Abstract
X-ray powder diffraction is a vital analytical tool that is used in pharmaceutical science. It is increasingly used to establish the crystal structure of a new pharmaceutical substance, in particular, cocrystal or its polymorphic forms. This review begins with a brief discussion of the reliability of the structural parameters retrieved from powder patterns. Recent examples of the successful determination of crystal structures of pharmaceutical cocrystals and salts from powder diffraction data are discussed. These examples show the increased capabilities of laboratory X-ray powder diffractometers and modern software in solving actual problems of pharmaceutical science.
1. Introduction
X-ray powder diffraction (XRPD) has occupied a special place among the analytical methods utilized by pharmaceutical scientists for many years, as evidenced by numerous publications [1,2,3,4,5]. Due to its nondestructive nature and the relatively easy sample preparation, XRPD is the methodology of choice for the detection of crystalline impurities, analysis of final dosage forms, detection of changes in morphology during production, and optimization of drug carriers and excipients. XRPD also allows for the quantification of the proportions of crystalline forms within a sample, characterization of amorphous pharmaceuticals [6], and optimization of the process parameters. X-ray powder pattern is also required for the registration of new products and patent applications.
It is necessary to emphasize the fundamental possibility of XRPD in solving new crystal structures of the active pharmaceutical ingredients (API) when they cannot be prepared as single crystals of the appropriate size and quality. The established crystal structure of pharmaceutical solids helps to uncover structure–activity relationships and therefore offers an opportunity for the selection of the most suitable form of API for development into a drug product. Moreover, a thorough analysis of crystal structures gives rise to crystal engineering in design of the novel pharmaceutical compounds with the desired physical and mechanical properties [7]. Modern X-ray diffractometers, powerful computers, and the continuously improving methods of structure determination from powder data, so called SDPD methods [8,9,10,11,12,13,14,15,16,17,18,19,20], provide a reliable basis for conducting such research in laboratories of any level.
The ability of XRPD to uncover new crystal structures turned out to be in high demand when dealing with pharmaceutical polymorphs. Back in 1969, Haleblian and McCrone [21] noted that XRPD, “one of the most widely used techniques”, has been repeatedly applied “to identify polymorphs in pharmaceuticals”. These words have not lost their relevance after five decades of the development of pharmaceutical sciences. Every new review devoted to polymorphism of organic compounds and related to pharmaceutical field underlines the unique opportunities of XRPD for obtaining reliable information about the crystal form of any ingredient of a pharmaceutical product [22,23,24,25]. Considerable interest in the study of API polymorphism features is created by the fact that different polymorphs usually have different properties that are important for a drug product, such as solubility, permeability, bioavailability, pharmacokinetics, and stability. Therefore, one can expect that it should be possible to obtain different crystal forms of a drug and, thus, modify its performance properties.
The same expectations fuel numerous studies of pharmaceutical cocrystals, which have gained significant popularity in the last two decades [26,27,28,29,30,31,32,33,34,35]. Cocrystals comprising several molecules in the unit cell—at least one of which is API, and the rest are pharmaceutical excipients—look attractive to the pharmaceutical industry because they allow companies to produce new drugs with improved properties, without capital investments in the search and design for new APIs. Examples of pharmaceutical cocrystals approved by the US-FDA as drugs are already known—Entresto®, Lexapro®, Steglatro®, and Seglentis®. Many more promising cocrystals are now being actively investigated by pharmaceutical scientists in the universities and companies. A prerequisite for the success of these studies is knowledge of the cocrystal structure, its stability at various temperatures, humidity, and contacts with the packaging material, and all this information can be provided by XRPD.
This article presents an overview of recent successful applications of X-ray powder diffraction methods in the structure elucidation of new pharmaceutical cocrystals. We discuss the results obtained with the help of laboratory X-ray equipment, which is available in many laboratories. Though synchrotron diffractometry represents a much more powerful instrument for analyzing pharmaceutical materials in comparison with the laboratory X-ray diffractometers [36], only a small number of scientific groups can afford to carry out daily routine measurements on synchrotron beamlines. These lucky groups are part of the scientific divisions of major international drug companies such as Abbott Laboratories, Bayer, Bristol-Myers Squibb, Glaxo Wellcome, Eli Lilly, Merck, Monsanto/Searle, Parke-Davis, Pharmacia and Upjohn, Proctor and Gamble, Schering-Plough, and Smith Kline Beecham, which have access to synchrotron facilities through the collaborative access team at the US Advanced Photon Source (IMCA-CAT [37]). However, the laboratory X-ray equipment that allows high-throughput measurements near the chemist’s table is also capable of providing reliable crystallographic information about new pharmaceutical cocrystals and salts, as is shown below.
2. Crystal Structures from Powder Diffraction—How Reliable Are They?
Before proceeding to discuss the details of structural features elucidation from the XRPD data, it is necessary to give yourself an answer to the following question—to what extent can the obtained structural data be trusted? In an attempt to establish a new crystal structure from the powder diffraction pattern, the researcher deals with a set of possible models of these structures and tries to choose the best one. The best structural model must meet several criteria, such as the following:
- (1)
- It should provide minimal discrepancies between the experimental and calculated patterns, i.e., minimal values of χ2 and profile R-factors, Rp and Rwp;
- (2)
- It must be in complete agreement with all experimental data on the other physicochemical studies;
- (3)
- The geometry of all molecular fragments, i.e., bond lengths and angles, and geometry of intermolecular contacts (H-bonds, π…π, dipole–dipole, and others weak interactions) must correspond to the known data, for example, collected in the Cambridge Structural Database (CSD [38]).
In 2014, van de Streek and Neumann [39] added one more useful quantitative benchmark for the validation of molecular crystal structures determined from XRPD data. Using the dispersion-corrected density functional theory (DFT-D), they optimized 215 published organic crystal structures, analyzed the root-mean-square Cartesian displacement (RMSCD) of the non-hydrogen atoms for each structure, and concluded the following:
- (4)
- For the reliable crystal structure determined from XRPD data, the upper RMSCD limit should not exceed the value of 0.35 A.
Among the four aforementioned Criteria (1)–(4), Criterion (1) is of particular value because, in the case of good crystallinity of the powder sample, it allows one to sharply limit the search area of possible structural models. Indeed, a pharmaceutical cocrystal consists of the predefined molecules in a ratio established by other analytical methods with good accuracy. It remains only to arrange these molecules in 3D space in such a way as to obtain a set of crystal structure models satisfying Criteria (3) and (4), and then select the best one in accordance with (1). The best structural model can be achieved with the help of crystal structure prediction (CSP) methodology [40]. However, without the unit cell parameters and without the limited set of possible space groups, the calculations are excessively time-consuming.
A well-crystallized powder sample provides an X-ray pattern, which can be indexed. Indexing means that the unit cell parameters are determined, and the positions of all observed diffraction peaks coincide with the calculated ones. Furthermore, the use of the Pawley [41] or Le Bail [42] fit is recommended, as both require no atomic coordinates and enable one to estimate the reliability of the unit cell parameters and limit the number of possible space groups, sometimes up to one. As a bonus, the researcher obtains χ2, Rp, and Rwp with the smallest values achievable with the unit cell and space group, which the researcher needs to strive for in the final Rietveld refinement of the solved crystal structure. Knowing the unit cell parameters, space group, and chemical content of a molecular crystal, one can estimate the number of moieties in the asymmetric unit. After that, it remains only to arrange the moieties in the asymmetric unit, using SDPD methods [8,9,10,11,12,13,14,15,16,17,18,19,20]. A sequence of steps in the search for the best structural model, which begins with the fulfillment of Criteria (1) and (2) and ends with (3) and (4), requires much less computation time, and that is why this approach has been most popular over the past two decades. Sometimes a few hours are enough to solve, refine, and validate a new crystal structure by using an X-ray powder pattern of good quality.
For molecular substances producing poorly crystalline powder samples and, as a consequence, low-quality X-ray patterns that resist indexing (unindexed powder patterns), the problem of crystal structure determination is a serious challenge. However, this problem does not seem hopeless. Habermehl, Schlesinger, and Schmidt [43] recently introduced a new method for ab initio crystal structure determination from XRPD for organic and metal–organic compounds which does not require the prior indexing of the powder pattern. The same authors, having a poor-quality unindexed powder data of 4,11-difluoroquinacridone, tried to determine its crystal structure by applying their new method and eventually obtained four different structural models which all led to the acceptable R-factors in the Rietveld refinement [44]. To select the correct structure among the four models, six methods were used. The results of this research bring to mind the “old” question to the focus of our attention—are the results of SDPD always reliable [45]? Before trying to find an unambiguous answer to this question, it is necessary to clarify the meanings of expressions “correct structure” and “reliable structure”.
In a retrospective view of the history of the XRPD, it is easy to see correlations with similar problems in the development of indexing methods. Considering the quantitative criterion, M20, which helps to assess the degree of reliability of a powder pattern indexing, Pieter M. de Wolff wrote that “a hypothesis such as the correctness of a unit cell can never be verified with absolute certainty” [46]. Therefore, when analyzing further examples of indexing and evaluating them as “correct” and “incorrect”, he explains that the “term ‘correct’ is used for results which have been confirmed by single-crystal investigations”. In modern publications, one can find examples of structures independently determined from powder and single-crystal samples. A few such examples related to pharmaceutical substances are presented in Table 1.

Table 1.
Pharmaceutical substances whose crystal structures have been independently determined from powder and single-crystal diffraction data.
These examples demonstrate good reproducibility of the main structural features in powder structures as compared with the single-crystal ones, and this adds confidence in the reliability of crystal structures derived from powder data. When we use the term “reliable” for any crystal structure, we mean that this structure is in accordance with all experimental data obtained for the solid-state sample, and, most important, for the substance with API, it provides a basis for quantitative assessments of important properties of the bulk material—solubility, stability, permeability, etc.—which can be evidenced experimentally.
The three following statements conclude this section:
- -
- When working with XRPD and a new polycrystalline sample, the search for its “correct” crystal structure is useless because nobody knows what it should look like. Instead, the search for the best structural model(s) suitable for Criteria (1)–(4) allows you to obtain a certain result;
- -
- Any additional information limiting the area of possible structural models has to be used;
- -
- The best structural model that has been obtained in several independent diffraction experiments with different samples of the substance, and which allows us to predict the properties of the bulk material, can be considered a reliable crystal structure.
It is necessary to understand that the earlier obtained models can be considered as a “best structural model” and a “reliable crystal structure” only until new experimental data contradicting our model structures are obtained.
3. Applications of SDPD Methods to Pharmaceutical Cocrystals and Salts
Recent examples of successful crystal structure determinations from laboratory X-ray powder patterns are presented below to demonstrate the “pervasive abilities of X-rays” in the in-house characterization of the multicomponent pharmaceutical solid-state substances.
3.1. Lamivudine Camphorsulfonate (1:1) Salt
The powder diffraction pattern of the title salt, C8H12N3O3S+·C10H15O4S− [59] (Figure 1), used for structure solution was recorded at 153 K in transmission geometry on a STOE Stadi-P diffractometer equipped with a curved Ge monochromator, using CuKα1 radiation. It was indexed with the program DICVOL [60] in a monoclinic unit cell with a volume of 2051 Å3. The Pawley fit [41] confirmed unit cell parameters and chiral space group P21, the asymmetric unit of which contains two formula units—two cations and two anions. The crystal structure was solved with the simulated annealing technique implemented in the DASH program [61] and refined by the Rietveld method, using the program TOPAS [62].
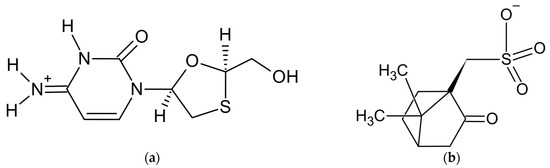
Figure 1.
Components of lamivudine camphorsulfonate (1:1) salt: (a) lamivudine cation and (b) camphorsulfonate anion.
In the original paper [59], the crystal structure of theophylline benzamide (1:1) cocrystal was successfully determined from synchrotron powder diffraction data. However, the application of SDPD methods to the crystal structure determination of the third compound, aminoglutethimide camphorsulfonate hemihydrate, failed, and its crystal structure was solved later, using a single crystal of suitable size. A detailed description of all the steps of SDPD for two compounds and an analysis of the reasons for failure when working with the third one make this article [59] very useful for scientists who are trying to apply SDPD methods to pharmaceutical substances.
3.2. Furosemide Urea (1:1) and Carbamazepine Indomethacin (1:1)
The powder diffraction patterns of the title cocrystals, C12H11ClN2O5S·CH4N2O and C15H12N2O·C19H16ClNO4, used for structure solution were recorded at 298 K in transmission geometry on a Bruker D8 Advance diffractometer equipped with a LynxEye detector, using CuKα1 radiation. Both powder patterns were indexed with the program DASH—in a triclinic unit cell with a volume of 836 Å3 for furosemide urea (FUR:URE) and in a monoclinic unit cell with a volume of 2823 Å3 for carbamazepine indomethacin (CBZ:IND). The Pawley fit confirmed unit cell parameters and space groups P-1 and P21/n for FUR:URE and CBZ:IND, respectively, with one formula unit per asymmetric part in each structure. The crystal structures were solved with the simulated annealing technique implemented in the DASH program and subjected to rigid-body Rietveld refinement, using TOPAS. Energy minimization, using the DFT-D theory, was used when finalizing both structures.
In the research of [63], a new polymorph of CBZ:IND (1:1) cocrystal was obtained and described as form II (CCDC ref. code LEZKEY01). Earlier, the same group of authors published the CBZ:IND cocrystal form I (CCDC ref. code LEZKEY, [64]), which crystallizes in a monoclinic unit cell with a volume of 2922 Å3 and space group P21/c. The crystal structure of form I was also determined from laboratory X-ray powder data with the use of SDPD methods. Therefore, two polymorphic modifications of carbamazepine indomethacin (1:1) cocrystal were revealed and structurally characterized by XRPD methods, thus demonstrating the modern abilities of the latter.
In both polymorphic forms, the carbamazepine and indomethacin molecules are paired via acid–amide heterosynthon [35], with slightly different conformations of the indomethacin molecule (Figure 2).
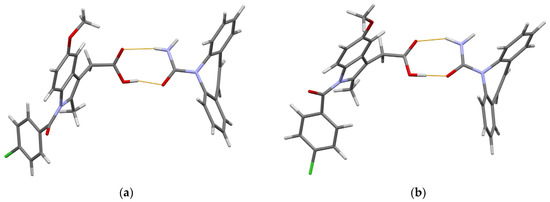
Figure 2.
(Prepared with Mercury [65]). The hydrogen-bonded pair of carbamazepine and indomethacin molecules in 1:1 cocrystal of (a) form I and (b) form II. Thin orange lines denote intermolecular N–H…O and O–H…O hydrogen bonds.
The O atom in the chlorobenzoyl (chlb) fragment acts as an H-bond acceptor in further intermolecular interactions. In form I, a N–H…Ochlb hydrogen bond with a N…Ochlb distance of 2.69 Å links these pairs into zigzag chains extended in the [101] direction. In form II, Ochlb acts as an acceptor of two weak hydrogen bonds, namely N–H… Ochlb [N… Ochlb 3.31 Å] and C–H… Ochlb [C… Ochlb 3.36 Å], which link the carbamazepine–indomethacin pairs into corrugated layers parallel to the ac plane. These differences in the crystal packing help us to understand why form II is more thermodynamically stable than form I [63].
3.3. Three Imidazole-Based (1:1) Salts of Salicylic Acid
To improve the poor solubility of salicylic acid (SA) in water, Emmerling and co-workers obtained its 1:1 salts with imidazole (IMI), 1-methylimidazole (1-MEIM), and 2-methylimidazole (2-MEIM). All three new salts, namely SA:IMI, SA:1-MEIM, and SA:2-MEIM, were mechanochemically synthesized using a vibratory ball mill. Their crystal structures were solved by powder X-ray diffraction.
The powder diffraction patterns that were used for structure solutions were recorded at room temperature in Debye–Scherrer geometry on a Bruker D8 Discover diffractometer equipped with a position-sensitive LynxEye detector and a Johansson monochromator, using CuKα1 radiation. Powder patterns were indexed with DICVOL [60] in orthorhombic unit cells with the volumes of 2020, 2192, and 2239 Å3 for SA:IMI, SA:1-MEIM, and SA:2-MEIM, respectively. The Pawley fits confirmed unit cell parameters and space groups Pbca, Pbca, and Pbcn, respectively, thus assuming one formula unit per asymmetric part in each structure. The crystal structures were solved with the simulated annealing technique implemented in the DASH program and refined by the Rietveld method, using the program TOPAS and restraints applied for all bond lengths, angles, and planar rings.
The subsequent physicochemical studies revealed that all three new cocrystal salts present remarkably higher solubility and a faster dissolution rate than free salicylic acid [66]. It is noteworthy that the orthorhombic crystal structure of SA:IMI (CCDC ref. code NIJDOB02) presents a new polymorph of this compound in addition to the earlier established tetragonal polymorph with the CCDC ref. code NIJDOB [67]. Different crystal packings in two polymorphs are shown in Figure 3.
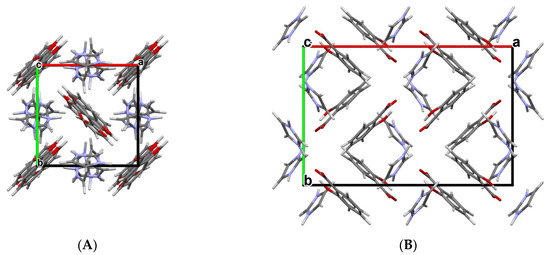
Figure 3.
Crystal packings of two polymorphs of SA:IMI (1:1) cocrystal viewed along the c-axis: (A) tetragonal polymorph (NIJDOB) and (B) orthorhombic polymorph (NIJDOB02).
The same SDPD methodology was used later by Martins and Emmerling in the crystal structure determination of three new 1:1 cocrystals of carbamazepine with 2,4-, 2,5-, and 2,6-dihydroxybenzoic acids as coformers [68].
3.4. Metaxalone Nicotinamide (1:1)
The powder diffraction pattern used for the structure solution of the title cocrystal, C12H15NO3·C6H6N2O [69], was recorded at 295 K in transmission geometry on a Guinier-Huber camera G670 equipped with a curved Ge monochromator, using CuKα1 radiation. It was indexed with the program ITO [70] in a triclinic unit cell, with a volume of 872 Å3. The Pawley fit confirmed the unit cell parameters. The crystal structure was solved in space group P-1 with the simulated annealing technique [71] and refined by the Rietveld method, using the program MRIA [72].
Metaxalone (MET) is a poor aqueous soluble drug, while its cocrystals with nicotinamide (NAM), salicylamide, and 4-hydroxybenzoic acid (hydrate) exhibit better solubility of the native drug, as followed from the dissolution experiments in pH 6.8 phosphate buffer [69]. Dissolution experiments indicated the superior solubility of the MET-NAM cocrystal, and this was correlated with its lower melting point, higher solubility of NAM, and weaker intermolecular interaction between MET and NAM. The improved solubility of MET–NAM is anticipated to enhance the drug’s bioavailability and decrease side effects [69].
In the MET-NAM crystal structure, the drug and coformer form hydrogen-bonded imide and amide homodimers of the R22(8) ring, respectively, which are further linked into chains in [110] via N–H…O hydrogen bonds (Figure 4).
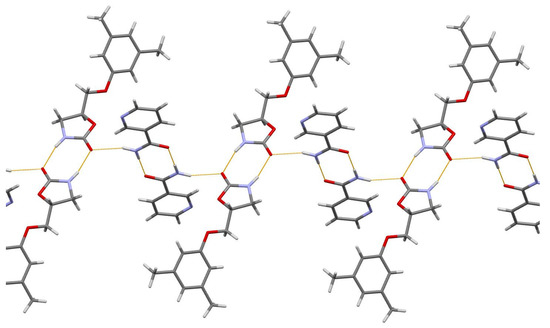
Figure 4.
The hydrogen-bonded chain extended in [110] of imide and syn-amide homodimers of MET and NAM molecules, respectively, in MET-NAM 1:1 cocrystal. Thin orange lines denote intermolecular N–H…O hydrogen bonds.
The aforementioned experimental facilities and SDPD methodology were used in the crystal structure determination of 1:1 cocrystals of dexamethasone with catechol and resorcinol [73], and cephalexin with serine [74].
3.5. Acridine Diclofenac (1:1) Salt
The powder diffraction pattern of the title salt, C13H10N+·C14H10Cl2NO2− [75], used for the structure solution was recorded at 295 K in transmission geometry on a Bruker D8 Advance diffractometer equipped with a LynxEye XE-T detector, using CuKα radiation. It was indexed with the programs EXPO2014 [76] and Topas Academic [62] in a monoclinic unit cell with a volume of 2247 Å3. The crystal structure was solved in space group P21/n, with the simulated annealing technique, and refined by the Rietveld method, using Topas Academic.
The authors [75] paid special attention to the correct positioning of the hydrogen atom between the N atom of acridine and O atom of diclofenac separated at 2.77(8) Å to make a choice between the salt (H atom attached to N) and cocrystal (H atom attached to O). The final choice in favor of salt was made after the energy framework calculations performed with CrystalExplorer 17.5 [77], which led to the following total lattice energies: −513.4 kJ mol−1 for the case of salt (N–H bond) and −215.2 kJ mol−1 for cocrystal (O–H bond). The content of asymmetric unit of the final structural model of the title salt is shown in Figure 5.
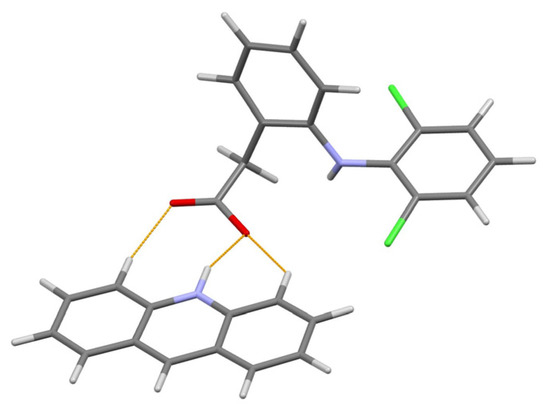
Figure 5.
The content of asymmetric unit of the acridine diclofenac (1:1) salt. Thin orange lines denote intermolecular N–H…O and weak non-classical C–H…O hydrogen bonds.
Among the advantages of this research, the quantitative phase analysis should also be noted, as it allowed one to estimate the excess of reactants in the sample (3.5% of acridine and a 4.5% of diclofenac) obtained with the liquid-assisted grinding, and thus define the yield of final product 92%.
The aforementioned experimental facilities and SDPD methodology were used in the crystal structure determination of orthorhombic form of naproxen acridine 1:1 cocrystal [78].
3.6. Acefylline Nicotinamide (1:2) Cocrystal
The powder diffraction pattern used for the structure solution of the title cocrystal, C9H10N4O4·2(C6H6N2O) [79], was recorded at 295 K in transmission geometry on a Guinier-Huber camera G670, using CuKα1 radiation. It was indexed with the program TREOR [80] in a triclinic unit cell with a volume of 1127 Å3. The Pawley fit confirmed the unit cell parameters. The crystal structure was solved in space group P-1 and refined using the program MRIA.
The asymmetric unit of the title 1:2 cocrystal contains one acefylline (ACF) and two independent isonicotinamide (INA) molecules. One independent INA molecule, labeled with the letter A, being translated along the short parameter c = 5.0288(7) Å, generates hydrogen-bonded chain, while another independent INA molecule, labeled with the letter B, forms centrosymmetric amide homodimer (Figure 4). Interestingly, the acefylline molecule acts as a bridge, which links the INA chains and dimers via intermolecular N–H…N and N–H…O hydrogen bonds (Figure 6).
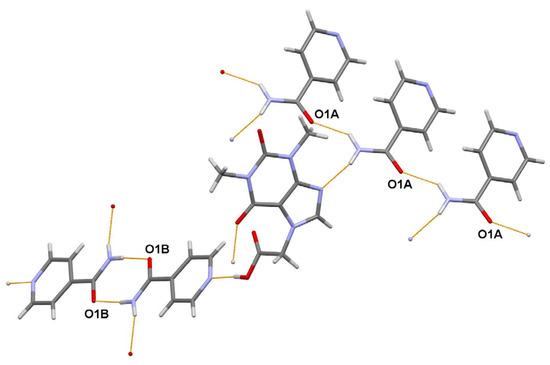
Figure 6.
A portion of the crystal packing of ACF:INA (1:2) cocrystal showing the intermolecular N–H…N and N–H…O hydrogen bonds as thin orange lines.
In the research [79] devoted to acefylline binary and ternary cocrystals, salt–cocrystals, and their polymorphs co-crystallized with different coformers, the crystal structure determination of the title ternary cocrystal from the laboratory X-ray powder pattern is a particular result, although it demonstrates the routine capabilities of SDPD methods.
3.7. Salt–Cocrystal Continuum of the Agomelatine: Phosphoric Acid (1:1) System
The existence of two polymorphic forms of the title compound, C15H17NO2·H3PO4 [81], referred to as RT and HT, was observed from the DSC analysis. The powder diffraction patterns of the RT and HT polymorphs were measured at 298 K in transmission geometry on a Bruker D8 Advance diffractometer, using CuKα radiation, and indexed with the program X-Cell [82] in monoclinic unit cells, with the volumes of 1644 and 1682 Å3, respectively. The Pawley fit confirmed unit cell parameters and space group P21/c, the asymmetric unit of which contains one formula unit. The crystal structures were solved with the simulated annealing technique implemented in the DASH program [61], refined by the Rietveld method, using the program TOPAS [60], and optimized using plane wave-periodic DFT-D2 calculations implemented in the MedeA software [83].
The authors of [81] concluded that the obtained crystal structures of RT and HT forms of the title compound in combination with DFT-D2 analysis allowed them to discriminate between the salt and cocrystal unambiguously. Moreover, detailed characterization using a set of complementary techniques, including DSC/TGA, solid state NMR, FTIR, and chemical crystallographic database analysis, confirmed that the nature of polymorphic form transformation is a solid-state phase transition, exhibiting an enantiotropic and reversible relationship. The portions of the DFT-D2 optimized crystal packings of RT and HT forms are shown in Figure 7.
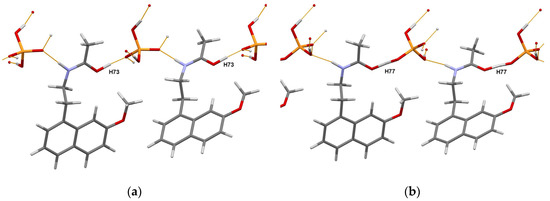
Figure 7.
Portions of the crystal packings in the DFT-D2 optimized RT (a) and HT (b) forms of the agomelatine:phosphoric acid (1:1) system showing the definite position of H73 atom in RT form, and intermediate position of H77 atom (migrating proton) in HT form. Thin orange lines denote intermolecular N–H…O and O–H…O hydrogen bonds.
This work [81] once again demonstrated the obstacles that researchers face in the choice between the salt and cocrystal. It also emphasized the special role of DFT-D optimization, which complements experimental methods such as X-ray photoelectron spectroscopy [84] or solid-state NMR [85,86], thus helping to make an informed choice.
4. Conclusions
The consistent development of the laboratory X-ray powder diffraction equipment and methods of processing experimental data allows one to significantly reduce the measurement time, increase the accuracy of the results obtained, and expand the range of tasks to be routinely solved. The factor of the time spent on solving a specific task is one of the most important in the pharmaceutical industry. This fully applies to the processes of design and launching the production of new substances with improved properties, such as pharmaceutical cocrystals. At the first exploratory stages of the development of a new cocrystal with improved properties, when the main API is screened to find new multicomponent substances by using a set of coformers or codrugs and various synthesis techniques, any new crystal structure established with the use of single-crystal or powder diffraction data shortens the path to the desired goal. The impressive development of SDPD methods and the availability of appropriate software provide an opportunity for many scientific groups to solve new crystal structures by using laboratory powder diffractometers, as evidenced by numerous examples. The need to search for and create new medicines, accelerate their journey from lab bench to patient bedside, urgently requires the automation of screening processes, including XRPD measurements and the subsequent selection of the powder patterns most suitable for crystal structure determination with the help of Artificial Intelligence [87]. There is no doubt that very soon we will see such automatic lines in action.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
I am grateful to K.A. Paseshnichenko for the useful discussions and valuable comments when preparing an article.
Conflicts of Interest
The author declares no conflict of interest.
References
- Brittain, H.G.; Bogdanowich, S.J.; Bugay, D.E.; DeVincentis, J.; Lewen, G.; Newman, A.W. Physical characterization of pharmaceutical solids. Pharm. Res. 1991, 8, 963–973. [Google Scholar] [CrossRef]
- Brittain, H.G. X-Ray diffraction of pharmaceutical materials. Profiles Drug Subst. Excip. Relat. Methodol. 2003, 30, 271–319. [Google Scholar]
- Randall, C.S.; Rocco, W.L.; Ricou, P. XRD in pharmaceutical analysis: A versatile tool for problem-solving. Am. Pharm. Rev. 2010, 13, 52–59. [Google Scholar]
- Thakral, N.K.; Zanon, R.L.; Kelly, R.C.; Thakral, S. Applications of powder X-ray diffraction in small molecule pharmaceuticals: Achievements and aspirations. J. Pharm. Sci. 2018, 107, 2969–2982. [Google Scholar] [CrossRef]
- Rodríguez, I.; Gautam, R.; Tinoco, A.D. Using X-ray diffraction techniques for biomimetic drug development, formulation, and polymorphic characterization. Biomimetics 2021, 6, 1. [Google Scholar] [CrossRef]
- Thakral, S.; Terban, M.W.; Thakral, N.K.; Suryanarayanan, R. Recent advances in the characterization of amorphous pharmaceuticals by X-Ray diffractometry. Adv. Drug Deliv. Rev. 2016, 100, 183–193. [Google Scholar] [CrossRef]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Disc. 2004, 3, 42–57. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M. Crystal structure determination from powder diffraction data. Chem. Mater. 1996, 8, 2554–2570. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M.; Kariuki, B.M. Contemporary advances in the use of powder X-ray diffraction for structure determination. Angew. Chem. Int. Ed. 2001, 40, 1626–1651. [Google Scholar] [CrossRef]
- Chernyshev, V.V. Structure determination from powder diffraction. Russ. Chem. Bull. 2001, 50, 2273–2292. [Google Scholar] [CrossRef]
- Harris, K.D.M. Modern applications of powder X-ray diffraction in pharmaceutical sciences. Am. Pharm. Rev. 2004, 7, 86–91. [Google Scholar]
- David, W.I.F.; Shankland, K.; McCusker, L.B.; Baerlocher, C. (Eds.) Structure Determination from Powder Diffraction Data; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Cerný, R.; Favré-Nicolin, V. Direct space methods of structure determination from powder diffraction: Principles, guidelines and perspectives. Z. Krist.-Cryst. Mater. 2007, 222, 105–113. [Google Scholar] [CrossRef]
- Tsue, H.; Horiguchi, M.; Tamura, R.; Fujii, K.; Uekusa, H. Crystal structure solution of organic compounds from X-ray powder diffraction data. J. Synth. Org. Chem. Jpn. 2007, 65, 1203–1212. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K. Structure determination from powder diffraction data. Acta Crystallogr. Sect. A 2008, 64, 52–64. [Google Scholar] [CrossRef]
- Dinnebier, R.E.; Billinge, S.J.L. (Eds.) Powder Diffraction: Theory and Practice; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Cerný, R. Crystal structures from powder diffraction: Principles, difficulties and progress. Crystals 2017, 7, 142. [Google Scholar] [CrossRef]
- Altomare, A.; Ciriaco, F.; Cuocci, C.; Falcicchio, A.; Fanelli, F. Combined powder X-ray diffraction data and quantum-chemical calculations in EXPO2014. Powder Diffr. 2017, 32, S123–S128. [Google Scholar] [CrossRef]
- Kabova, E.A.; Cole, J.C.; Korb, O.; López-Ibáñez, M.; Williams, A.C.; Shankland, K. Improved performance of crystal structure solution from powder diffraction data through parameter tuning of a simulated annealing algorithm. J. Appl. Crystallogr. 2017, 50, 1411–1420. [Google Scholar] [CrossRef]
- Kabova, E.A.; Blundell, C.D.; Shankland, K. Pushing the limits of molecular crystal structure determination from powder diffraction data in high-throughput chemical environments. J. Pharm. Sci. 2018, 107, 2042–2047. [Google Scholar] [CrossRef]
- Haleblian, J.; McCrone, W. Pharmaceutical applications of polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef]
- Ainurofiq, A.; Dinda, K.E.; Pangestika, M.W.; Himawati, U.; Wardhani, W.D.; Sipahutar, Y.T. The effect of polymorphism on active pharmaceutical ingredients: A review. Int. J. Res. Pharm. Sci. 2020, 11, 1621–1630. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.-S.; Hong, S.; Ha, E.-S.; Nie, H.; Zhou, Q.T.; Kim, M.-S. Tableting process-induced solid-state polymorphic transition. J. Pharm. Investig. 2022, 52, 175–194. [Google Scholar] [CrossRef]
- Braga, D.; Casali, L.; Grepioni, F. The relevance of crystal forms in the pharmaceutical field: Sword of Damocles or innovation tools? Int. J. Mol. Sci. 2022, 23, 9013. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, S.; Wang, L.; Tao, X. Recent advances in polymorph discovery methods of organic crystals. Cryst. Growth Des. 2023, 23, 637–654. [Google Scholar] [CrossRef]
- Almarsson, Ö.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical co-crystals represent a new path to improved medicines? Chem. Commun. 2004, 1889–1896. [Google Scholar] [CrossRef]
- Vishweshwar, P.; McMahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef]
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef]
- Kumar, S.; Nanda, A. Pharmaceutical cocrystals: An overview. Indian J. Pharm. Sci. 2017, 79, 858–871. [Google Scholar] [CrossRef]
- Vioglio, P.C.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Duggirala, N.K.; LaCasse, S.M.; Zaworotko, M.J.; Krzyzaniak, J.F.; Arora, K.K. Pharmaceutical cocrystals: Formulation approaches to develop robust drug products. Cryst. Growth Des. 2020, 20, 617–626. [Google Scholar] [CrossRef]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical cocrystals: A review of preparations, physicochemical properties and applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Teoh, Y.; Ayoub, G.; Huskić, I.; Titi, H.M.; Nickels, C.W.; Herrmann, B.; Friščić, T. SpeedMixing: Rapid tribochemical synthesis and discovery of pharmaceutical cocrystals without milling or grinding media. Angew. Chem. Int. Ed. 2022, 61, e202206293. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Nangia, A. Crystal engineering of pharmaceutical cocrystals in the discovery and development of improved drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef]
- Munjal, B.; Suryanarayanan, R. Applications of synchrotron powder X-ray diffractometry in drug substance and drug product characterization. Trends Anal. Chem. 2021, 136, 116181. [Google Scholar] [CrossRef]
- Available online: https://www.imca.aps.anl.gov/IMCA-CAT (accessed on 20 February 2023).
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Van de Streek, J.; Neumann, M.A. Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D). Acta Crystallogr. Sect. B 2014, 70, 1020–1032. [Google Scholar] [CrossRef]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta Crystallogr. Sect. B 2016, 72, 439–459. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Habermehl, S.; Schlesinger, C.; Schmidt, M.U. Structure determination from unindexed powder data from scratch by a global optimization approach using pattern comparison based on cross-correlation functions. Acta Crystallogr. Sect. B 2022, B78, 195–213. [Google Scholar] [CrossRef]
- Schlesinger, C.; Fitterer, A.; Buchsbaum, C.; Habermehl, S.; Chierotti, M.R.; Nervi, C.; Schmidt, M.U. Ambiguous structure determination from powder data: Four different structural models of 4,11-difluoroquinacridone with similar X-ray powder patterns, fit to the PDF, SSNMR and DFT-D. IUCrJ 2022, 9, 406–424. [Google Scholar] [CrossRef]
- Altomare, A. Solving molecular compounds from powder diffraction data: Are results always reliable? IUCrJ 2022, 9, 403–405. [Google Scholar] [CrossRef]
- De Wolff, P.M. A simplified criterion for the reliability of a powder pattern indexing. J. Appl. Crystallogr. 1968, 1, 108–113. [Google Scholar] [CrossRef]
- Chan, F.C.; Anwar, J.; Cernik, R.; Barnes, P.; Wilson, R.M. Ab initio structure determination of sulfathiazole polymorph V from synchrotron X-ray powder diffraction data. J. Appl. Crystallogr. 1999, 32, 436–441. [Google Scholar] [CrossRef]
- Hughes, D.S.; Hursthouse, M.B.; Threlfall, T.; Tavener, S. A new polymorph of sulfathiazole. Acta Crystallogr. Sect. C 1999, 55, 1831–1833. [Google Scholar] [CrossRef]
- Llinas, A.; Box, K.J.; Burley, J.C.; Glen, R.; Goodman, J.M. A new method for the reproducible generation of polymorphs: Two forms of sulindac with very different solubilities. J. Appl. Crystallogr. 2007, 40, 379–381. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Matzger, A.J. New form discovery for the analgesis flurbiprofen and sulindac facilitated by polymer-induced heteronucleation. J. Pharm. Sci. 2007, 96, 2978–2986. [Google Scholar] [CrossRef]
- Chernyshev, V.V.; Machula, A.A.; Kukushkin, S.Y.; Velikodny, Y.A. Carvedilol dihydrogen phosphate hemihydrate: A powder study. Acta Crystallogr. Sect. E 2009, 65, o2020–o2021. [Google Scholar] [CrossRef]
- Vogt, F.G.; Copley, R.C.B.; Mueller, R.L.; Spoors, G.P.; Cacchio, T.N.; Carlton, R.A.; Katrincic, L.M.; Kennady, J.M.; Parsons, S.; Chetina, O.V. Isomorphism, disorder, and hydration in the crystal structures of racemic and single-enantiomer carvedilol phosphate. Cryst. Growth Des. 2010, 10, 2713–2733. [Google Scholar] [CrossRef]
- Bruning, J.; Alig, E.; Schmidt, M.U. Ezetimibe anhydrate, determined from laboratory powder diffraction data. Acta Crystallogr. Sect. C 2010, 66, o341–o344. [Google Scholar] [CrossRef]
- Shimpi, M.R.; Childs, S.L.; Bostrom, D.; Velaga, S.P. New cocrystals of ezetimibe with L-proline and imidazole. CrystEngComm 2014, 16, 8984–8993. [Google Scholar] [CrossRef]
- Bortolotti, M.; Lonardelli, I.; Pepponi, G. Determination of the crystal structure of nifedipine form C by synchrotron powder diffraction. Acta Crystallogr. Sect. B 2011, 67, 357–364. [Google Scholar] [CrossRef]
- Gunn, E.; Guzei, I.A.; Cai, T.; Yu, L. Polymorphism of nifedipine: Crystal structure and reversible transition of the metastable β polymorph. Cryst. Growth Des. 2012, 12, 2037–2043. [Google Scholar] [CrossRef]
- Prohens, R.; Barbas, R.; Portell, A.; Font-Bardia, M.; Alcobe, X.; Puigjaner, C. Polymorphism of cocrystals: The promiscuoud behavior of Agomelatine. Cryst. Growth Des. 2016, 16, 1063–1070. [Google Scholar] [CrossRef]
- Lee, M.-J.; Aitipamula, S.; Choi, G.J.; Chow, P.S. Agomelatine-hydroquinone (1:1) cocrystal: Novel polymorphs and their thermodynamic relationship. Acta Crystallogr. Sect. B 2019, 75, 969–977. [Google Scholar] [CrossRef]
- Schlesinger, C.; Bolte, M.; Schmidt, M.U. Challenging structure determination from powder diffraction data: Two pharmaceutical salts and one cocrystal with Z′ = 2. Z. Krist.-Cryst. Mater. 2019, 234, 257–268. [Google Scholar] [CrossRef]
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Rahal, O.A.; Majumder, M.; Spillman, M.J.; van de Streek, J.; Shankland, K. Co-crystal structures of furosemide: Urea and carbamazepine:indomethacin determined from powder X-ray diffraction data. Crystals 2020, 10, 42. [Google Scholar] [CrossRef]
- Majumder, M.; Buckton, G.; Rawlinson-Malone, C.; Williams, A.C.; Spillman, M.J.; Shankland, N.; Shankland, K. A carbamazepine: Indomethacin (1:1) cocrystal produced by milling. CrystEngComm 2011, 13, 6327–6328. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Al-Sabbagh, D.; Meyer, K.; Maiwald, M.; Scholz, G.; Emmerling, F. Insight into the structure and properties of novel imidazole-based salts of salicylic acid. Molecules 2019, 24, 4144. [Google Scholar] [CrossRef]
- Nabulsi, N.A.R.; Gandour, R.D.; Fronczek, F.R. CSD Communication (Private Communication). 2013. [Google Scholar]
- Martins, I.C.B.; Emmerling, F. Carbamazepine dihydroxybenzoic acid cocrystals: Exploring packing interactions and reaction kinetics. Cryst. Growth Des. 2021, 21, 6961–6970. [Google Scholar] [CrossRef]
- Gohel, S.K.; Palanisamy, V.; Sanphui, P.; Prakash, M.; Singh, G.P.; Chernyshev, V. Isostructural cocrystals of metaxalone with improved dissolution characteristics. RSC Adv. 2021, 11, 30689–30700. [Google Scholar] [CrossRef]
- Visser, J.W. A fully automatic program for finding the unit cell from powder data. J. Appl. Crystallogr. 1969, 2, 89–95. [Google Scholar] [CrossRef]
- Zhukov, S.G.; Babaev, E.V.; Chernyshev, V.V.; Rybakov, V.B.; Sonneveld, E.J.; Schenk, H. Crystal structure determination of 2-oxo-3-benzoyloxazolo[3,2-a]pyridine from X-ray powder data. Z. Krist.-Cryst. Mater. 2000, 215, 306–308. [Google Scholar] [CrossRef]
- Zlokazov, V.B.; Chernyshev, V.V. MRIA—A program for a full profile analysis of powder multiphase neutron-diffraction time-of-flight (direct and Fourier) spectra. J. Appl. Crystallogr. 1992, 25, 447–451. [Google Scholar] [CrossRef]
- Varsa, S.R.B.; Sanphui, P.; Chernyshev, V. Polymorphs and isostructural cocrystals of dexamethasone: Towards the improvement of aqueous solubility. CrystEngComm 2022, 24, 6045–6058. [Google Scholar] [CrossRef]
- Fayaz, T.K.S.; Palanisamy, V.; Sanphui, P.; Chernyshev, V. Multicomponent solid forms of antibiotic cephalexin towards improved chemical stability. CrystEngComm 2023, 25, 1252–1262. [Google Scholar] [CrossRef]
- Mirocki, A.; Conterosito, E.; Palin, L.; Sikorski, A.; Milanesio, M.; Lopresti, M. Crystal structure of a new 1:1 acridine-diclofenac salt, obtained with high yield by a mechanochemical approach. Crystals 2022, 12, 1573. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Mirocki, A.; Lopresti, M.; Palin, L.; Conterosito, E.; Sikorski, A.; Milanesio, M. Exploring the molecular landscape of multicomponent crystals formed by naproxen drug and acridines. CrystEngComm 2022, 24, 6839–6853. [Google Scholar] [CrossRef]
- Allu, S.; Garai, A.; Chernyshev, V.V.; Nangia, A.K. Synthesis of ternary cocrystals, salts, and hydrates of acefylline with enhanced dissolution and high permeability. Cryst. Growth Des. 2022, 22, 4165–4181. [Google Scholar] [CrossRef]
- Werner, P.-E.; Eriksson, L.; Westdahl, M. TREOR, a semi-exhaustive trial-and-error powder indexing program for all symmetries. J. Appl. Crystallogr. 1985, 18, 367–370. [Google Scholar] [CrossRef]
- Voguri, R.S.; Ranga, S.; Dey, A.; Ghosal, S. Solid-state phase transition of agomelatine-phosphoric acid molecular complexes along the salt-cocrystal continuum: Ab initio powder X-ray diffraction structure determination and DFT-D2 analysis. Cryst. Growth Des. 2020, 20, 7647–7657. [Google Scholar] [CrossRef]
- Neumann, M.A. X-Cell: A novel indexing algorithm for routine tasks and difficult cases. J. Appl. Crystallogr. 2003, 36, 356–365. [Google Scholar] [CrossRef]
- MedeA, Materials Design, Inc.: San Diego, CA, USA, 2015.
- Stevens, J.S.; Byard, S.J.; Schroeder, S.L.M. Salt or co-crystal? Determination of protonation state by X-ray photoelectron spectroscopy (XPS). J. Pharm. Sci. 2010, 99, 4453–4457. [Google Scholar] [CrossRef]
- Harris, K.D.M. NMR Crystallography as a vital tool in assisting crystal structure determination from powder XRD data. Crystals 2022, 12, 1277. [Google Scholar] [CrossRef]
- Smalley, C.J.H.; Logsdail, A.J.; Hughes, C.E.; Iuga, D.; Young, M.T.; Harris, K.D.M. Solid-state structural properties of alloxazine determined from powder XRD data in conjunction with DFT-D calculations and solid-state NMR spectroscopy: Unraveling the tautomeric identity and pathways for tautomeric interconversion. Cryst. Growth Des. 2022, 22, 524–534. [Google Scholar] [CrossRef]
- Heng, T.; Yang, D.; Wang, R.; Zhang, L.; Lu, Y.; Du, G. Progress in research on Artificial Intelligence applied to polymorphism and cocrystal prediction. ACS Omega 2021, 6, 15543–15550. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).