Abstract
Scientists have been interested in hybrid coumarin derivatives due to their wide clinical anticancer use. Herein, ethyl 8-methoxycoumarin-3-carboxylate (Compound 1) served as the starting material for the synthesis of a series of novel hybrid coumarin derivatives (Compounds 3–6). Their structure was determined using 13C NMR, 1H NMR, elemental analysis, and mass spectrometry. The in vitro cytotoxic activities of coumarin derivatives (Compounds 3, 5, and 6) and brominated coumarin derivatives (Compounds 4, 8, and 9) against MCF-7 and MDA-MB-231 were evaluated. Several substances have been identified as promising candidates for future study, especially Compound 6 due to its potent activity against β-tubulin (TUB) polymerization, sulfatase, and aromatase enzymes. It also has a role in inducing cell-cycle arrest at the S phase in the MCF-7 cell line, as well as apoptosis.
1. Introduction
Hybrid compounds are often designed and synthesized utilizing molecular hybridization procedures, which include the identification of pharmacophoric subunits in two or more known physiologically active derivatives’ molecular structures. Increasing numbers of scientists are seeking hybrids that can simultaneously attack many biological targets [1,2,3]. Hybrid compounds also provide depth by reducing the potential for adverse medication reactions and limiting the development of drug resistance [4]. Well-known examples of such compounds include sunitinib and lapatinib for the treatment of cancer and ziprasidone, duloxetine, and ladostigil for the treatment of multiple central nervous system illnesses. Metabolic disorders, malaria, inflammation, organophosphorus poisoning, and ischemia are only a few of the multifactorial illnesses that have been treated by hybrid molecules [5,6,7,8].
Novel hybrid compounds with vasorelaxant, platelet anti-aggregating, anticancer, monoamine oxidase- (MAO-B) inhibitory, antioxidant, and anti-inflammatory characteristics have been created by hybridizing or coupling various coumarin derivatives with a wide variety of bioactive chemicals [9,10,11,12]. Thus, the molecular hybridization strategy is crucial in the creation of new molecules for the treatment of a wide range of multifactorial disorders. It is significant to note that building drug-like compounds by combining the coumarin template with other heterocycles is an established technique that allows for the creation of novel pharmacological profiles, an increase in action, or a decrease in toxicity [13]. This work contributes to an ongoing effort to identify novel, effective anticancer agents [14,15,16] (Figure 1).
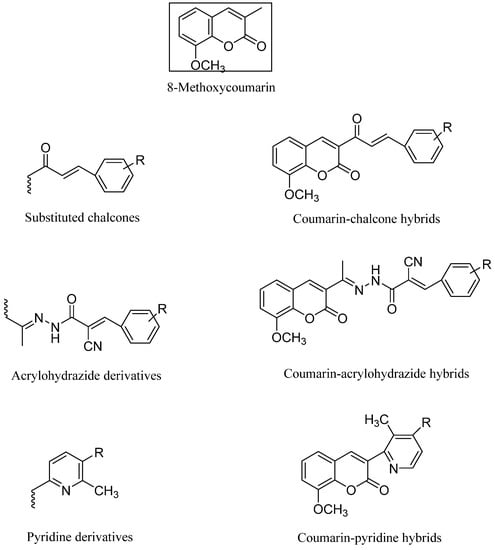
Figure 1.
Hybrid coumarin molecules containing chalcone, acyl hydrazide, and pyridine derivatives as anticancer agents.
Cancer, which is characterized by the uncontrolled multiplication and accumulation of cells in an organism, is a major global health problem due to its multifactorial nature; the World Health Organization (WHO) predicts that 13.5 million people will die from cancer by the year 2030 [17,18,19]. Due to the complexity of the illness and the fact that several proteins and enzymes are dysregulated, therapies focusing on a single target have been unsuccessful thus far. On the basis of our previous work, we have found that some coumarin derivatives have shown very promising activities against carcinoid cells [20,21,22]. Therefore, we have continued this research on certain new hybrid coumarin compounds.
In the current work, we aimed to design, synthesize, and study the impact of oxazole, carboxamide, and hydroxyl methylideneamino-phenol with an 8-methoxy-5-substituted coumarin mixed scaffolds impact on breast cancer management. By repositioning the oxazole, carboxamide, and methylideneaminophenol derivatives in Position 3, we enabled the introduction of a bromine atom substituent in Position 5 of the 8-methoxycoumarin moiety, we were able to investigate the ensuing structural changes (Figure 2).
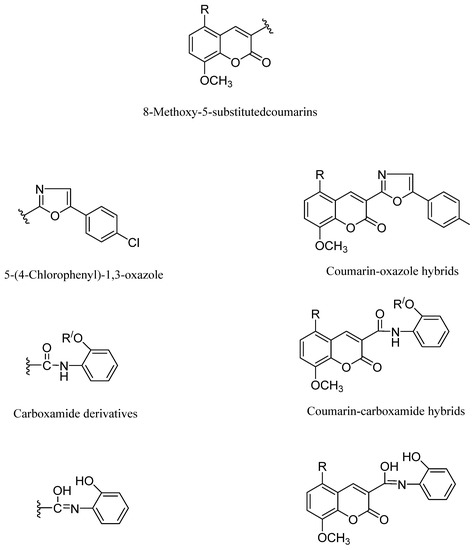
Figure 2.
Coumarin-hydroxymethylideneaminophenol hybrids.
2. Results and Discussion
2.1. Chemistry
Scheme 1 and Scheme 2 demonstrate the overarching procedures for synthesizing the desired hybrid 8-methoxy-5-substituted coumarins with oxazole and carboxamide moieties at Position 3. The cyclo-condensation of 3-methoxy-2-hydroxybenzaldehyde and diethyl malonate with piperidine as a catalyst base yielded ethyl 8-methoxy coumarin-3-carboxylate (Compound 1). Ammonolysis was conducted for the latter compound via the reaction of ester coumarin (Compound 1) with NH3 from the CH3COONH4 and/or HCONH2 under fusion to yield 8-methoxycoumarin-3-carboxamide (Compound 2) as a starting material in the syntheses of a target hybrid coumarin bearing an oxazole moiety. When 8-methoxycoumarin-3-carboxamide (Compound 2) was reacted with 4-chlorophenyl bromide in the presence of CH3COONa fused in a refluxing dimethyl formamide, 3-(5-(4-chlorophenyl)oxazol-2-yl)-8-methoxycoumarin (Compound 3) was produced. Compound 4, 5-Bromo-3-(5-(4-chlorophenyl)oxazol-2-yl)-8-methoxycoumarin, was obtained by the halogenation of Compound 3 with bromine in glacial acetic acid while stirring at 60 °C. In addition, the condensation of ethyl 8-methoxycoumarin-3-carboxylate (Compound 1) with 2-aminophenol in dimethyl formamide under reflux afforded the corresponding N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carboxamide (Compound 5) and N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carbimidic acid (Compound 6, Scheme 1).
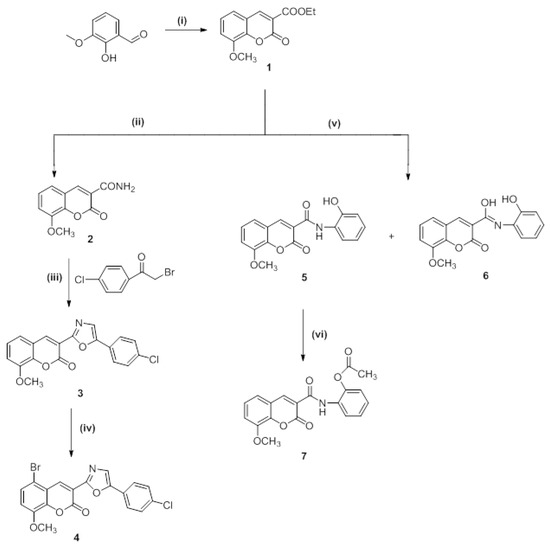
Scheme 1.
Synthesis of target compounds (Compounds 1–7). Reagents and reaction conditions: (i) Diethylmalonate, piperidine, ethanol, fusion 2–3 min at 110–120 °C, then reflux for 2 h at 75 °C; (ii) ammonium acetate, fusion then reflux for 2 h, 120–140 °C; (iii) sodium acetate, DMF, reflux for 2–3 h, 150 °C; (iv) Br2, AcOH, stirring for 5–10 min at 60 °C, then 2 h at room temperature; (v) 2-aminophenol, DMF, reflux for 3 h, 150 °C; (vi) Ac2O, reflux for 2 h, 115 °C.
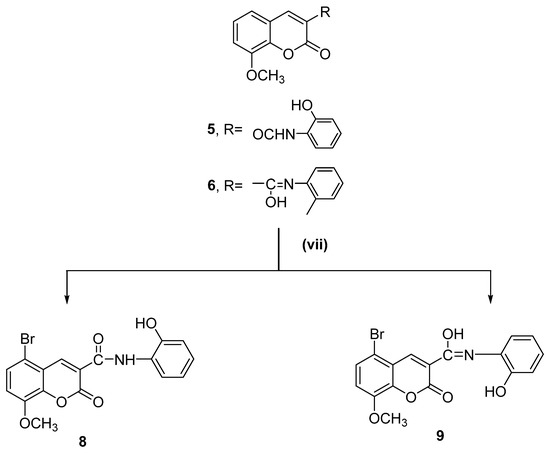
Scheme 2.
Synthesis of target compounds (Compounds 8 and 9). Reagents and reaction conditions: (vii) Br2, AcOH, stirring for 5–10 min at 60 °C, then 2 h at room temperature.
The acetoxy derivative (Compound 7) was produced by treating Compound 5 with acetic anhydride in a refluxing solution. The product of the acetylation reaction confirmed the structure of Compound 5. When bromine was added to glacial acetic acid alongside Compounds 5 and 6, the reaction produced 5-bromo-N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carboxamide (Compound 8) and 5-bromo-N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carbimidic acid (Compound 9, Scheme 2).
The characterization information for the newly synthesized hybrid coumarins (Compounds 3–9) in the experimental phase is presented herein. The structure of the produced compounds was validated by analytical and spectral data (1H NMR, 13C NMR, and EI-MS) (in Supplementary Materials).
2.2. NMR Spectra Investigation of 8-Methoxycoumarin-3-Carboxamide Derivatives (Compounds 3–9)
Both 1H and 13C NMR spectra were used to analyze the structure of the target molecules (Compounds 3 and 4). Within the 1H NMR spectra, Compound 3 showed a characteristic singlet peak at δ 3.94 ppm for the methoxy group. Other aromatic multiplet signals appeared within the expected region of δ 7.34–7.99 ppm. The singlet signals for both H-oxazole and H-4 of the coumarin ring were visible in the region of δ 8.10–8.84 ppm. The bromo derivative of Compound 3 was confirmed by the appearance of the expected signals within the expected regions, as the methoxy signal appeared at δ 3.92 ppm and aromatic hydrogen signals along with H-4 of the coumarin ring appeared at δ 7.38–8.77 ppm. The 13C NMR spectra for Compounds 3 and 4 provided further confirmation of their chemical structure, as the carbonyl (C=O) signals for both compounds were recorded at δ 162.98 and 159.80 ppm, respectively. The 13C NMR spectra also exhibited distinctive signals related to aromatic carbons at δ 143.80–116.47 ppm for Compound 3 and at δ 144.83–112.63 ppm for Compound 4. Signals attributed to OCH3 groups for both compounds were visible at δ 56.64 and 56.95 ppm, respectively. In addition, the structure of the target Compound 5 was investigated by 1H and 13C NMR spectra. The fragment methoxy group of Compounds 5–9 which are derivatives of 3,8-disubstituted coumarin derivatives appeared on the 1H NMR spectra as a singlet peak in the area of δ 3.82–3.99 ppm. For the Compounds 5–9, signals of protons from the phenolic hydroxyl (OH) group were visible as singlet signals in the region of δ 9.70–10.50 ppm. The protons of the NH from the amide group for Compounds 5, 7, and 8 were observed in the 1H NMR spectra at δ 11.13, 11.19, and 11.30 ppm as singlet signals, respectively. The protons of the OH group appeared in the spectra as singlet signals at δ 14.10, 13.77, and 13.87 ppm for Compounds 6 and 9. Additionally, proton H-4 of the coumarin rings of Compounds 5–9 appeared in the spectra as a singlet peak in the area of δ 8.75–9.10 ppm. The aromatic ring (CH=CH) protons were noticed in the anticipated area of δ 8.49–6.71 ppm as multiplet signals of Compounds 5–9.
The presence of all carbon atoms for Compounds 5–9 is supported by the 13C NMR spectra. For Compounds 5–9, which are derivatives of 8-methoxycoumarin-3-carboxamides, carbon signals of all methoxy (OCH3) groups appeared in the δ 57.05–56.30 ppm region. The signals of all the C=O bonds of the coumarin ring and amide were visible in the δ 165.50–159.00 ppm range for the Compounds 5, 7, and 8, while the carbon signals of the carbonyl groups of the coumarin ring for Compounds 6 and 9 were observed in the region of δ 163.02–156.23 ppm. The carbon signals of N=C-OH groups for Compounds 6 and 9 were observed at δ 192.46 and 190.35 ppm. The carbon signals of three C-O and C-4 of the coumarin ring for Compounds 5–9 were observed in the δ 152.61–143.78 ppm range.
From the spectral data of NMR for Compounds 6, 8, and 9 containing two isomer forms, the compounds Br79 and Br81 are presented as the two stereoisomers for Compounds 6 and 9 in Chart 1. The detailed results of the 1H NMR and 13C NMR spectra are presented in the experimental section.
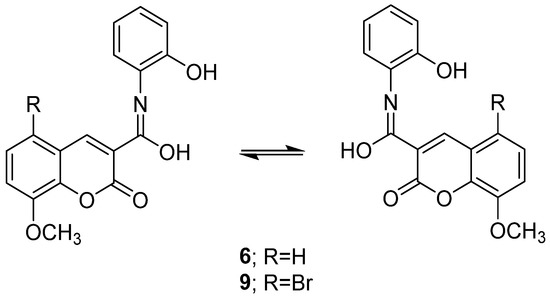
Chart 1.
Two stereoisomers of Compounds 6 and 9.
2.3. Mass Spectrometry Investigation
To clarify the chemical structures of the synthesized Compounds 3, 4, 5, 7, and 9, the mass spectral fragmentation modes of these compounds were investigated. Intense molecular ion peaks at m/z 353 and m/z 411 (unstable) were visible in the mass spectra of Compounds 3 and 4, which were identified by their respective molecular formulas of C19H12NClO4 and C19H11NBrClO4. Peaks at m/z 219 and m/z 297 were created as a result of the fragmentation and rearrangement of the molecular ion peaks at m/z 353 and m/z 411. The ions at m/z 203 and 281 were produced as a result of oxygen atoms being lost from the ion peaks at m/z 219 and 297. By removing a bromine atom and a carbon monoxide (C=O), the ion peak at m/z 297 was disrupted to generate ion peaks at m/z 218 and m/z 190. Additionally, the ion peak at m/z 219 underwent fragmentation by losing hydrogen cyanide, C=O, and formaldehyde to create, respectively, ion peaks at m/z 176, 148, and 118 (Scheme 3). By fragmenting and rearranging the molecular ion peaks of Compounds 3 and 4, the fragment ion m/z 139 was produced. While Compound 4’s mass spectrum displayed a stable ion peak at m/z 297, Compound 3’s mass spectrum displayed a base peak at m/z 139.
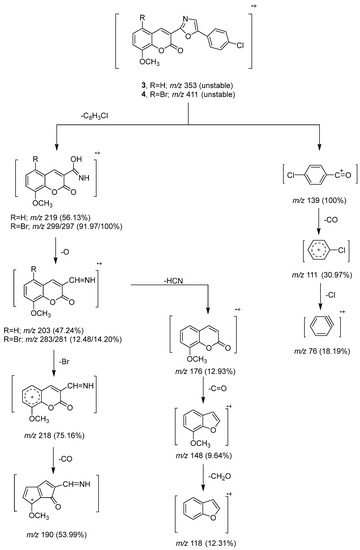
Scheme 3.
Mass fragmentation pathway of Compounds 3 and 4.
The molecular ion peak of Compound 7 (Scheme 4) underwent fragmentation by losing a ketene fragment (CH2CO) to produce a peak ion at m/z 311 for Compound 5. It further underwent the loss of C6H6NO and water molecules to give the base peak at m/z 203 and another peak ion at m/z 293. The rearrangement of the stable peak produced a peak ion at m/z 119, which was followed by the loss of the alkoxy group (OCH2) to produce a peak ion at m/z 89 related to cyclohepta-2,4-dien-6-yn-1-ylium.
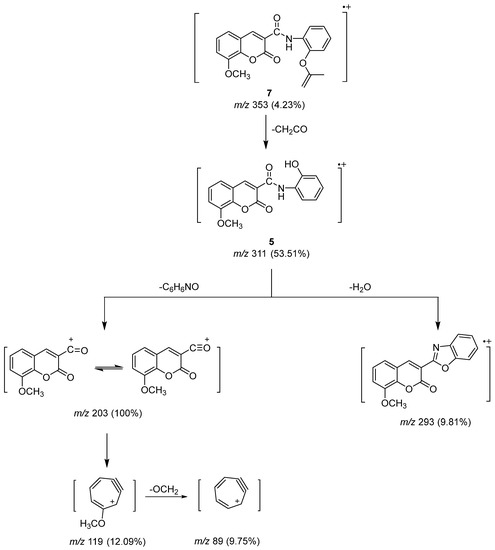
Scheme 4.
Mass fragmentation pathway of Compounds 5 and 7.
The unstable molecular ion peak at m/z 389 for Compound 8 was broken to produce another unstable peak ion at m/z 371 by the loss of a water molecule (Scheme 5). The loss of a carboxyl group from the unstable fragment gave base peaks at m/z 328 and 236 due to the Br79 and Br81 isomers. The two isomers of the bromine atom formed ion peaks at m/z 299 and 297 of equal intensity as a result of the loss of the C6H4O fragment from the molecular ion peak. An oxygen atom was lost from the ion peak to create the next two ion peaks at m/z 283 and 281, and then a cyanohydrin molecule (HCN) was lost to create the ion peaks at m/z 256 and m/z 245. The loss of a bromine atom from the subsequent ion peaks allowed for the observation of the ion peak at m/z 145 [23].
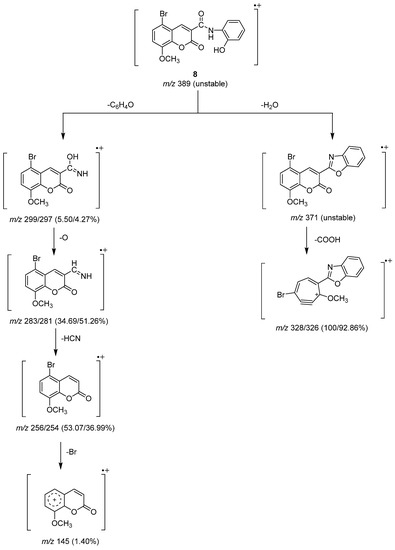
Scheme 5.
Mass fragmentation pathway of Compound 8.
2.4. In Vitro Antitumor Activity
2.4.1. In Vitro Cytotoxic Effects against Breast Cancer Cell Lines
The newly synthesized Compounds 3–6, 8, and 9 were tested for their anti-proliferative effects against breast cancer cell lines (MCF-7 and MDA-MB-231) via an MTT assay, comparing the compounds against the known chemotherapy medicine of choice STU under the same circumstances. Table 1 and Figure 3 showed the IC50 amounts resulting from the fabricated compounds. Several of the novel compounds were shown to have strong anticancer effects against the tested cell line. The strongest cytotoxic activity was shown by Compounds 3 and 6 compared to 4.086 μM of STU against MCF-7 and 7.03 μM against MDA-MB-231, with IC50 values of 9.165 and 12.65 μM for Compound 3 and 6.621 and 9.62 μM for Compound 6, respectively. It is notable that within the tested compounds, Compound 6 was the most efficient compound versus the MCF-7 and MDA-MB-231 cultures. Compound 6 was thus selected for further biological analysis due to its promising effect against breast tumor cells.

Table 1.
Compounds 3–6 and 8–9 and their cytotoxic effects on the proliferation of various breast cancer cell lines and the epithelial breast cell line.
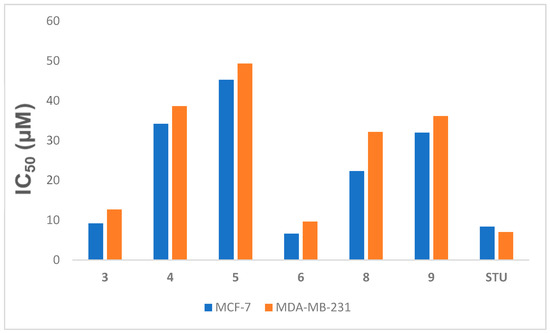
Figure 3.
IC50 (µM) of Compounds 3–6, 8, and 9 versus the MCF-7 and MDA-MB-231 cell lines related to STU. Data expressed as the mean ± SEM from the dose–response curve of at least three experiments.
2.4.2. Cell Cycle Analysis
In order to investigate how Compound 6 affected the MCF-7 cell line’s ability to move through the cell cycle, the DNA content of the sample was measured using flow cytometry. The results reported in Figure 4 showed that the coumarin derivative Compound 6 was able to induce cell-cycle arrest at the S phase in the MCF-7 cell line when compared to controls, from 29.66% to 45.12%. Compound 6 also produced a decrease in the number of cells during G2-M transition and G0-G1 phases from 13.30% to 7.56% and from 57.04% to 47.32%, respectively, which confirmed apoptosis induction after 48 h.
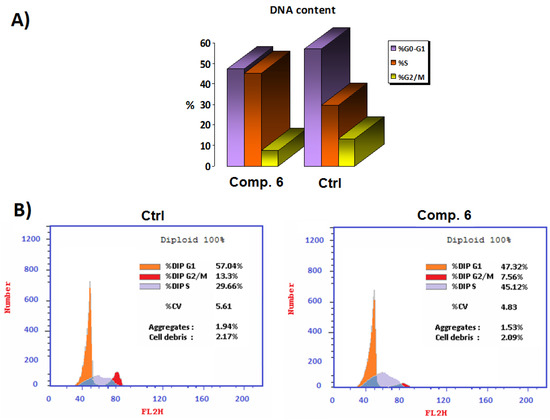
Figure 4.
(A) Comparison of the half-maximal (IC50) concentration (µM) of Compound 6 to the untreated control cell cycle in panel. (B) Cell-cycle analysis at the IC50 concentration (µM) of Compound 6 compared to the untreated control.
2.4.3. Annexin V-FITC/PI Screening
Component 6 of the synthetic peptide blocked the progress of MCF-7 cells, suggesting that it may have anticancer properties. By use of a highly specific test, it was discovered that the novel chemical could cause apoptosis in MCF-7 cells. The compound’s death and/or inhibitory impact on the proliferation of cancer cells was thoroughly explained using the apoptosis/necrosis technique, Annexin-V-FITC/PI. Figure 5B shows that apoptosis rates increased from 2.41 to 35.79 percent overall and from 0.46 to 24.28 percent in the early stages when compared to the control group. Furthermore, the late-stage apoptosis percentage ranged from 0.21% in comparison with the control. As a result, it is reasonable to regard the newly identified chemical Compound 6 to be an effective inducer of apoptosis in the MCF-7 cell line.
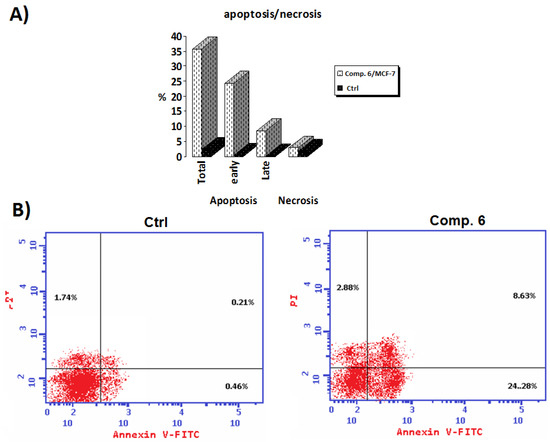
Figure 5.
(A) Annexin V-FITC at the 50% inhibitory concentration (IC50) of Compound 6 in comparison to an untreated control; (B) Compound 6’s IC50 (µM) for Annexin-V FITC against untreated control.
2.4.4. Tubulin Polymerization Inhibition Assay
One of the main targets in anticancer therapy is the microtubules because they serve a leading role in the cells’ division, as well as in the maintenance of their shape. Many anticancer drugs exhibit their activity by sabotaging microtubule dynamics, leading to the deregulation of mitotic spindles, which causes tumor cell-cycle arrest and prompts the apoptosis of malignant cells [24,25,26,27]. A functional group substitution at a bioactive molecule’s appropriate position was found to have a significant pharmacological impact [28]. In MCF-7 cells, the newly synthesized chemical Compound 6 was tested for its IC50 inhibitory impact on β-tubulin polymerization within 24 h. The results showed that the new derivative Compound 6 displayed a strong inhibition activity against β-tubulin polymerization by decreasing the concentration of β-tubulin polymerization by 0.206-fold when compared with the positive control. These results confirmed that Compound 6 represents quite a powerful anti-β-tubulin (TUB) polymerization agent. The results of a 24-h IC50 measurement of β -tubulin polymerization inhibition in MCF-7 cells treated with Compound 6 and a control arhe shown in Figure 6.
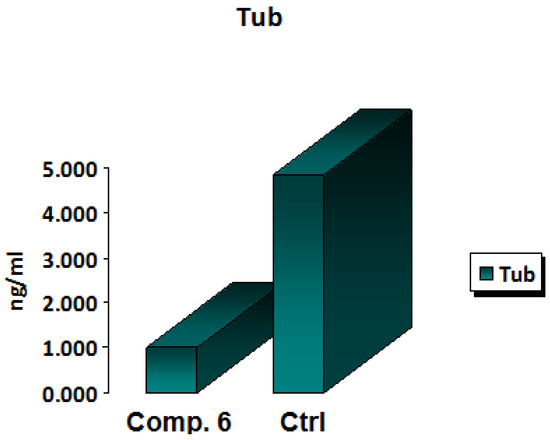
Figure 6.
In vitro β-tubulin polymerization inhibition for Compound 6 and control cells at their IC50 (µM).
2.4.5. In Vitro Sulfatase Polymerization Inhibition Assay
The hydrolysis of sulfate esters from a variety of biological substances, including steroids, carbohydrates, and proteins, is catalyzed by sulfatases, enzymes of the esterase class [29]. These are dispersed in a variety of cells and tissues and can be discovered in extracellular and intracellular areas. The enzyme steroid sulfatase (STS) is in charge of producing active estrogens in postmenopausal women’s breast tissue, which may subsequently lead to breast cancer [30]. Thus, sulfatase inhibition is a promising target in cancer therapy [31] because estrogens are one of the primary causes of breast cancer, according to the World Health Organization (WHO) [32]. As shown in Figure 7, Compound 6 expressed a decrease in the activity of sulfatase by 0.636-fold compared to the positive control, indicating a promising role of this derivative as an anti-breast cancer agent.
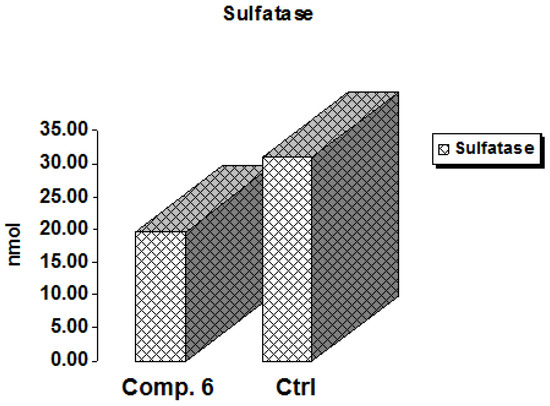
Figure 7.
In vitro inhibitory effect of sulfatase by Compound 6 and control cells at their IC50 (µM).
2.4.6. Aromatase Enzyme Assay of Compound 6
Breast cancer is one of the most common forms of female cancer [33,34]. Estrogens play a critical role in initiating cancer cell proliferation in hormone-dependent breast carcinoma [34,35]. Given that elevated estrogen levels in the body are a known risk factor for breast cancer [36], medicinal chemists have focused on two major approaches to counteract this malignancy-inducing hormone. The first inhibited estrogen receptor function [37], while the second was associated with blocking estrogen production by inhibiting aromatase (AR) [38]. AR converts inactive sulfated estrone into active nonsulfated estrone; thus, steroid sulfatase is another key enzyme in estrogen’s ability to suppress cancer development activity [39]. Here, we set out to create novel AR inhibitors with a coumarin ring as their active ingredient. One of the most potent aromatase inhibitors is a properly substituted coumarin derivative [40]. Compound 6 was selected because it showed the greatest in vitro aromatase inhibition efficacy when related to the reference blocker letrozole and the positive control cells. As shown in Figure 8, the data confirmed that the examined Compound 6 had a worthy AR blocking action, as it decreased the concentration of the aromatase enzyme by 0.401-fold compared to positive control cells, while the letrozole drug decreased the concentration by 0.139-fold. Thus, Compound 6 could be considered a potent inhibitor for aromatase enzyme activity.
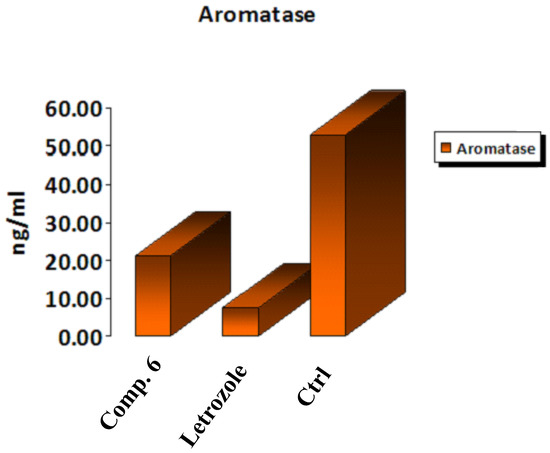
Figure 8.
In vitro inhibitory effect of aromatase by Compound 6, letrozole, and control cells at their IC50 (µM).
2.4.7. Molecular Docking
In order to learn more about the binding interactions and amino-acid residues responsible for triggering the biological activity of Compound 6, molecular docking experiments were conducted. In this research work, a docking simulation study was performed between Compound 6 and the binding pocket of the progesterone and aromatase cytochrome P450 protein receptors in order to assess the affinity scores and mode of interaction (hydrophilic and hydrophobic interactions); the results are presented in Table 2. Compound 6 is shown to interact with the receptor-binding site for both progesterone and aromatase cytochrome P450. Figure 9, Figure 10, Figure 11 and Figure 12 depict the 2D and 3D binding characteristics of Compound 6 in the binding pockets of the progesterone and aromatase cytochrome P450 receptors, respectively.
Table 2.
The affinity scores and mode of interaction (hydrophilic and hydrophobic interactions) of Compound 6 with progesterone and aromatase cytochrome P450 receptors.
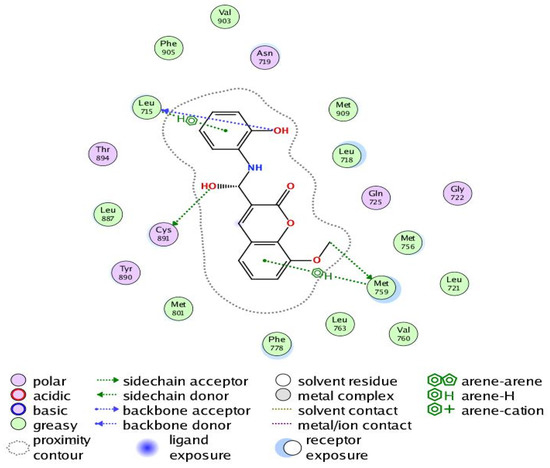
Figure 9.
Two-dimensional interaction of Compound 6 with the amino acids of the active site of the human progesterone receptor.
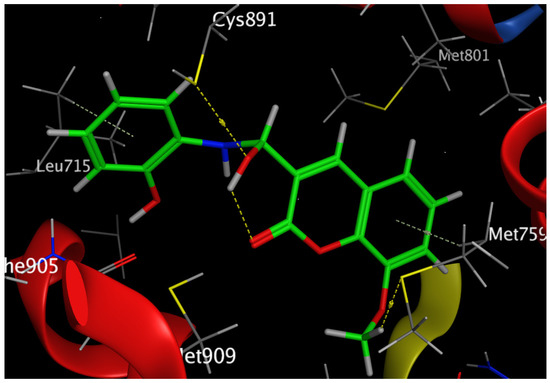
Figure 10.
Three-dimensional interaction of Compound 6 with the amino acids of the active site of the human progesterone receptor.
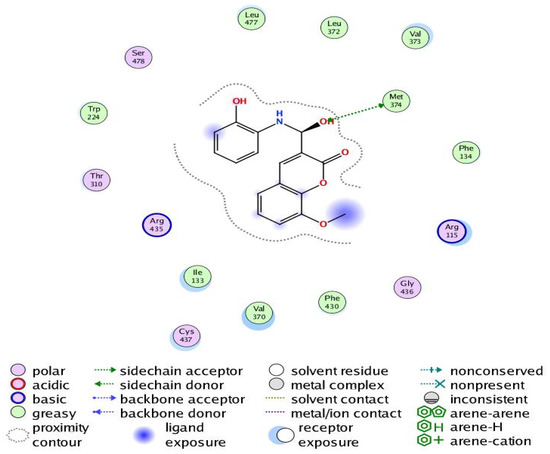
Figure 11.
Two-dimensional interaction of Compound 6 with the amino acids of the human aromatase cytochrome P450 (CYP19A1).
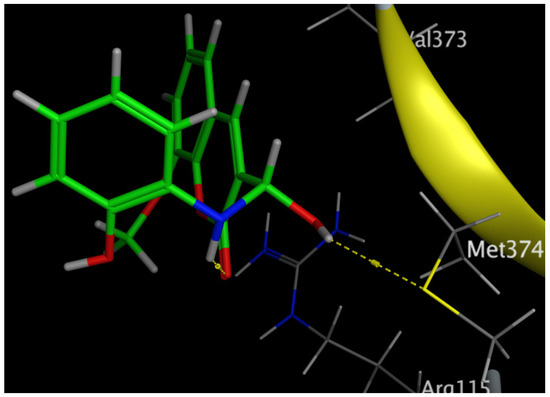
Figure 12.
Three-dimensional interaction of Compound 6 with the amino acids of the human aromatase cytochrome P450 (CYP19A1).
The Human Progesterone Receptor
The progesterone receptor gene (PR) is controlled by estrogen. Breast cancer and the growth of the mammary gland are both mediated by the progesterone receptor. It has been proposed that progesterone receptor levels in breast cancer could be an indicator of a functioning estrogen receptor (ER) signal transduction system, adding further independent prognostic data [41].
In breast cancer, PR is particularly helpful as a predictive biomarker for hormone-positive tumors. Previous research has linked elevated progesterone levels to a higher probability of developing breast cancer and the growth of both hormones receptor-positive and -negative cancers. Women past menopause who have greater than average amounts of circulating progesterone are 16% more likely to develop breast cancer. PR is a direct estrogen-inducing gene in most target tissues, where it can either cooperate with or oppose ER to modulate biological processes. Increased RANKL protein is produced when progesterone binds to PR in luminal cells and stabilizes its mRNA. Afterwards, RANKL promotes proliferation or differentiation by elevating its receptor RANK. Tumorigenesis in hormone-induced mammary epithelium is directly suppressed by RANKL suppression [42]. Our results revealed that Compound 6 bound through the phenolic-OH groups to the amino-acid residues Cys891 and Leu715. Additionally, Compound 6 bound through the ether group to the amino-acid residue Met759. In addition, the amino-acid residues Met759 and Leu715 were bound to Compound 6 by arene–hydrogen interaction.
The Human Aromatase Cytochrome P450 (CYP19A1)
The aromatization of androstenedione and testosterone to estrone and estradiol, respectively, is the last and rate-limiting step in estrogen biosynthesis. This process is catalyzed by aromatase, which is mostly encoded by the CYP19A1 gene [43]. Several studies have shown that breast tumors overexpress aromatase in comparison to healthy breast epithelial cells, which has been associated with poor survival and the course of the illness [44]. Increased aromatase expression in breast carcinomas may also be associated with DNA polymorphisms within the CYP19A1 gene. Aromatase inhibitors have been utilized extensively in the treatment of postmenopausal breast cancer, further demonstrating the role of aromatase in the development of this disease [45,46]. Our results revealed that Compound 6 bound through the phenolic-OH group to the amino-acid residue Met374.
3. Materials and Methods
3.1. Chemistry
All melting points were measured using electrothermal equipment in open capillary tubes without correction. The Microanalytical Unit, Central Services Laboratory, National Research Center, Dokki, Cairo, Egypt, using Vario Elemental (Berlin, Germany), carried out the elemental analysis. Using the KBr disc method, infrared spectra were captured using a Jasco FT/IR-6100 Fourier transform infrared spectrometer (Tokyo, Japan). 1H NMR (400 MHz) and 13C NMR (200 MHz) spectra were run with a Bruker 400 DRX-Avance NMR spectrometer. Chemical shifts were measured using tetramethylsilane as an internal standard and expressed in parts per million downfield. At the central services laboratory of the National Research Center in Dokki, Cairo, Egypt, mass spectra were obtained using a Finnigan MATSSQ-7000 mass spectrometer (Olympia, WA, USA) with an ionizing potential of 70 eV. No additional purification was performed on any of the reagents after they were obtained from Acros, Aldrich Chimie, and Fluka France.
3.1.1. Ethyl 8-Methoxycoumarin-3-Carboxylate (Compound 1)
Condensation of (0.01 mol, 1.52 g) 3-methoxy-2-hydroxybenzaldehyde with (0.01 mol, 1.60 g) diethyl malonate under fusion in the presence of 1.0 mL piperidine at 110–120 °C for 2–3 min, followed by the addition of ethanol, yielded the desired starting chemical. The final product was cooled, poured into water, and neutralized with 2% diluted hydrochloric acid after being heated for two hours under reflux. Ethyl 8-methoxycoumarin-3-carboxylate (Compound 1) was produced by separating the precipitated product through filtration, washing it with water, and drying it as a powder that was used without further purification. The characteristics of Compound 1 are as follows: yield (2.07 g, 85%), m.p. 99 °C; 1H NMR(DMSO-d6) δ: 1.32 (t, 3H, CH3), 3.92 (s, 3H, OCH3), 4.31 (q, 2H, OCH2), 7.30–7.45 (m, 3H, Ar-H), 8.71 (s, 1H, H-4 of coumarin ring) ppm; 13C NMR (DMSO-d6) δ: 164.45, 163.04 (C=O), 157.15, 154.41 (C-O), 149.19 (C-4 of coumarin ring), 134.95, 130.75, 125.30, 118.76, 118.24 (C-aromatic), 116.60 (C-3 of coumarin ring), 61.72 (OCH2), 56.60 (OCH3), 14.53 (CH3) ppm.
3.1.2. Synthesis of 8-Methoxycoumarin-3-Carboxamide (Compound 2)
A mixture of ester coumarin (Compound 1, 0.01 mol, 2.48 g) and CH3COONH4 (0.03 mol, 2.31 g) was combined and fused at 120–140 °C for 2 h on a hot plate, followed by cooling and pouring into water with stirring. After filtration, washing with water, drying, and recrystallization from CH3COOH, crystals of Compound 2 were obtained. The characteristics of Compound 2 are as follows: yield (1.16 g, 53%), m.p. 253 °C; IR (KBr) υmax: 3315, 3187 (NH2), 1725, 1689 (C=O of coumarin and amide), 1611, 1585 (C=C), 1083, 1045 (C-O) cm−1; 1H NMR(DMSO-d6) δ: 3.94 (s, 3H, OCH3), 7.35–7.52 (m, 3H, Ar-H), 7.95–8.09 (d, J = 5.6 Hz, 2H, NH2), 8.84 (s, 1H, H-4 of coumarin ring) ppm; 13C NMR (DMSO-d6) δ: 162.98, 160.52 (C=O), 148.50, 146.72 (C-O), 143.80 (C-4 of coumarin ring), 125.50, 121.62, 119.85, 119.46 (C-aromatic), 116.49 (C-3 of coumarin ring), 56.66 (OCH3) ppm; MS: m/z (%) = 220 (M++1, 14.50), 219 (M+, 100), 203 (35.33), 176 (11.18), 175 (10.51), 174 (6.21), 173 (5.06), 133 (32.07), 131 (3.02), 120 (7.96), 119 (14.36), 118 (12.96), 117 (3.92), 105 (29.26), 104 (9.60), 102 (5.94), 91 (12.96), 90 (11.88), 89 (31.65), 77 (45.22), 76 (35.40), 75 (16.45), 74 (16.85), 65 (10.09), 63 (22.72), 62 (17.43), 51 (23.99), 50 (18.49). Analytically calculated for C11 H9NO4 (M. wt. = 219): C, 60.27; H, 4.11; N, 6.39. Found: C, 60.01; H, 3.95; N, 6.09.
3.1.3. Synthesis of 3-(5-(4-Chlorophenyl)oxazol-2-yl)-8-Methoxycoumarin (Compound 3)
Heating a mixture of coumarin-3-carboxamide (Compound 2, 0.01 mol, 2.19 g), 4-chlorophenacyl bromide (0.01 mol, 2.33 g), and fused sodium acetate (0.03 mol, 2.46 g) in 30 mL dimethyl formamide under reflux for 2–3 h. After cooling, the reaction mixture was transferred into water where it was neutralized with 2% hydrochloric acid. The solid residue was then filtered out, rinsed, and dried. In the end, Compound 3 was obtained when the crystallization was performed from ethanol. The characteristics of Compound 3 are as follows: yield (2.16 g, 61%), m.p. 183 °C; IR (KBr) υmax: 1728 (C=O), 1625 (C=N), 1605, 1583 (C=C), 1125, 1093, 1035 (C-O) cm−1; 1H NMR (DMSO-d6) δ: 3.94 (s, 3H, OCH3), 7.34–7.65 (m, 5H, Ar-H), 7.95–7.99 (dd, J = 8 Hz, 2H, Ar-H), 8.10 (s, 1H, H-oxazole), 8.84 (s, 1H, H-4 of coumarin ring) ppm; 13C NMR (DMSO-d6) δ: 162.98 (C=O), 160.51 (C-2 of oxazole ring), 148.49, 146.71 (C-O), 143.80 (C-4 of coumarin ring), 130.55, 130.16, 129.52, 129.24, 125.49, 121.61, 119.82, 119.45, 116.47 (C-aromatic, C-4 of oxazole and C-3 of coumarin ring), 56.64 (OCH3) ppm; MS: m/z (%) = 353 (M+, unstable), 333 (1.90), 331 (2.35), 303 (0.63), 241 (0.90), 219 (56.13), 220 (42.85), 221 (0.56), 204 (3.67), 203 (47.24), 202 (3.60), 191 (1.24), 177 (1.64), 176 (12.93), 175 (3.97), 174 (6.49), 173 (5.24), 161 (4.17), 148 (9.64), 147 (8.05), 146 (3.47), 141 (19.39), 140 (5.52), 139 (100), 134 (2.09), 133 (33.02), 120 (2.87), 119 (8.75), 118 (12.31), 117 (5.85), 116 (2.16), 115 (2.70), 113 (3.09), 111 (30.69), 110 (1.45), 106 (1.15), 105 (40.19), 104 (9.09), 103 (5.92), 102 (4.66), 101 (3.68), 91 (16.22), 90 (14.51), 89 (36.64), 77 (43.95), 76 (18.19), 75 (12.94), 65 (3.08), 64 (1.38), 63 (6.99), 51 (6.18), 50 (2.66). Analytically calculated for C19 H12NClO4 (M. wt. = 353.5): C, 64.50; H, 3.39; N, 3.96. Found: C, 64.33; H, 3.13; N, 3.63.
3.1.4. Condensation of Ester Coumarin (Compound 1) with 2-Aminophenol: Formation of Compounds 5 and 6
A mixture of ethyl 8-methoxycoumarin-3-carboxylate (Compound 1, 0.01 mol, 2.48 g) and 2-aminophenol (0.01 mol, 1.09 g) in 30 mL dimethyl formamide was heated under reflux for 3 h, followed by cooling and pouring into water. This was followed by filtration of the resulting product to be separated, washing with water, drying, and recrystallization from ethanol to produce Compound 5. The filtrate was concentrated and left for 24 h. The solid obtained was filtered, washed with water, dried, and recrystallized from ethanol to produce Compound 6.
N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carboxamide (Compound 5) was produced as crystals of an orange hue: yield (1.58 g, 51%) m.p. 293 °C. IR (KBr) υmax: 3452 (br. OH), 3194 (NH), 1746, 1706 (C=O), 1612, 1569 (C=C), 1244, 1102, 1028 (C-O) cm−1. 1H NMR(DMSO-d6) δ: 3.93, 3.97 (s, 3H, OCH3 of two isomers), 6.83–6.87 (t, J = 7.2 Hz, 1H, Ar-H), 6.92–6.99 (m, 2H, Ar-H), 7.39–7.49 (m, 2H, Ar-H), 7.47–7.49 (d, J = 6.8 Hz, 1H, Ar-H), 7.59–7.60 (d, J = 6.4 Hz, 1H, Ar-H), 8.40–8.42 (d, J = 8 Hz, 1H, Ar-H), 9.03 (s, 1H, H-4 of coumarin ring), 10.26 (s, 1H, phenolic OH), 11.13 (s, 1H, NH of amide group) ppm. 13C NMR (DMSO-d6) δ: 161.19, 159.28 (C=O of amide and coumarin ring), 148.98, 147.12, 146.78 (C-O), 143.78 (C-4 of coumarin ring), 126.99, 125.69, 124.82, 121.84, 120.41, 119.67, 119.65, 119.34, 116.81 (C-aromatic), 115.10 (C-3 of coumarin ring), 56.73, 56.64 (OCH3 of two isomers) ppm. MS: m/z (%) = 312 (M++1, 8.09), 311 (M+, 53.01), 294 (1.14), 293 (9.81). 204 (14.61), 203 (100), 202 (47.05), 201 (6.37), 119 (12.09), 117 (9.11), 116 (4.78), 105 (1.68), 104 (1.82), 89 (9.75), 77 (1.59), 76 (2.33). Analytically calculated for C17H13NO5 (M. wt. = 311): C, 65.59; H, 4.18; N, 4.50. Found: C, 65.33; H, 4.01; N, 4.28.
N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carbimidic acid (Compound 6) was produced as pale orange crystals: yield (1.04 g, 33.5%), m.p. 168 °C. IR (KBr) υmax: 3430–3415 (br. OH), 1743 (C=O), 1632 (C=N), 1615, 1567 (C=C), 1210, 1109, 1032 (C-O) cm−1. 1H NMR(DMSO-d6) δ: 3.82 (s, 3H, OCH3 of three isomers), 6.84–6.91 (m, 2H, Ar-H), 6.97–6.99 (d, J = 8 Hz, 1H, Ar-H), 7.08–7.10 (d, J = 8 Hz, 1H, Ar-H), 7.12–7.16 (t, J = 7.6 Hz, 1H, Ar-H), 7.18–7.20 (d, J = 7.6 Hz, 1H, Ar-H), 7.38–7.40 (d, J = 8 Hz, 1H, Ar-H), 8.97 (s, 1H, H-4 of coumarin), 9.82 (s, 1H, phenolic OH), 14.10 (s, 1H, OH) ppm. 13C NMR (DMSO-d6) δ: 192.46 (O-C=N), 163.02, 161.97, 156.23 (C=O of three isomers of coumarin), 152.30, 151.45, 151.18, 149.42, 148.82, 148.63, 146.69, 144.75, 144.34 (C-O and C-4 of coumarin ring), 134.85, 128.55, 125.27, 124.25, 122.94, 121.64, 120.54, 120.10, 119.97, 119.91, 119.69, 119.64, 118.79, 118.41, 118.28, 118.02, 117.51, 116.96, 116.88 (C-aromatic), 115.59, 115.35, 114.90 (C-3 of coumarin for the three isomers), 56.62, 56.54, 56.28 (OCH3 of three isomers) ppm. Analytically calculated for C17H15NO5 (M. wt. = 313): C, 65.17; H, 4.83; N, 4.47. Found: C, 64.98; H, 4.51; N, 4.34.
3.1.5. Synthesis of 2-(8-Methoxycoumarin-3-Carboxamido)phenyl Acetate (Compound 7)
A solution of Compound 5 (0.01 mol, 3.11 g) in 20 mL acetic anhydride was refluxed for 2 h, then chilled in ice water. The reaction amalgam was dropped for a full day, and the precipitate produced underwent separation, water cleaning, and dehydration. Finally, crystallization was carried out from benzene to produce Compound 7 as pale orange crystals: yield (2.15 g, 61%), m.p. 198 °C. IR (KBr) υmax: 3189 (NH), 1752–1743 (C=O of ester and pyranone ring), 1716 (C=O of amide), 1612, 1583 (C=C), 1215, 1109, 1032 (C-O) cm−1. 1H NMR(DMSO-d6) δ: 2.44 (s, 3H, COCH3), 3.97 (s, 3H, OCH3), 7.19–7.22 (t, J = 7.2 Hz, 1H, Ar-H), 7.279–7.281 (d, J = 0.8 Hz, 1H, Ar-H), 7.32–7.36 (t, J = 8 Hz, 1H, Ar-H), 7.41–7.45 (t, J = 8 Hz, 1H, Ar-H), 7.49–7.51 (d, J = 8 Hz, 1H, Ar-H), 7.60–7.62 (d, J = 7.6 Hz, 1H, Ar-H), 8.47–8.49 (d, J = 8 Hz, 1H, Ar-H), 9.05 (s, 1H, H-4 of coumarin), 11.10 (s, 1H, NH of amide) ppm. 13C NMR (DMSO-d6) δ: 168.32, 165.31, 161.63 (C=O), 149.61, 146.62 (C-O), 143.79 (C-4 of coumarin), 134.98, 130.75, 126.98, 125.52, 124.63, 123.36, 121.95, 120.98, 117.31 (C-aromatic), 116.20 (C-3 of coumarin), 56.77 (OCH3), 21.09 (COCH3) ppm. MS: m/z (%) = 354 (M++1, 0.57), 353 (M+, 4.23), 312 (8.09), 311 (53.51), 310 (2.40), 294 (1.14), 293 (9.81), 204 (14.61), 203 (100), 202 (47.05), 119 (12.09), 117 (9.11), 116 (4.78), 105 (1.68), 104 (1.82), 101 (1.05), 91 (2.10), 89 (9.75), 77 (1.59), 76 (2.33). Analytically calculated for C19H15NO6 (M. wt. = 353): C, 64.59; H, 4.25; N, 3.97. Found: C, 64.22; H, 4.01; N, 3.63.
3.1.6. General Procedure for the Bromination Reactions of Compounds 3, 5, and 6: Formation of Bromo Derivatives Compounds 4, 8, and 9
The coumarin derivatives Compounds 3, 5, and 6 (0.01 mol, 3.53 g, 3.11 g, 3.11g) were dissolved in 25 mL of glacial acetic acid, and then 10 mL of bromine in CH3COOH (0.01 mol, 0.60 g) was added dropwise to the mixture while stirring at 60 °C. After five to ten minutes, the bromine color was discharged, leaving a yellowish solution. For the next two hours, stirring of the mixture was performed at room temperature while 0.5–1.0 mL of bromine-AcOH solution was added. The reaction mixture was poured into ice water with stirring and the resulting product was filtered, rinsed, and dried. Crystals of Compounds 4, 8, and 9 were obtained when the product was crystallized from the appropriate solvent.
5-Bromo-3-(5-(4-chlorophenyl)oxazol-2-yl)-8-methoxycoumarin (Compound 4) was produced as colorless crystals: yield (2.67 g, 62%), m.p. 225 °C. IR (KBr) υmax: 1735 (C=O), 1631 (C=N), 1605, 1601, 1582 (C=C), 1104, 1038 (C-O) cm−1. 1H NMR(DMSO-d6) δ: 3.92 (s, 3H, OCH3), 7.38–8.09 (m, 7H, Ar-H and H-oxazole), 8.77 (s, 1H, H-4 of coumarin ring) ppm. 13C NMR (DMSO-d6) δ: 162.46 (N=C-O), 159.80 (C=O of coumarin ring), 146.70, 145.92 (C-O), 144.83 (C-4 of coumarin ring), 132.32, 131.53, 131.23, 129.14, 128.86, 121.14, 118.62, 117.35 (C-aromatic and oxazole ring), 112.63 (C-3 of coumarin ring), 56.95 (OCH3) ppm. MS: m/z (%) = 431 (M+, unstable), 396 (0.29), 381 (0.36), 375 (0.47), 350 (0.26), 346 (0.24), 314 (0.25), 300 (6.44), 299 (91.97), 298 (17.10), 297 (100), 283 (12.48), 281 (14.20), 271 (0.75), 269 (0.42), 256 (3.21), 255 (1.23), 254 (2.08), 227 (0.88), 226 (1.33), 219 (5.89), 218 (75.16), 217 (13.78), 213 (13.70), 211 (24.40), 210 (1.50), 203 (1.80), 199 (4.67), 197 (11.17), 190 (53.99), 189 (25.82), 188 (2.58), 185 (16.49), 183 (12.10), 182 (7.96), 175 (1.91), 174 (3.44), 173 (1.88), 162 (5.00), 159 (2.34), 158 (7.47), 157 (8.19), 155 (6.68), 154 (5.37), 141 (2.28), 139 (23.24), 132 (2.94), 131 (1.20), 120 (1.57), 119 (13.60), 118 (6.83), 117 (7.45), 116 (2.04), 113 (0.74), 111 (3.16), 104 (8.85). 103 (40.80), 102 (5.99), 91 (8.91), 90 (4.15), 89 (13.68), 88 (7.50), 87 (12.27), 86 (6.34), 77 (3.39), 76 (23.98), 75 (48.04), 74 (32.63), 73 (2.76), 63 (2.78), 62 (3.33), 50 (2.75). Analytically calculated for C19H11BrClNO4 (M. wt. = 431.5): C, 52.84; H, 2.55; N, 3.24. Found: C, 52.63; H, 2.31; N, 3.08.
5-bromo-N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carboxamide (Compound 8) was produced as crystals of an orange hue: yield (2.45 g, 63%), m.p. 325 °C. IR (KBr) υmax: 3426 (br. OH), 3189 (NH), 1747, 1715 (C=O), 1611, 1585 (C=C), 1210, 1107, 1031 (C-O) cm−1. 1H NMR(DMSO-d6) δ: 3.93, 3.97 (s, 3H, OCH3 of two isomers), 6.97–7.64 (m, 6H. Ar-H), 8.39, 8.57 (s, 2H, H-4 of coumarin ring of two isomers), 8.84, 9.02 (s, 1H, phenolic OH of two isomers), 11.11, 11.39 (s, 1H, NH of amide group of two isomers) ppm. 13C NMR(DMSO-d6) δ: 162.65, 161.17 (C=O), 155.45, 148.96, 147.12 (C-O), 146.84 (C-4 of coumarin ring), 146.79, 146.67, 145.44, 143.84, 128.62, 127.01, 125.67, 121.83, 120.40, 119.64, 119.56, 119.33, 117.94, 117.73, 116.79, 115.11, 112.60 (C-aromatic), 56.92, 56.47 (OCH3 of two isomers) ppm. MS: m/z (%) = 389 (M+, unstable), 328 (100), 327 (29.86), 326 (92.86), 325 (36.36), 324 (4.58), 311 (4.03), 299 (5.50), 297 (4.27), 284 (2.58), 283 (34.43), 281 (51.36), 280 (6.16), 257 (3.85), 256 (53.07), 255 (13.58), 254 (36.99), 253 (28.83), 247 (2.26), 239 (1.17), 227 (1.18), 226 (2.60), 219 (15.79), 218 (3.45), 213 (4.05), 211 (3.85), 202 (9.51), 197 (1.60), 174 (4.44), 169 (3.93), 157 (7.29), 156 (3.72), 155 (4.79), 154 (4.67), 147 (8.11), 145 (1.40), 141 (5.13), 140 (4.97), 133 (1.66), 132 (2.22), 131 (1.94), 120 (1.22), 119 (8.81), 118 (3.84), 117 (5.41), 104 (2.51), 103 (24.49), 102 (2.14), 90 (1.38), 89 (4.38). 88 (5.51), 87 (2.64), 86 (1.86), 82 (1.00), 80 (2.19), 76 (4.82), 75 (17.79), 74 (9.15). Analytically calculated for C17H12BrNO5 (M. wt. = 389): C, 52.33; H, 3.10; N, 3.59. Found: C, 51.45; H, 2.91; N, 3.44.
5-bromo-N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carbimidic acid (Compound 9) was produced as crystals of an orange hue: yield (2.37 g, 61%), m.p. 180 °C. IR (KBr) υmax: 3467 (br. OH), 1732 (C=O), 1633 (C=N), 1605, 1568 (C=C), 1210, 1117, 1031 (C-O) cm−1. 1H NMR(DMSO-d6) δ: 3.82, 3.84 (s, 3H, OCH3 of two isomers), 6.88–6.90 (d, J = 6 Hz, 1H, Ar-H), 7.10–7.42 (m, 5H, Ar-H), 8.97 (s, 1H, H-4 of coumarin ring), 10.24–10.40 (br. s., 1H, phenolic OH), 13.77, 13.87 (s, 1H, OH of two isomers) ppm. 13C NMR(DMSO-d6) δ: 192.46, 190.35 (-N=C-OH), 162.78, 160.94 (C=O), 152.61, 152.59, 151.88, 151.77, 151.17, 150.70, 150.30, 149.91, 148.53 (C-O and C-4 of coumarin ring), 125.53, 124.46, 124.28, 124.04, 122.90, 122.84, 122.37, 121.66, 121.48, 120.60, 120.57, 120.13, 120.10, 119.69, 119.51, 119.46, 118.70, 118.02, 117.87, 117.33, 116.12, 115.83 (C-aromatic), 110.95, 109.15 (C-3 of coumarin ring), 57.05, 56.65, 56.55, 56.30 (OCH3 of different isomers). Analytically calculated for C17H12BrNO5 (M. wt. = 389): C, 52.44; H, 3.08; N, 3.60. Found: C, 52.11; H, 2.93; N, 3.33.
3.2. Biological Assay
3.2.1. In Vitro Cytotoxicity Assay against (MCF-7) and (MDA-MB-231) Cell Lines
The cytotoxicity of Compounds 3–6 and 8–9 was evaluated using the MCF-7 and MDA-MB-231 breast cancer cell cultures. The cell lines were developed in culture flasks with a 10% fetal bovine serum addition at 5% CO2. Before being treated, cell lines were cultured in 96-well plates (10,000 cells/well) for 24 h to allow for attachment. Each substance was introduced to wells with cell lines at a concentration series of 0.39–1.56–6.25–25–100 μg/mL. For 48 h, chemicals were left in a 37 °C incubator with the cells. After 48 h, the cells were fixed, cleaned, and MTT stained. The color intensity was detected by an ELISA reader at 570 nm [47].
3.2.2. Cell Cycle Analysis
MCF-7 cells (2 × 105/well) were subjected to Compound 6 treatments for 24 h at respective IC50 values. After treatment, the cells were centrifuged, washed twice with ice-cold PBS, fixed in ice-cold 66% (v/v) ethanol at 4 °C for two hours, and then washed once more with PBS at 37 °C for 30 min. After being separated by centrifugation at 2000 rpm for 5 min, the cells were stained with PI buffer. The samples were then gently blended and left to incubate for 20 min at room temperature in the dark. Following this, the DNA content was determined using a BD FACS CALIBER flow cytometer. All tests were run in duplicate [48].
3.2.3. Annexin V-FITC/PI Assay of Compound 6
Using a fluorescent Annexin V-FITC/PI detection kit (BioVision Annexin V-FITC Apoptosis Detection Kit, Catalog #: K101by) and flow cytometry test, apoptosis in MCF-7 cultures was investigated. Compound 6 (IC50 (μM)) was added to a well containing 2 × 105 MCF-7 cells for 48 h. The cells were then removed and stained for 15 min in low light under ambient conditions with Annexin V-FITC/PI dye. A FACS Calibur flow cytometer (Becton and Dickinson, Heidelberg, Germany) was used to evaluate the samples immediately [49].
3.2.4. In Vitro Evaluation of β-Tubulin Inhibition Assay
According to the manufacturer’s recommendations, Compound 6 was tested using the Human Beta-tubulin Easy Step ELISA Kit ab245722 (Abcam) to determine its tubulin inhibitory activity. DMEM (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 μg/mL of insulin (Sigma), and 1% penicillin–streptomycin was used for the breast cancer cell lines’ cultivation. Prior to the tubulin enzyme assay, 100 μL of the tumor cell suspension and the test substance were dispensed into each well of a 96-well plate for 18–24 h. The given microtiter was pre-coated with a TUB β-specific antibody. Biotin combined with antibody specific for TUB β was then added to the appropriate microtiter plate wells along with the samples or standards. Each microplate well was next treated to the addition of avidin coupled to horseradish peroxidase (HRP), which was followed by incubation. Next, TMB substrate solution was added—color change will be noticed only in those wells that contain biotin combined with antibody, TUB β, and enzyme-coupled avidin. After adding sulfuric acid solution to stop the enzyme–substrate reaction, the color change was measured spectrophotometrically at a wavelength of 450 nm +± 10 nm. The O.D. of the samples was compared to the standard curve in order to determine the concentration of TUB β in the samples. Every experiment was carried out twice [50].
3.2.5. Sulfatase Enzyme Inhibitory Assay
The screening of Compound 6 for sulfatase inhibitory activity was accomplished according to the manufacturer’s instructions using the sulfatase activity assay colorimetric kit ab204731. The first step involves washing and resuspension tissues in cold PBS with protease inhibitors, followed by incubation, centrifugation, and collection of supernatants in a clean tube. In the second step, the enzyme was dissolved in ddH2O, 0.2% NaCl, PBS, or an appropriate buffer. All samples were prepared in duplicate. After adding the sulfatase mix to the positive control and sample cells, incubation at 37 °C for 30 min was performed, followed by adding 90 μL of developing/stop solution to each well and measuring the plate at OD = 515 nm.
3.2.6. In Vitro Aromatase Enzyme Inhibitory Assay
The instructions were provided by the manufacturer for the SEC319Ra 96 tests. The Aromatase Enzyme-Linked Immunosorbent Assay Kit (ARO) has aided us in our examination of the chemical ability of Compound 6 to block the aromatase enzyme, following the manufacturer’s instructions. All reagents, samples, and working standards were prepared prior to experiment. Each well received 100 μL of sample and standard before being incubated at 37 °C for two hours. The liquid from each well was discarded. After that, each well received the addition of 120 μL of biotinylated detection antibody before being incubated for 60 min at 37 °C. After removing the media using an aspirator, a washing buffer was used three times. Each well was filled with 100 μL of horseradish peroxidase (HRP, avidin), and then incubated at 37 °C for almost an hour. The aspiration and washing procedure was conducted five times. Each well received 90 μL of 3,3’,5,5’-tetramethylbenzidine (TMB) substrate before being incubated for 15 min at 37 °C. Each well received around 50 μL of stop solution just before the optical density determination of every well using a ROBONEK P2000 ELISA reader set to 450 nm. Non-linear regression analysis of a sigmoidal dose–response curve was used to plot the values of % activity versus a range of chemical concentrations (2.5 μM, 5 μM, 10 μM, 15 μM). Finally, a comparison of the data was conducted with letrozole as a reference drug. All experiments were conducted in triplicate [51].
3.2.7. Preparation of Protein and Molecular Docking
The structures considered were the human progesterone receptor co-crystallized with metribolone (PDB: 1E3K) [52] and the human aromatase cytochrome P450 (CYP19A1) co-crystallized with polyethylene glycol (PDB: 5JKV) [53]. Three-dimensional X-ray crystallized structures of the progesterone and aromatase cytochrome P450 protein receptors were obtained from the Protein Data Bank (https://www.rcsb.org/, accessed on 10 June 2023). Three-dimensional protonation, hydrogen addition, energy minimization, and ligand active site prediction were used to obtain receptor proteins suitable for molecular docking. Next, Compound 6 was docked with the target protein (progesterone or aromatase cytochrome P450) using the Triangle Matcher placement method while using MOE software. While performing docking, the ligand was selected, and rescoring was set at “London dG” and rescoring at “GBVI/WSA dG”, operating to detect ligand–protein interactions. All of the data collected during the protein-ligand docking process, including the score, ligand characteristics, and 2D and 3D structures, were stored [54].
4. Conclusions
In this study, several new hybrid 8-methoxy coumarin derivatives bearing oxazole and N-arylcarboxamide moieties (Compounds 3–9) were synthesized through the condensation of a coumarin ester with ammonia and 2-aminophenol to produce 8-methoxycoumarin-3-carboxamide (Compound 2) and N-(2-hydroxyphenyl)-8-methoxycoumarin-3-carboxamide (Compound 5) and its analog (Compound 6). Two 8-methoxy-5-substituted coumarin derivatives (Compounds 3 and 4) bearing an oxazole core were obtained through the cyclization of coumarin-3-carboxamide (Compound 2) with 4-chlorophenacyl bromide to produce Compound 3, followed by the halogenation of Compound 3 with bromine to produce Compound 4. The halogenation of Compounds 5 and 6 with bromine yielded two bromo derivatives (Compounds 8 and 9). Spectroscopic methods were used to confirm the synthesized substances’ structures (IR, NMR, and MS spectrometry), as well as elemental analysis. The synthesized compounds, Compounds 3, 4, 5, 6, 8, and 9, exhibited a good anti-proliferative activity against the breast cancer cell line, especially Compound 6, which has been chosen for further anticancer analysis. This compound has shown the ability to induce cell-cycle arrest at the S phase in the MCF-7 cell line, as well as apoptosis. This compound also represents a potent anti-β-tubulin (TUB) polymerization, sulfatase, and aromatase enzyme agent.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13071037/s1.
Author Contributions
Conceptualization, T.A.-W., O.A.A.A., L.S.A., E.A.-E., M.E.B., A.E.A.E.-B., M.S.A.Z., E.F. and E.M.R.; methodology; software, validation, formal analysis, investigation, resources, data curation, writing—original draft preparation, writing—review and editing, visualization, supervision, project administration, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through large group Research Project under (grant number RGP2/40/44) and the Princess Nourah bint Abdulrahman University Researchers Supporting Project (number PNURSP2023R25), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through large group Research Project under (grant number RGP2/40/44) and the Princess Nourah bint Abdulrahman University Researchers Supporting Project (number PNURSP2023R25), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors would also like to thank Science Shake Inc. for conducting proofreading and English language editing (https://science-shake.com/, accessed on 10 June 2023).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sharma, S.; Singh, J.; Ojha, R.; Singh, H.; Kaur, M.; Bedi, P.M.S.; Nepali, K. Design Strategies, Structure Activity Relationship and Mechanistic Insights for Purine as Kinase Inhibitors. Eur. J. Med. Chem. 2016, 112, 298–346. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharn, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.I. Rational Approaches, Design Strategies, Structure Activity Relationship and Mechanistic Insights for Anticancer Hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Nepali, K.; Ojha, R.; Sharma, S.; Bedi, P.M.S.; Dahr, K.I. Tubulin Inhibitors: A Patent Survey. Recent Pat. Anticancer Drug Discov. 2014, 9, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Soriano, E. The Medicinal Chemistry of Hybrid-based Drugs Targeting Multiple Sites of Action. Curr. Med. Chem. 2011, 11, 2714–2715. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31. [Google Scholar] [CrossRef] [PubMed]
- Geldenhys, W.J.; Youdim, M.R.H.; Carroll, R.T.; Van der Schyf, C. The emergence of designed multiple ligands for neurodegenerative disorders. J. Prog. Neurobiol. 2011, 94, 347. [Google Scholar] [CrossRef]
- Zhang, H.Y. One-compound-multiple-targets strategy to combat Alzheimer’s disease. FEBS Lett. 2005, 579, 5260. [Google Scholar] [CrossRef]
- Geldenhys, W.J.; Van der Schyf, C. Multimodal drugs and their future for Alzheimer’s and Parkinson’s disease. J. Int. Rev. Neurobiol. 2011, 100, 107. [Google Scholar]
- Flores-Morales, V.; Villasana-Ruíz, A.P.; Garza-Veloz, I.; González-Delgado, S.; Martinez-Fierro, M.L. Therapeutic Effects of Coumarins with Different Substitution Patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef]
- Vilar, S.; Quezada, E.; Santana, L.; Uriarte, E.; Yanez, M.; Fraiz, N.; Alcaide, C.; Cano, E.; Orallo, F. Design, synthesis, and vasorelaxant and platelet antiaggregatory activities of coumarin–resveratrol hybrids. Bioorg. Med. Chem. Lett. 2006, 16, 257. [Google Scholar] [CrossRef]
- Song, H.Y.; Ngai, M.H.; Song, Z.Y.; MacAry, P.A.; Hobley, J.; Lear, M. Practical synthesis of maleimides and coumarin-linked probes for protein and antibodylabellingviareduction of native disulfides. J. Biomol. Chem. 2009, 7, 3400. [Google Scholar] [CrossRef]
- Melagraki, G.; Afantitis, A.; Igglesst, M.O.; Detsi, A.; Koufaki, M.; Kontogiorgis, C.; Hadjipavlou, L.D. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur. J. Med. Chem. 2009, 44, 3020. [Google Scholar] [CrossRef]
- Kamath, P.R.; Sunil, D.; Josheph, M.M.; Salam, A.A.A.; Sreelekha, T.T. Indole-coumarin-thiadiazole hybrid: An appraisal of their MCF-7 cell growth inhibition, apoptic, antimetastatic and computational BCL-2 binding potential. Eur. J. Med. Chem. 2017, 136, 442–451. [Google Scholar] [CrossRef]
- Mokale, S.N.; Begum, A.; Sakle, N.S.; Shelke, V.R.; Bhavale, S.A. Design, synthesis and anticancer screening of 3-(3-(substitutedphenyl)acryloyl)-2H-chromen-2-ones as selective anti-brast cancer agent. Biomed. Pharmacother. 2017, 89, 966–972. [Google Scholar] [CrossRef]
- Falcon, I.H.; Amesty, A.; Afonso, L.A.; Castrillejo, I.L.; Machin, F.; Estevez Braun, A. Synthesis and biological evaluation of naphthoquinpne-coumarin conjugates as topoisomerase II inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 484–489. [Google Scholar] [CrossRef]
- Elshemy, H.A.H.; Zaki, M.A. Design and synthesis of new coumarin hybrids and insight into their mode of antiproliferative action. Bioorg. Med. Chem. 2017, 25, 1066–1075. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global Cancer Statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Kim, S.-K. Handbook of Anticancer Drugs from Marine Origin; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- WHO. Cancer Fact Sheet No. 297; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Radwan, E.M.; Elsayed, E.H.; El-Moniem, M.A.; Moustafa, A.M.Y. Design, Synthesis, and Anti-Breast Cancer Activity of Some Hybrid Molecules Containing Coumarin Moiety. Russ. J. Bioorg. Chem. 2021, 47, 149–157. [Google Scholar] [CrossRef]
- Radwan, E.M.; Elsayed, E.H.; El-Moniem, M.A.; Moustafa, A.M.Y. Synthesis and Biological Evaluation of Some New 3,4-Disubstituted Coumarin Derivatives as Anticancer Agents. Indian J. Heterocycl. Chem. 2021, 31, 101–111. [Google Scholar]
- Zaki, I.; Elsayed, E.H.; Radwan, E.M. Synthesis and Antiproliferative Activity of Some New Coumarin Derivatives Derived from 8-Hydroxycoumarin. Russ. J. Bioorg. Chem. 2021, 47, 514–523. [Google Scholar] [CrossRef]
- Salem, M.G.; Abu-El Maaty, D.M.; El-Deen, Y.I.M.; Elesawy, B.H.; El Askary, A.; Saleh, A.; Saied, E.M.; El Behery, M. Novel 1,3-Thiazole Analogues with Potent Activity against Breast Cancer: A Design, Synthesis, In Vitro, and In Silico Study. Molecules 2022, 27, 4898. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Abel, A.-C.; Maruca, A.; Montalbano, A.; Raimondi, M.V.; Tarantelli, C.; Gaudio, E.; et al. Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: Further chemical modifications and potential therapeutic effects against lymphomas. Eur. J. Med. Chem. 2022, 243, 114744. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kaku, K.; Robles, A.J.; Hamel, E.; Mooberry, S.L.; Gangjee, A. Simple monocyclic pyrimidine analogs as microtubule targeting agents binding to the colchicine site. Bioorg. Med. Chem. 2023, 82, 117217. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spano, V.; Raimondi, M.V.; Tarantelli, C.; Spriano, F.; Bertoni, F.; Barraja, P.; Montalbano, A. Recurrence of the oxazole motif in tubulin colchicine site inhibitors with anti-tumor activity. Eur. J. Med. Chem. 2021, 1, 100004. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Xu, R.-M.; Zheng, G.-H.; Jin, Y.-Z.; Li, Y.; Chen, X.-Y.; Tian, Y.-S. Development of Combretastatin A-4 Analogues as Potential Anticancer Agents with Improved Aqueous Solubility. Molecules 2023, 28, 1717. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Jain, P.K.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef]
- Hettle, A.G.; Vickers, C.J.; Boraston, A.B. Sulfatases: Critical Enzymes for Algal Polysaccharide Processing. Front. Plant Sci. 2022, 13, 837636. [Google Scholar] [CrossRef]
- Lenne, P.F.; Trivedi, V. Sculpting tissues by phase transitions. Nat. Commun. 2022, 13, 664. [Google Scholar] [CrossRef]
- Huang, L.; Huang, J.; Nie, H.; Li, Y.; Song, L.; Wu, F. Design, synthesis and biological evaluation of combretastatin A-4 sulfamate derivatives as potential anti-cancer agents. RSC Med. Chem. 2021, 12, 1374–1380. [Google Scholar] [CrossRef]
- Kozak, W.; Daśko, M.; Masłyk, M.; Pieczykolan, J.S.; Gielniewski, B.; Rachon, J.; Demkowicz, S. Phosphate tricyclic coumarin analogs as steroid sulfatase inhibitors: Synthesis and biological activity. RSC Adv. 2014, 4, 44350–44358. [Google Scholar] [CrossRef]
- Brueggemeier, R.W. Aromatase inhibitors in breast cancer therapy. Expert. Rev. Anticancer Ther. 2002, 2, 181–191. [Google Scholar] [CrossRef]
- Brueggemeier, R.W.; Richards, J.A.; Joomprabutra, S.; Bhat, A.S.; Whetstone, J.L. Molecular pharmacology of aromatase and its regulation by endogenous and exogenous agents. J. Steroid Biochem. Mol. Biol. 2001, 79, 75–84. [Google Scholar] [CrossRef]
- Miller, W.R. Aromatase inhibitors in the treatment of advanced breast cancer. Cancer Treat. Rev. 1989, 16, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Banting, L.; Nicholls, P.J.; Shaw, M.A.; Smith, H.J. Recent developments in aromatase inhibition as a potential treatment for oestrogen-dependent breast cancer. Prog. Med. Chem. 1989, 26, 253–298. [Google Scholar] [PubMed]
- Park, W.C.; Jordan, V.C. Selective estrogen receptor modulators (SERMs) and their roles in breast cancer prevention. Trends Mol. Med. 2002, 8, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R. Biological rationale for endocrine therapy in breast cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; James, K.; Owen, C.P. Inhibition of estrone sulfatase (ES) by derivatives of 4-[(aminosulfonyl)oxy] benzoic acid. Biorg. Med. Chem. Lett. 2002, 12, 2391–2394. [Google Scholar] [CrossRef]
- Ratre, P.; Kulkarni, S.; Das, S.; Liang, C.; Mishra, P.K.; Thareja, S. Medicinal chemistry aspects and synthetic strategies of coumarin as aromatase inhibitors: An overview. Med. Oncol. 2022, 40, 41. [Google Scholar] [CrossRef]
- Brandão, M.; Aftimos, P.; Azim, H.A.; Sotiriou, C. Chapter 26-Molecular Biology of Breast Cancer, 2nd ed.; Coleman, W.B., Tsongalis, E., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 449–461. [Google Scholar]
- Li, Z.; Wei, H.; Li, S.; Wu, P.; Mao, X. The Role of Progesterone Receptors in Breast Cancer. Drug Des. Dev. Ther. 2022, 16, 305–314. [Google Scholar] [CrossRef]
- Barros-Oliveira, M.d.C.; Costa-Silva, D.R.; dos Santos, A.R.; Pereira, R.O.; Soares-Júnior, J.M.; da Silva, B.B. Influence of CYP19A1 gene expression levels in women with breast cancer: A systematic review of the literature. Clinics 2021, 76, e2846. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Breast Cancer: A Molecularly Heterogenous Disease Needing Subtype-Specific Treatments. Med. Sci. 2020, 8, 18. [Google Scholar] [CrossRef]
- Blackburn, H.L.; Ellsworth, D.L.; Shriver, C.D.; Ellsworth, R.E. Role of cytochrome P450 genes in breast cancer etiology and treatment: Effects on estrogen biosynthesis, metabolism, and response to endocrine therapy. Cancer Causes Control 2015, 26, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Kataoka, N.; Shu, X.; Wen, W.; Gao, Y.; Cai, Q.; Zheng, W. Genetic Polymorphisms of the CYP19A1 Gene and Breast Cancer Survival. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Talorete, T.P.N.; Yamada, P.; Isoda, H. Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology 2009, 59, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Zaki, I.; Abdelhameid, M.K.; El-Deen, I.M.; Wahab, H.A.A.; Ashmawy, A.M.; Mohamed, K.O. Design, synthesis and screening of 1, 2, 4-triazinone derivatives as potential antitumor agents with apoptosis inducing activity on MCF-7 breast cancer cell line. Eur. J. Med. Chem. 2018, 156, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Chen, Y.-J.; Chen, J.J.; Shieh, J.-J.; Huang, C.-H.; Lin, P.-S.; Chang, G.-C.; Chang, J.-T.; Lin, C.-C. Terpinen-4-ol induces apoptosis in human nonsmall cell lung cancer in vitro and in vivo. Evid. Based Complement. Altern. Med. 2012, 2012, 121–134. [Google Scholar]
- Mourad, A.A.E.; Rizzk, Y.W.; Zaki, I.; Mohammed, F.Z.; El Behery, M. Synthesis and cytotoxicity screening of some synthesized hybrid nitrogen molecules as anticancer agents. J. Mol. Struct. 2021, 1242, 130722. [Google Scholar] [CrossRef]
- AboulWafa, O.M.; Daabees, H.M.G.; Badawi, W.A. 2-Anilinopyrimidine derivatives: Design, synthesis, in vitro anti-proliferative activity, EGFR and ARO inhibitory activity, cell cycle analysis and molecular docking study. Bioorg. Chem. 2020, 99, 103798. [Google Scholar] [CrossRef]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor: Implications for pathogenic gene mutations. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef]
- Ghosh, D.; Egbuta, C.; Lo, J. Testosterone complex and non-steroidal ligands of human aromatase. J. Steroid Biochem. Mol. Biol. 2018, 181, 11–19. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).