SAW steps, N, for all lattices. The green dashed line is the best linear fit on the data for the HEX crystal.
Table 5.
Critical amplitudes, A and D, critical exponents, γ and v, and connective constant, μ, as calculated from best linear fits using the SAW enumeration data as reported in Table 3 and Table 4 and Figure 2 for the HCP, BCC, and HEX lattices. Due to the minimal differences between the HCP and FCC crystals in Section 4 (Discussion), we present the correlation of the difference.
The distribution of the discrete bending angles, which are compatible with each lattice studied here, is given in Figure 3 as a function of the number of SAW steps. For all crystal types, as N grows, the population of obtuse angles experiences small decreases in favor of the acute ones because of the self-avoidance condition. The fraction of bending angles with 90°, where available (HCP, FCC, and HEX), rapidly reaches a stable plateau. Extrapolating the current trends, to longer N, the most probable bending angle is 60°, 60°, 70.53°, and 90° for the FCC, HCP, BCC, and HEX crystals, respectively.
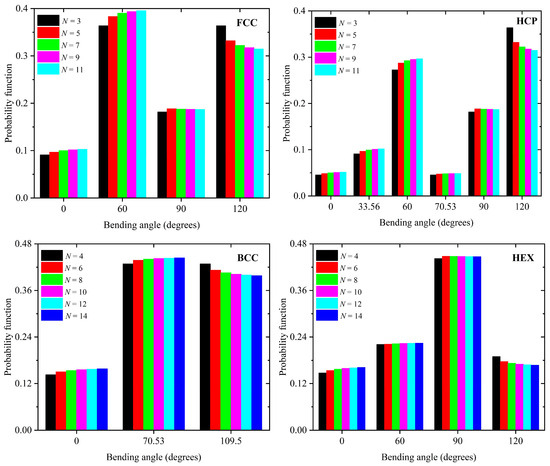
Figure 3.
Distribution of discrete bending angles of the SAWs on the (top left) FCC, (top right) HCP, (bottom left) BCC, and (bottom right) HEX crystals as a function of total number of SAW steps, N. All SAW bending angles compatible with each crystal are reported in Table 1. For a given angle, different colors correspond to SAWs of different lengths as indicated in the legend.
Figure 4 hosts the corresponding results for the distribution of discrete torsion angles which are compatible with each lattice crystal. For FCC, as N increases, the populations at 54.7, 125.3, and 180° increase, while the one at 90° shows a significant reduction. Compared to other crystals, the SAWs on the HCP lattice show a richer behavior with the primary angles resting at 54.7°, 70.5°, 109.5°, and 180°. The HEX crystal is the one that shows the smallest variation with increasing number of SAW steps.
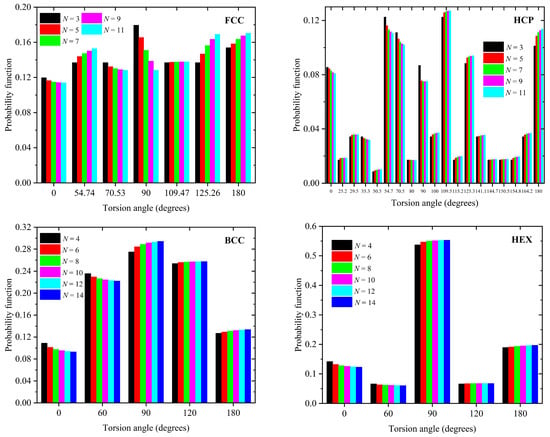
Figure 4.
Distribution of discrete torsion (dihedral) angles of the SAWs on the (top left) FCC, (top right) HCP, (bottom left) BCC, and (bottom right) HEX lattices as a function of the total number of SAW steps, N. All SAW torsion angles compatible with each lattice are reported in Table 2. For a given angle, different colors correspond to SAWs of different lengths as indicated in the legend.
The distribution of SAW size, as quantified by the square end-to-end distance, |ωN|2, is shown in Figure 5 and Figure 6 for the HCP–FCC and BCC–HEX pairs, respectively. The probability distribution function (PDF) is presented in the main figure while the cumulative distribution function (CDF) is shown in the inset. Given the major differences in the bonded geometry between HCP and FCC lattices, it is not surprising that the distribution of the SAW size shows significant deviations. However, it is interesting to notice that in spite of these variations, the global SAW size, on average, is indistinguishable between the close-packed FCC and HCP crystals, as shown in Table 1, and further confirmed by the CDF trends in the inset of Figure 5. On the other hand, SAWs on the BCC lattice have a smaller size than the ones on the HEX as indicated by the concentrated maxima of the BCC distribution at small values of the square end-to-end distance.
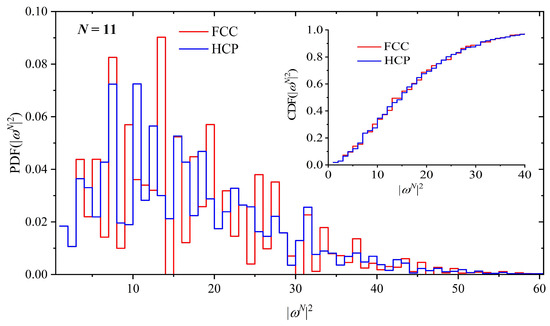
Figure 5.
Probability distribution function (PDF) of the square end-to-end SAW distance, |ωN|2, for the HCP and FCC crystals for a SAW of N = 11 steps. Inset: cumulative distribution function (CDF) for the same systems.
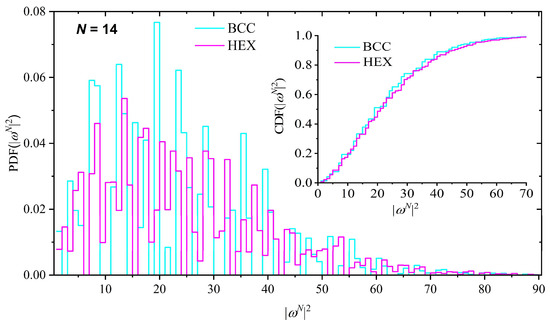
Figure 6.
Probability distribution function (PDF) of the square end-to-end SAW distance, |ωN|2, for the BCC and HEX crystals for a SAW of N = 14 steps. Inset: cumulative distribution function (CDF) for the same systems.
4. Discussion
We enumerate and describe the self-avoiding random walks, SAWs, grown on different crystals, characterized by the same coordination number (ncoord = 12 or 8). We observe that for the HCP and FCC pair, once a critical number of steps is reached (N = 6) the number of distinct SAWs becomes different. In relative numbers, this difference is minute but definitely non-zero with the HCP crystal showing a larger number of SAWs compared to FCC (cN(HCP) > cN(FCC) for N > 5). The present finding is clear evidence that the SAW behavior in the HCP and FCC crystals should not be considered as identical even though both are characterized by the same coordination number and packing density. In parallel, the average size of SAWs on these two crystals is indistinguishable for the whole range of studied SAW steps even if the internal chain/SAW bonded geometry, as quantified by the bending and torsion angles, is significantly different: the HCP crystal shows a much richer variety in bond geometry and same is true for the size distribution as quantified by the end-to-end distance.
The data presented here in Table 3 and Table 4, and Figure 2 for the HCP-FCC and BCC-HEX pairs clearly identify a difference in the number of SAWs once a critical number of steps is reached. The ratio of the SAWs in Equation (5) can be fitted using an asymptotic formula:
where X and Y are the two crystals to be compared and A1, B, and d are the fitting parameters. Fittings using the exponential formula of Equation (6) on available SAW enumeration data are reported in Figure 7.
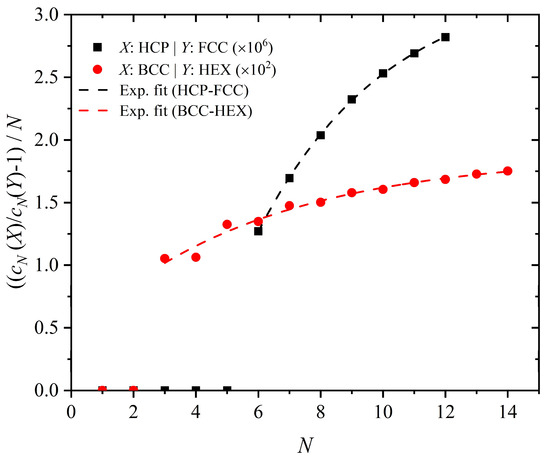
Figure 7.
((cN(X)-cN(Y)) − 1)/N versus number of steps, N, as obtained from direct SAW enumeration for the HCP/FCC and BCC/HEX pairs. Fittings with the asymptotic formula of Equation (6) are also shown.
Both pairs suggest an asymptotic behavior of the conformational entropy difference per monomer. For the HCP and FCC crystals, the established behavior is far from the asymptotic regime and significantly longer SAWs are required, pointedly increasing the computational complexity of the problem. However, application of the exponential formula (Equation (6)) allows for the prediction of the ratio for very long SAWs from short and intermediate values as the ones presented here. The fitting parameters for the HCP/FCC and BCC/HEX are summarized in Table 6. Although the maximum chain length for which we could exhaustively enumerate SAWs is moderate, the quality of the fit in Figure 7 suggests that the values of the parameter A1 in Table 6 are a quite accurate approximation to the asymptotic value, i.e., for infinite chain length, which is the physically relevant quantity for long polymers.
Table 6.
Fitting parameters A1, B, and d of the asymptotic formula (Equation (6)) applied on the current SAW enumeration data for the HCP/FCC and BCC/HEX pairs of crystal lattices.
Concentrating on the HCP-FCC pair, which is the main focus of the present study, placing the asymptotic formula in Equation (5) and considering the limit of very long chains provides an upper-bound estimate, k, as the HCP polymer crystal has a higher conformational entropy than the FCC one. However, this value is significantly lower than the translational entropy k. Accordingly, the FCC is the most stable crystal among the polymorphs for freely jointed polymers of tangent hard spheres.
5. Conclusions
Prediction of the thermodynamic stability of crystals made of athermal polymers is significantly more complicated than for monomeric analogs due to the presence of constraints imposed by chain connectivity. Here, we demonstrate that once a critical number of steps is reached, the HCP crystal has more self-avoiding random walks than the FCC one. Accordingly, a tight upper-bound estimate of the conformational entropic advantage of the HCP crystal suggests that this lead is not sufficient to overcome the significantly larger translational advantage of the FCC crystal [70]. Hence, the FCC crystal is the thermodynamically most stable athermal polymer crystal and should prevail among competing polymorphs, as confirmed by recent off-lattice simulations of freely jointed chains of hard spheres [71]. The present work is currently being extended to tackle freely rotating chains on crystals in two and three dimensions.
Author Contributions
Conceptualization, N.C.K. and M.L.; methodology, N.C.K. and M.L.; software, J.B. and U.U.; validation, U.U.; data curation, J.B.; writing—original draft preparation, J.B. and N.C.K.; writing—review and editing, U.U. and M.L.; funding acquisition, N.C.K. and M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICINN/FEDER (Ministerio de Ciencia e Innovación. Fondo Europeo de Desarrollo Regional), grant numbers “PID2021-127533NB-I00” and “RTI2018-097338-B-I00”.
Data Availability Statement
Data and SAW enumeration codes are fully available upon request.
Acknowledgments
The authors deeply thank Katerina Foteinopoulou, Miguel Herranz, Patricia Jimenez, and Ines del Caz for their very helpful interactions. The authors acknowledge support through projects “PID2021-127533NB-I00” and “RTI2018-097338-B-I00” of MINECO/FEDER (Ministerio de Ciencia e Innovación. Fondo Europeo de Desarrollo Regional). The authors gratefully acknowledge the Universidad Politécnica de Madrid (www.upm.es, accessed on August 2023) for providing computing resources on Magerit Supercomputer through projects “r727” and “s341”.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Doi, M. Soft Matter Physics; Oxford University Press: New York, NY, USA, 2017. [Google Scholar]
- Madras, N.; Slade, G. The Self-Avoiding Walk; Birkhauser: Boston, MA, USA, 1996. [Google Scholar]
- Weiss, G.H.; Rubin, R.J. Random-walks—Theory and selected applications. Adv. Chem. Phys. 1983, 52, 363–505. [Google Scholar]
- Webb, B.Z.; Cohen, E.G.D. Self-avoiding modes of motion in a deterministic Lorentz lattice gas. J. Phys. A Math. Theor. 2014, 47, 315202. [Google Scholar] [CrossRef][Green Version]
- Triampo, D.; Shobsngob, S.; Triampo, W.; Pongkitiwanichkul, P. Modified self-avoiding walk in a polymerization process. J. Korean Phys. Soc. 2005, 46, 1429–1432. [Google Scholar]
- Sykes, M.F. Self-avoiding walks on simple cubic lattice. J. Chem. Phys. 1963, 39, 410–412. [Google Scholar] [CrossRef]
- Brydges, D.; Frohlich, J.; Spencer, T. The random-walk representation of classical spin systems and correlation inequalities. Commun. Math. Phys. 1982, 83, 123–150. [Google Scholar] [CrossRef]
- Alvarez, J.; van Rensburg, E.J.J.; Soteros, C.E.; Whittington, S.G. Self-avoiding polygons and walks in slits. J. Phys. A Math. Theor. 2008, 41, 185004. [Google Scholar] [CrossRef]
- Fisher, M.E.; Sykes, M.F. Excluded-volume problem and the ising model of ferromagnetism. Phys. Rev. 1959, 114, 45–58. [Google Scholar] [CrossRef]
- Stauffer, D.; Aharony, A. Introduction to Percolation Theory: Revised Second Edition; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Janse van Rensburg, E.J. The Statistical Mechanics of Interacting Walks, Polygons, Animals and Vesicles, 2nd ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Ottinger, H.C. Stochastic Processes in Polymeric Fluids; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Rubin, R.J. The excluded volume effect in polymer chains and the analogous random walk problem. J. Chem. Phys. 1952, 20, 1940–1945. [Google Scholar] [CrossRef]
- Rubin, R.J. Random-Walk Model of Chain-Polymer Adsorption at a Surface. J. Chem. Phys. 1965, 43, 2392–2407. [Google Scholar] [CrossRef]
- Fisher, M.E. Shape of a self-avoiding walk or polymer chain. J. Chem. Phys. 1966, 44, 616–622. [Google Scholar] [CrossRef]
- Helfand, E. Theory of inhomogeneous polymers—Fundamentals of gaussian random-walk model. J. Chem. Phys. 1975, 62, 999–1005. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics (Chemistry); Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- de Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1980. [Google Scholar]
- Guyeux, C.; Nicod, J.M.; Philippe, L.; Bahi, J.M. The study of unfoldable self-avoiding walks—Application to protein structure prediction software. J. Bioinform. Comput. Biol. 2015, 13, 1550009. [Google Scholar] [CrossRef]
- Guyeux, C.; Charr, J.C.; Abdo, J.B.; Demerjian, J. Advances in the enumeration of foldable self-avoiding walks. Int. J. Comput. Sci. Eng. 2020, 22, 365–375. [Google Scholar] [CrossRef]
- Flory, P.J. The configuration of real polymer chains. J. Chem. Phys. 1949, 17, 303–310. [Google Scholar] [CrossRef]
- Orr, W.J.C. Statistical treatment of polymer solutions at infinite dilution. Trans. Faraday Soc. 1947, 43, 12–27. [Google Scholar] [CrossRef]
- Janse Van Rensburg, E.J.; Whittington, S.G. Self-avoiding walks adsorbed at a surface and pulled at their mid-point. J. Phys. A Math. Theor. 2017, 50, 055001. [Google Scholar] [CrossRef][Green Version]
- Pereira, G.G. Internal structure of polymer-chains. Phys. A 1995, 219, 290–304. [Google Scholar] [CrossRef]
- Rosa, A.; Everaers, R. Conformational statistics of randomly branching double-folded ring polymers. Eur. Phys. J. E 2019, 42, 7. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.; Clarke, J.H.R. Investigation of the end-to-end distance distribution function for random and self-avoiding walks in 2 and 3 dimensions. J. Chem. Phys. 1991, 94, 3936–3942. [Google Scholar] [CrossRef]
- Clisby, N.; Conway, A.R.; Guttmann, A.J. Three-dimensional terminally attached self-avoiding walks and bridges. J. Phys. A Math. Theor. 2016, 49, 015004. [Google Scholar] [CrossRef]
- Yang, Q.H.; Yang, X.; Luo, M.B. Adsorption of polymer chains on heterogeneous surfaces with random adsorption sites. Polymer 2019, 180, 121677. [Google Scholar] [CrossRef]
- Domb, C.; Gillis, J.; Wilmers, G. On shape and configuration of polymer molecules. Proc. Phys. Soc. Lond. 1965, 85, 625. [Google Scholar] [CrossRef]
- Beaton, N.R.; Flajolet, P.; Garoni, T.M.; Guttmann, A.J. Some New Self-avoiding Walk and Polygon Models. Fundam. Inform. 2012, 117, 19–33. [Google Scholar] [CrossRef]
- Bosi, G.; Campanino, M. Random Walk on a Randomly Oriented Honeycomb Lattice. Markov Process. Relat. Fields 2019, 25, 75–99. [Google Scholar]
- Adler, J. The self-avoiding walk on the honeycomb lattice. J. Phys. A Math. Gen. 1983, 16, L515–L517. [Google Scholar] [CrossRef]
- Beaton, N.R.; Guttmann, A.J.; Jensen, I. A numerical adaptation of self-avoiding walk identities from the honeycomb to other 2D lattices. J. Phys. A Math. Theor. 2012, 45, 035201. [Google Scholar] [CrossRef]
- Deforcrand, P.; Koukiou, F.; Petritis, D. Self-avoiding random-walks on the hexagonal lattice. J. Stat. Phys. 1986, 45, 459–470. [Google Scholar] [CrossRef]
- Guttmann, A.J. On the critical-behavior of self-avoiding walks. J. Phys. A Math. Gen. 1987, 20, 1839–1854. [Google Scholar] [CrossRef]
- Macdonald, D.; Hunter, D.L.; Kelly, K.; Jan, N. Self-avoiding walks in 2 to 5 dimensions—Exact enumerations and series study. J. Phys. A Math. Gen. 1992, 25, 1429–1440. [Google Scholar] [CrossRef]
- Jensen, I. Self-avoiding walks and polygons on the triangular lattice. J. Stat. Mech. Theory Exp. 2004, P10008. [Google Scholar] [CrossRef]
- Guttmann, A.J. On the critical-behavior of self-avoiding walks. ll. J. Phys. A Math. Gen. 1989, 22, 2807–2813. [Google Scholar] [CrossRef]
- MacDonald, D.; Joseph, S.; Hunter, D.L.; Moseley, L.L.; Jan, N.; Guttmann, A.J. Self-avoiding walks on the simple cubic lattice. J. Phys. A Math. Gen. 2000, 33, 5973–5983. [Google Scholar] [CrossRef]
- Schram, R.D.; Barkema, G.T.; Bisseling, R.H. Exact enumeration of self-avoiding walks. J. Stat. Mech. Theory Exp. 2011, P06019. [Google Scholar] [CrossRef]
- Schram, R.D.; Barkema, G.T.; Bisseling, R.H.; Clisby, N. Exact enumeration of self-avoiding walks on BCC and FCC lattices. J. Stat. Mech. Theory Exp. 2017, 083208. [Google Scholar] [CrossRef]
- McKenzie, D.S. End-to-end length distribution of self-avoiding walks. J. Phys. A Math. Gen. 1973, 6, 338–352. [Google Scholar] [CrossRef]
- Bahi, J.M.; Guyeux, C.; Mazouzi, K.; Philippe, L. Computational investigations of folded self-avoiding walks related to protein folding. Comput. Biol. Chem. 2013, 47, 246–256. [Google Scholar] [CrossRef]
- Duminil-Copin, H.; Hammond, A. Self-Avoiding Walk is Sub-Ballistic. Commun. Math. Phys. 2013, 324, 401–423. [Google Scholar] [CrossRef]
- Duminil-Copin, H.; Glazman, A.; Hammond, A.; Manolescu, I. On the probability that self-avoiding walk ends at a given point. Ann. Probab. 2016, 44, 955–983. [Google Scholar] [CrossRef]
- Duminil-Copin, H.; Ganguly, S.; Hammond, A.; Manolescu, I. Bounding the number of self-avoiding walks: Hammersley-welsh with polygon insertion. Ann. Probab. 2020, 48, 1644–1692. [Google Scholar] [CrossRef]
- Caracciolo, S.; Pelissetto, A.; Sokal, A.D. Dynamic critical exponent of the bfacf algorithm for self-avoiding walks. J. Stat. Phys. 1991, 63, 857–865. [Google Scholar] [CrossRef]
- Caracciolo, S.; Causo, M.S.; Ferraro, G.; Papinutto, M.; Pelissetto, A. Bilocal dynamics for self-avoiding walks. J. Stat. Phys. 2000, 100, 1111–1145. [Google Scholar] [CrossRef]
- Caracciolo, S.; Gherardi, M.; Papinutto, M.; Pelissetto, A. Geometrical properties of two-dimensional interacting self-avoiding walks at the theta-point. J. Phys. A Math. Theor. 2011, 44, 115004. [Google Scholar] [CrossRef]
- Hooper, W.; Klotz, A.R. Trapping in self-avoiding walks with nearest-neighbor attraction. Phys. Rev. E 2020, 102, 032132. [Google Scholar] [CrossRef] [PubMed]
- Brydges, D.C.; Imbrie, J.Z. Green’s function for a hierarchical self-avoiding walk in four dimensions. Commun. Math. Phys. 2003, 239, 549–584. [Google Scholar] [CrossRef]
- Gherardi, M. Exact Sampling of Self-avoiding Paths via Discrete Schramm-Loewner Evolution. J. Stat. Phys. 2010, 140, 1115–1129. [Google Scholar] [CrossRef][Green Version]
- Grimmett, G.R.; Li, Z.Y. Self-avoiding walks and amenability. Electron. J. Comb. 2017, 24, P4.38. [Google Scholar] [CrossRef]
- Lindorfer, C. A general bridge theorem for self-avoiding walks. Discret. Math. 2020, 343, 112092. [Google Scholar] [CrossRef]
- Zbarsky, S. Asymptotically faster algorithm for counting self-avoiding walks and self-avoiding polygons. J. Phys. A Math. Theor. 2019, 52, 505001. [Google Scholar] [CrossRef]
- James, E.W.; Soteros, C.E. New pattern theorems for square lattice self-avoiding walks and self-avoiding polygons. J. Phys. A Math. Theor. 2007, 40, 8621–8634. [Google Scholar] [CrossRef]
- Schram, R.D.; Barkema, G.T.; Bisseling, R.H. SAWdoubler: A program for counting self-avoiding walks. Comput. Phys. Commun. 2013, 184, 891–898. [Google Scholar] [CrossRef]
- Duminil-Copin, H.; Smirnov, S. The connective constant of the honeycomb lattice equals root 2+root 2. Ann. Math. 2012, 175, 1653–1665. [Google Scholar] [CrossRef]
- Clisby, N.; Dunweg, B. High-precision estimate of the hydrodynamic radius for self-avoiding walks. Phys. Rev. E 2016, 94, 052102. [Google Scholar] [CrossRef]
- Clisby, N. Accurate Estimate of the Critical Exponent nu for Self-Avoiding Walks via a Fast Implementation of the Pivot Algorithm. Phys. Rev. Lett. 2010, 104, 055702. [Google Scholar] [CrossRef] [PubMed]
- Conway, A.R.; Enting, I.G.; Guttmann, A.J. Algebraic techniques for enumerating self-avoiding walks on the square lattice. J. Phys. A-Math. Gen. 1993, 26, 1519–1534. [Google Scholar] [CrossRef]
- Conway, A.R.; Guttmann, A.J. Square lattice self-avoiding walks and corrections to scaling. Phys. Rev. Lett. 1996, 77, 5284–5287. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P. Physics of Disordered Solids; Mittal Publications: Delhi, India, 1982. [Google Scholar]
- Benito, J.; Karayiannis, N.C.; Laso, M. Confined Polymers as Self-Avoiding Random Walks on Restricted Lattices. Polymers 2018, 10, 1394. [Google Scholar] [CrossRef]
- Parreno, O.; Miguel Ramos, P.; Karayiannis, N.C.; Laso, M. Self-Avoiding Random Walks as a Model to Study Athermal Linear Polymers under Extreme Plate Confinement. Polymers 2020, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.M.; Karayiannis, N.C.; Laso, M. Off-lattice simulation algorithms for athermal chain molecules under extreme confinement. J. Comput. Phys. 2018, 375, 918–934. [Google Scholar] [CrossRef]
- Ramos, P.M.; Herranz, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Entropy-Driven Heterogeneous Crystallization of Hard-Sphere Chains under Unidimensional Confinement. Polymers 2021, 13, 1352. [Google Scholar] [CrossRef]
- Ramos, P.M.; Herranz, M.; Martinez-Fernandez, D.; Foteinopoulou, K.; Laso, M.; Karayiannis, N.C. Crystallization of Flexible Chains of Tangent Hard Spheres under Full Confinement. J. Phys. Chem. B 2022, 126, 5931–5947. [Google Scholar] [CrossRef]
- Ostwald, W. Studien uber die bildung and umwandlung fester korper. Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Herranz, M.; Benito, J.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Polymorph Stability and Free Energy of Crystallization of Freely-Jointed Polymers of Hard Spheres. Polymers 2023, 15, 1335. [Google Scholar] [CrossRef] [PubMed]
- Herranz, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Polymorphism and Perfection in Crystallization of Hard Sphere Polymers. Polymers 2022, 14, 4435. [Google Scholar] [CrossRef] [PubMed]
- Herranz, M.; Pedrosa, C.; Martínez-Fernández, D.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Fine-tuning of colloidal polymer crystals by molecular simulation. Phys. Rev. E 2023, 107, 064605. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, P.G.; Frenkel, D.; Mau, S.C.; Huse, D.A. Entropy difference between crystal phases. Nature 1997, 388, 235–236. [Google Scholar] [CrossRef]
- Bruce, A.D.; Wilding, N.B.; Ackland, G.J. Free energy of crystalline solids: A lattice-switch Monte Carlo method. Phys. Rev. Lett. 1997, 79, 3002–3005. [Google Scholar] [CrossRef]
- Mau, S.C.; Huse, D.A. Stacking entropy of hard-sphere crystals. Phys. Rev. E 1999, 59, 4396–4401. [Google Scholar] [CrossRef]
- Pronk, S.; Frenkel, D. Can stacking faults in hard-sphere crystals anneal out spontaneously? J. Chem. Phys. 1999, 110, 4589–4592. [Google Scholar] [CrossRef]
- Herranz, M.; Martínez-Fernández, D.; Ramos, P.M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Simu-D: A Simulator-Descriptor Suite for Polymer-Based Systems under Extreme Conditions. Int. J. Mol. Sci. 2021, 22, 12464. [Google Scholar] [CrossRef]
- Herranz, M.; Santiago, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Crystal, Fivefold and Glass Formation in Clusters of Polymers Interacting with the Square Well Potential. Polymers 2020, 12, 1111. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. Entropy-Driven Crystallization in Dense Systems of Athermal Chain Molecules. Phys. Rev. Lett. 2009, 103, 045703. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. The role of bond tangency and bond gap in hard sphere crystallization of chains. Soft Matter 2015, 11, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Ramos, P.M.; Herranz, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Identification of Local Structure in 2-D and 3-D Atomic Systems through Crystallographic Analysis. Crystals 2020, 10, 1008. [Google Scholar] [CrossRef]
- Frank, F.C.; Kasper, J.S. Complex alloy structures regarded as sphere packings. 1. definitions and basic principles. Acta Crystallogr. 1958, 11, 184–190. [Google Scholar] [CrossRef]
- Frank, F.C.; Kasper, J.S. COMPLEX Alloy structures regarded as sphere packing. 2. analysis and classification of representative structures. Acta Crystallogr. 1959, 12, 483–499. [Google Scholar] [CrossRef]
- Serrano-Illán, J.; Navascués, G.; Velasco, E. Noncompact crystalline solids in the square-well potential. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2006, 73, 011110. [Google Scholar] [CrossRef]
- Armas-Pérez, J.C.; Quintana-H, J.; Chapela, G.A.; Velasco, E.; Navascués, G. Phase diagram of a square-well model in two dimensions. J. Chem. Phys. 2014, 140, 064503. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Modell. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Clisby, N. Scale-free Monte Carlo method for calculating the critical exponent. of self-avoiding walks. J. Phys. A Math. Theor. 2017, 50, 264003. [Google Scholar] [CrossRef]
- Sykes, M.F.; Watts, M.G.; Roberts, P.D.; Guttmann, A.J. Asymptotic behavior of selfavoiding walks and returns on a lattice. J. Phys. Part A Gen. 1972, 5, 653. [Google Scholar] [CrossRef]
- Clisby, N.; Liang, R.; Slade, G. Self-avoiding walk enumeration via the lace expansion. J. Phys. A Math. Theor. 2007, 40, 10973–11017. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).