

The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study


Abstract
1. Introduction
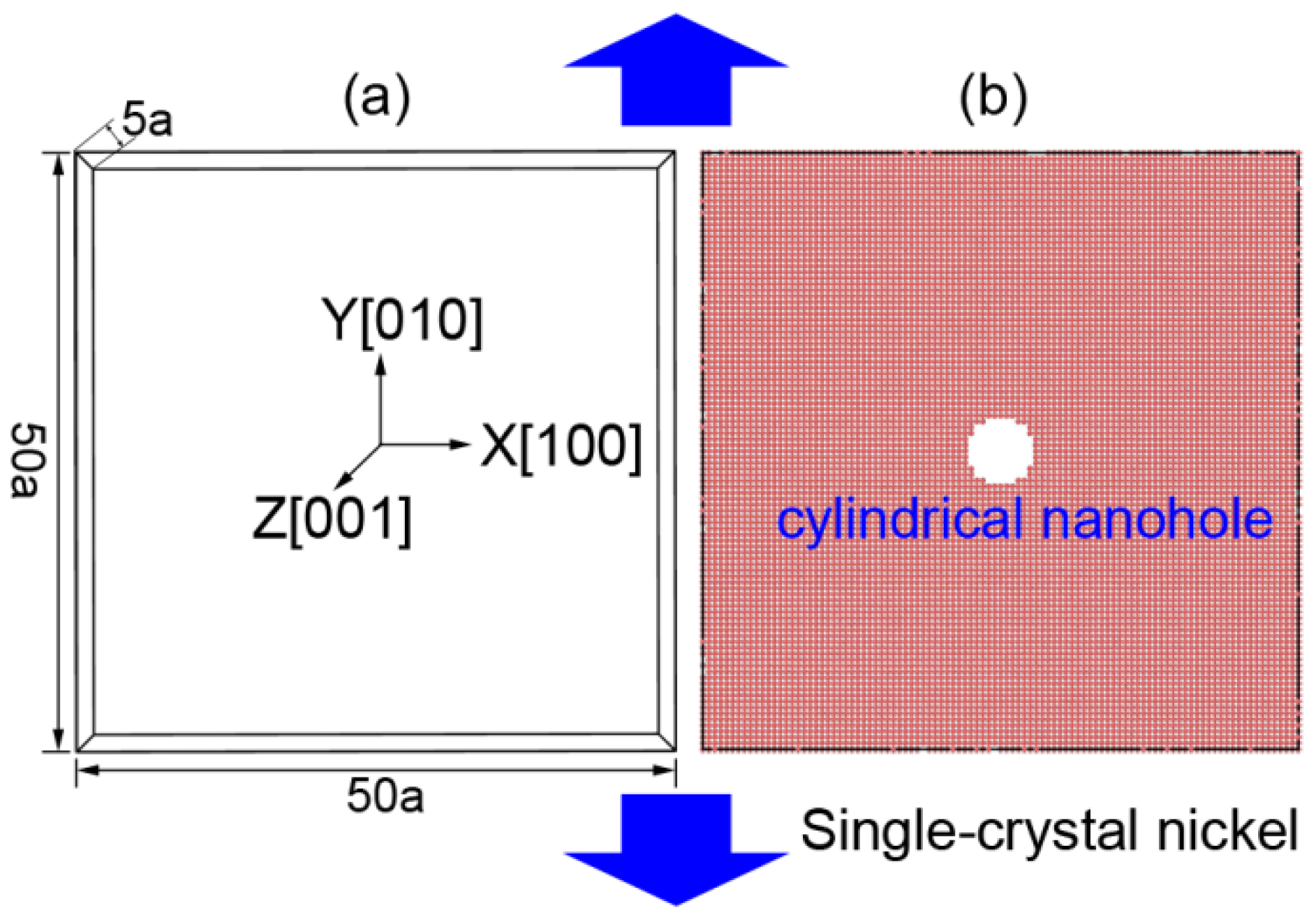
2. Simulation Conditions
2.1. Initial Conditions
2.2. Potential between Atoms
3. Simulation Results and Discussion
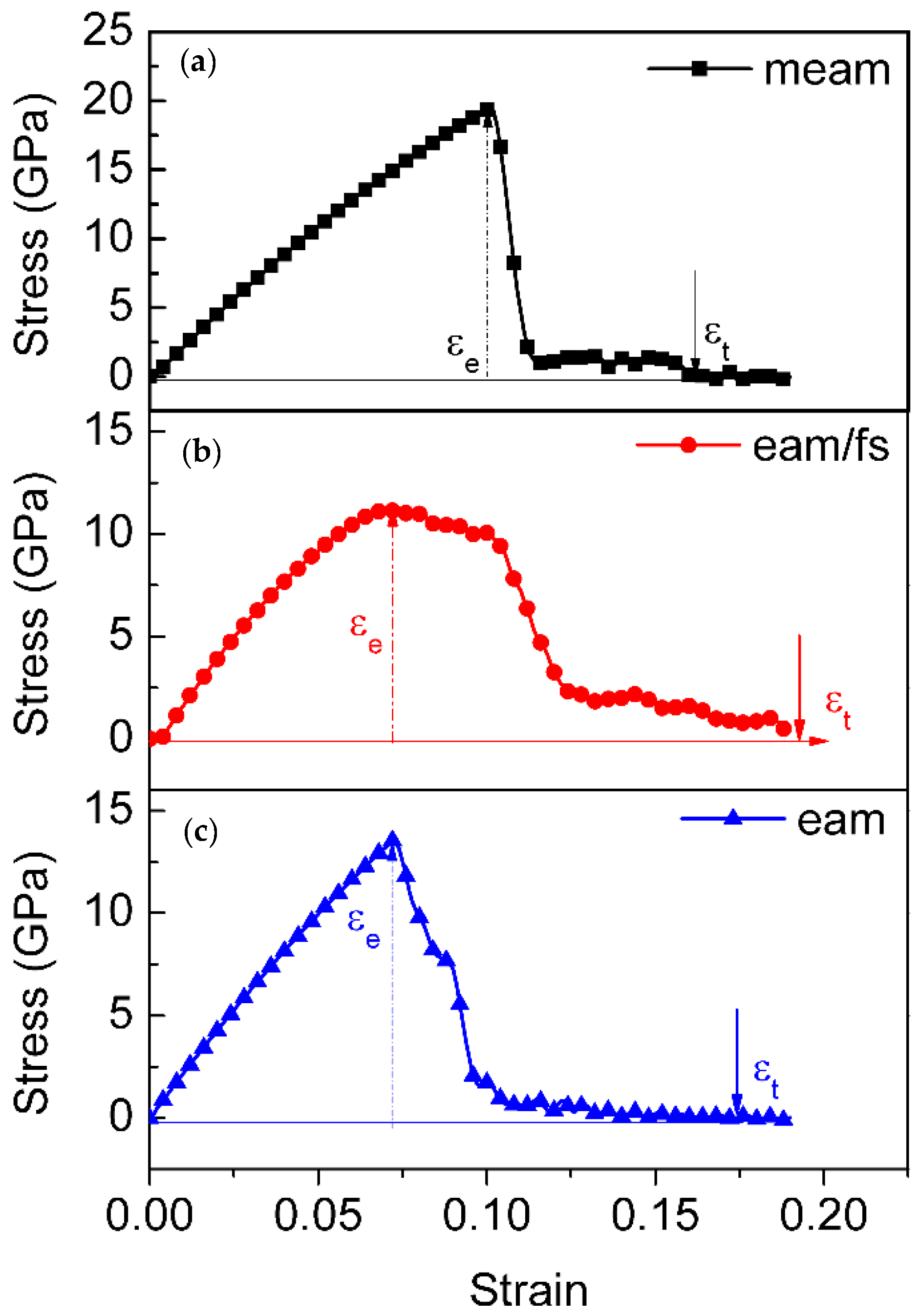
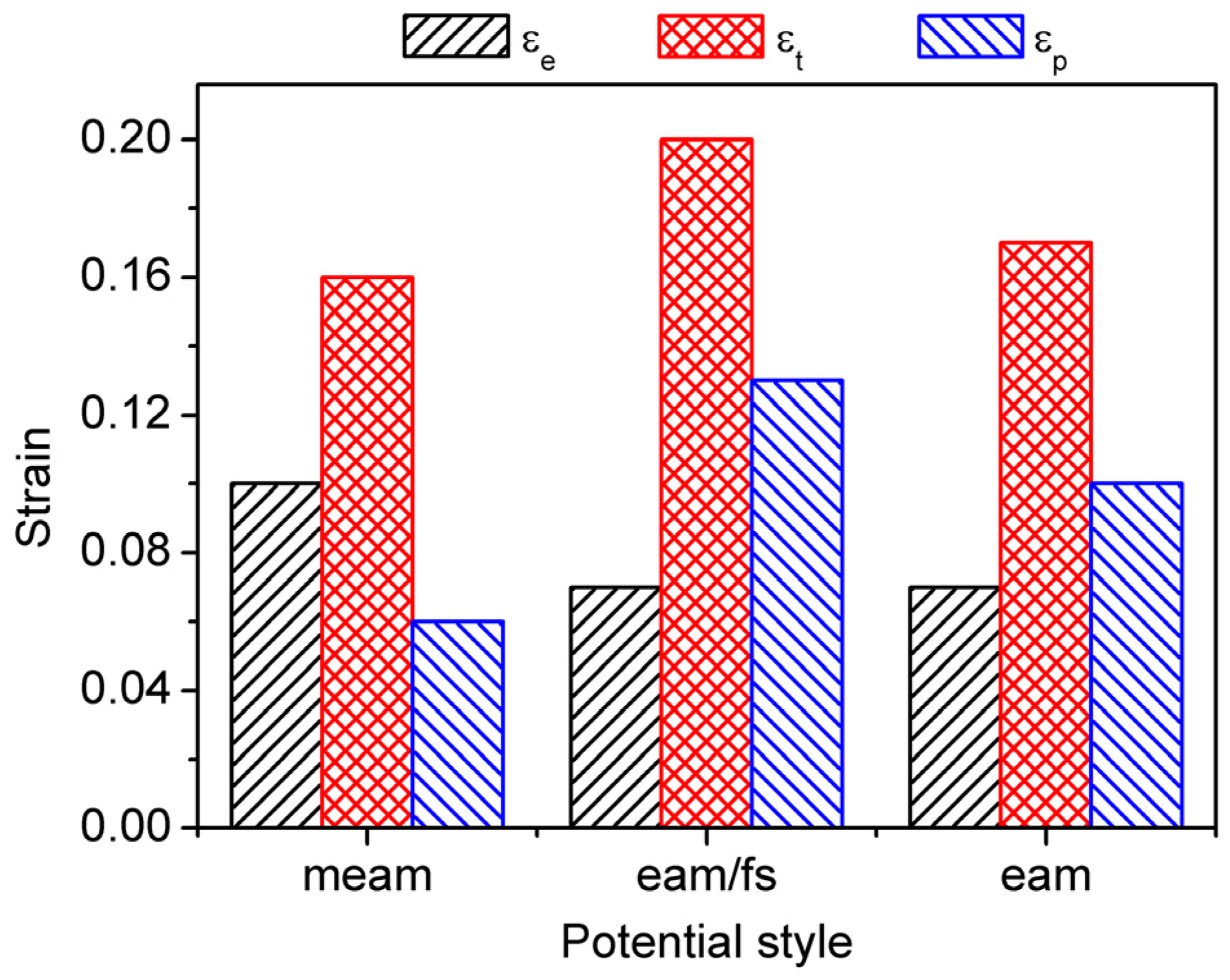
3.1. Stress–Strain Behavior
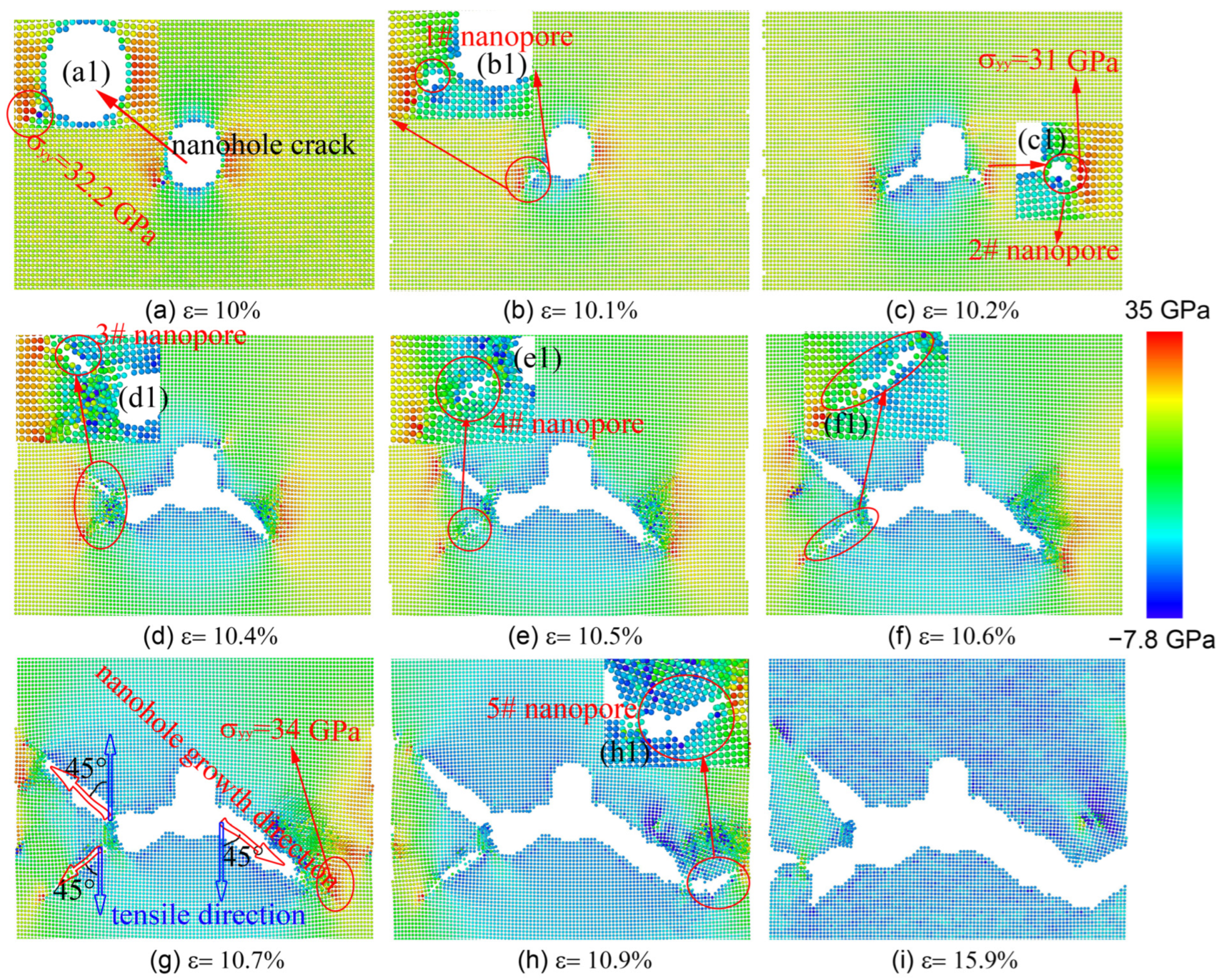
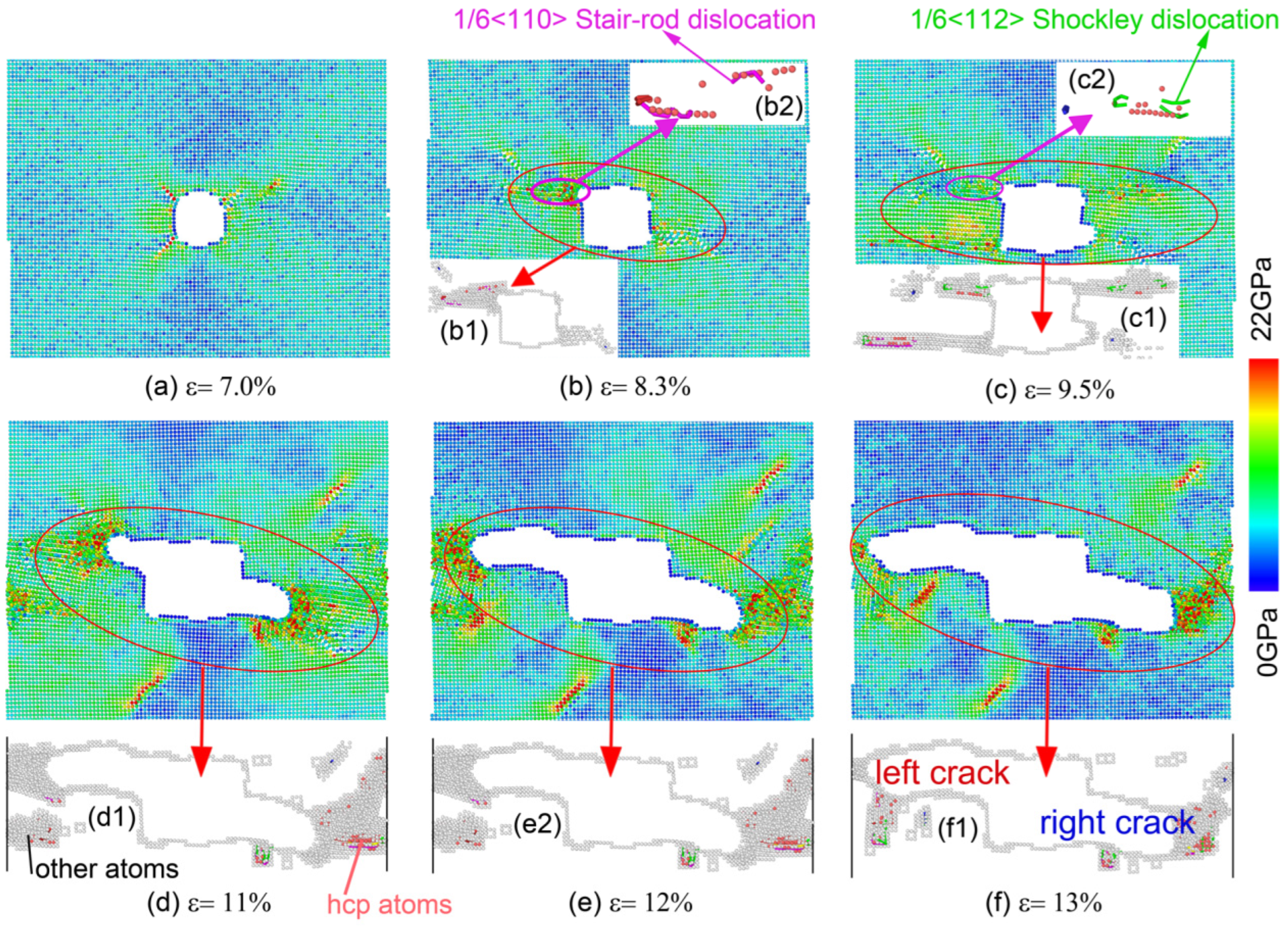
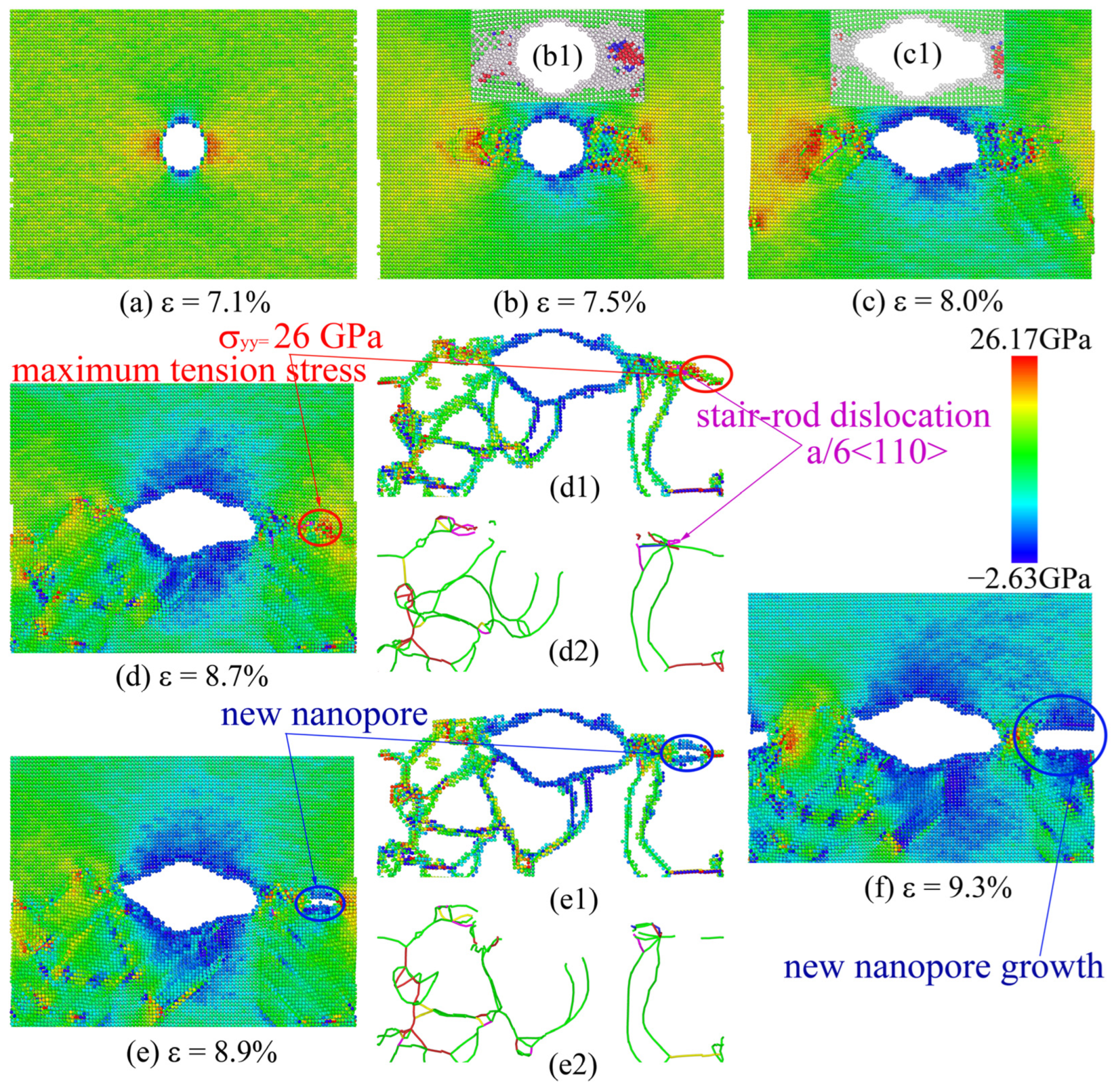
3.2. Nanohole Propagation Behavior
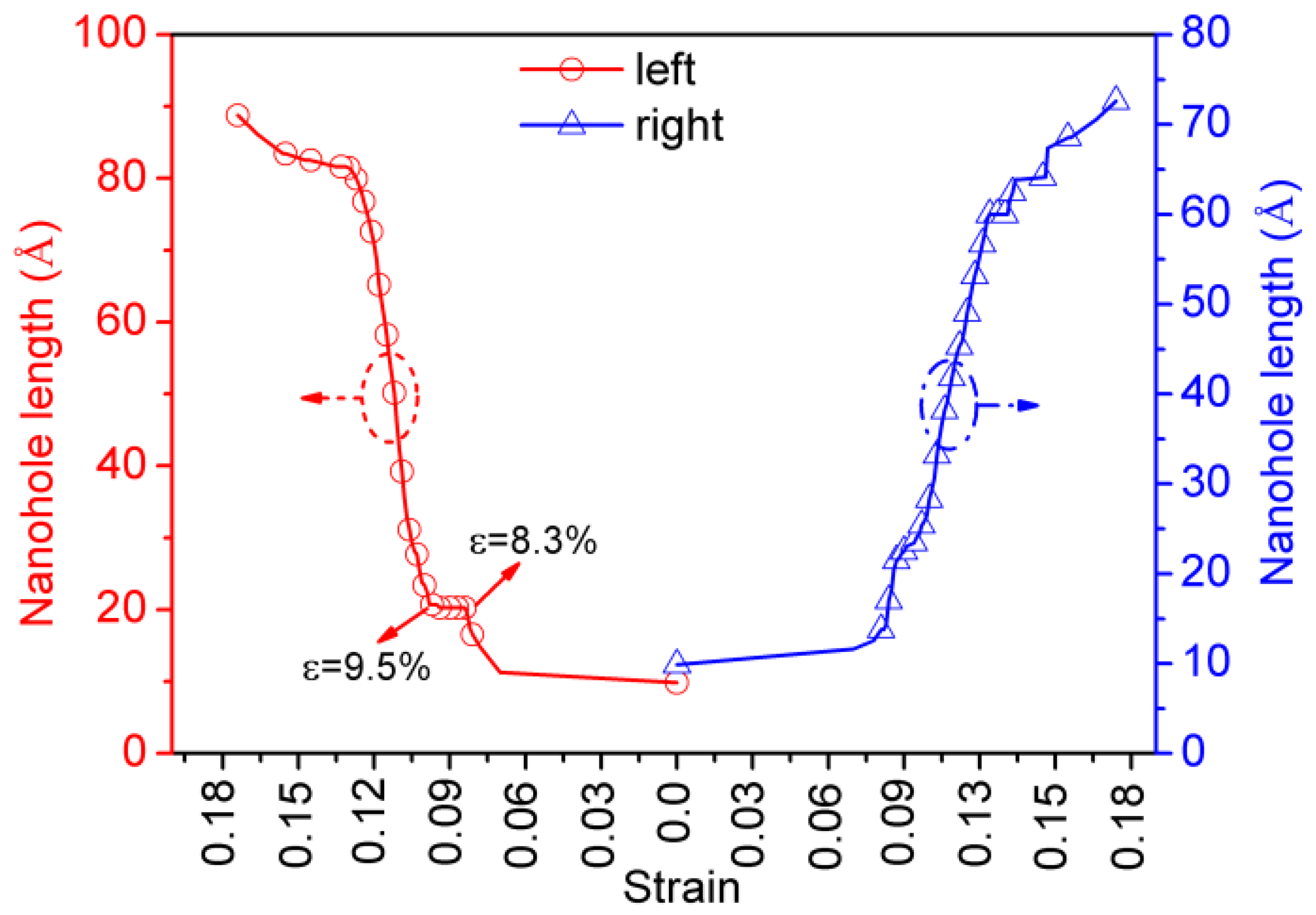
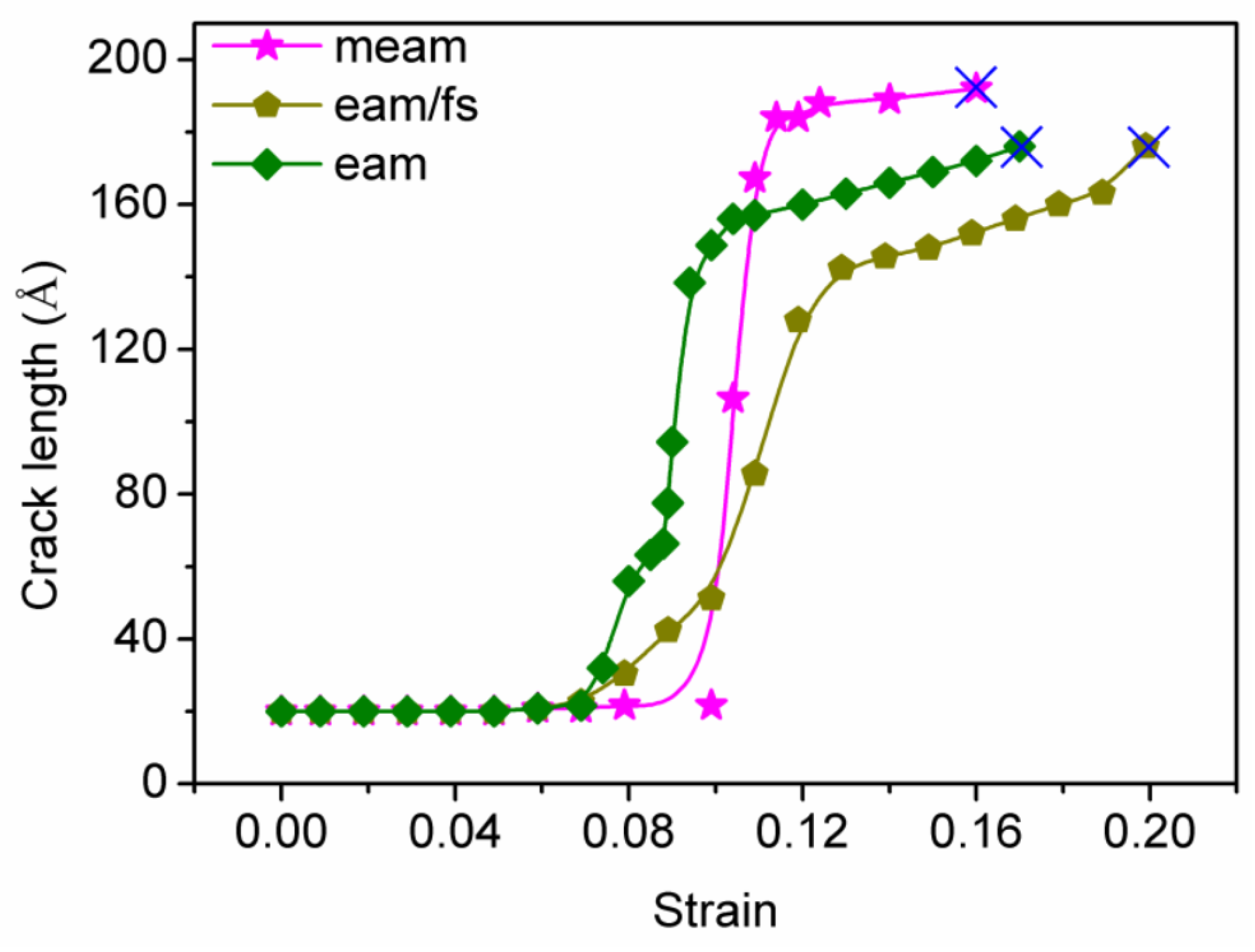
3.3. Relationship between Crack Length and Tensile Strain
3.4. Discussion
4. Conclusions
- (1)
- The MEAM potential is best suited to describe the brittle propagation behavior of nanoholes in single-crystal Ni.
- (2)
- The EAM/FS potential is effective in characterizing the plastic growth behavior of nanoholes in single-crystal Ni.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Proudhon, H.; Li, J.; Wang, F.; Roos, A.; Chiaruttini, V.; Forest, S. 3D simulation of short fatigue crack propagation by finite element crystal plasticity and remeshing. Int. J. Fatigue 2016, 82, 238–246. [Google Scholar] [CrossRef]
- Lin, B.; Zhao, L.G.; Tong, J. A crystal plasticity study of cyclic constitutive behavior, crack-tip deformation and crack-growth path for a polycrystalline nickel-based superalloy. Eng. Fract. Mech. 2011, 78, 2174–2192. [Google Scholar] [CrossRef]
- Li, L.; Shen, L.; Proust, G. Fatigue crack initiation life prediction for aluminum alloy7075 using crystal plasticity finite element simulations. Mech. Mater. 2015, 81, 84–93. [Google Scholar] [CrossRef]
- Yang, S.; Ma, G.; Ren, X.; Ren, F. Cover refinement of numerical manifold method for crack propagation simulation. Eng. Anal. Bound. Elem. 2014, 43, 37–49. [Google Scholar] [CrossRef]
- Özden, U.A.; Mingard, K.P.; Zivcec, M.; Bezold, A.; Broeckmann, C. Mesoscopical finite element simulation of fatigue crack propagation in WC/Co-hard metal. Int. J. Refract. Met. Hard Mater. 2015, 49, 261–267. [Google Scholar] [CrossRef]
- Dewang, Y.; Hora, M.S.; Panthi, S.K. Prediction of crack location and propagation in stretch flanging process of aluminum alloy AA-5052 sheet using FEM simulation. Trans. Nonferrous Met. Soc. China 2015, 25, 2308–2320. [Google Scholar] [CrossRef]
- Özden, U.A.; Bezold, A.; Broeckmann, C. Numerical simulation of fatigue crack propagation in WC/Co based on a continuum damage mechanics approach. Prog. Mater. Sci. 2014, 3, 1518–1523. [Google Scholar] [CrossRef]
- Keyhani, A.; Goudarzi, M.; Mohammadi, S.; Roumina, R. XFEM–dislocation dynamics multi-scale modeling of plasticity and fracture. Comput. Mater. Sci. 2015, 104, 98–107. [Google Scholar] [CrossRef]
- Calvo, F.; Yurtsever, E. The quantum structure of anionic hydrogen clusters. J. Chem. Phys. 2018, 148, 102305. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Wang, D.; Qu, X.; Wu, J. Using a molecular dynamics simulation to investigate asphalt nano-cracking under external loading conditions. Appl. Sci. 2017, 7, 770. [Google Scholar] [CrossRef]
- Ramezani, M.G.; Golchinfar, B. Mechanical properties of cellulose nanocrystal (CNC) bundles: Coarse-grained molecular dynamic simulation. J. Compos. Sci. 2019, 3, 57. [Google Scholar] [CrossRef]
- Liu, C.; Yao, Y. Study of crack-propagation mechanism of Al0.1CoCrFeNi high-entropy alloy by molecular dynamics method. Crystals 2023, 13, 11. [Google Scholar] [CrossRef]
- Lee, S.; Kang, H.; Bae, D. Molecular dynamics study on crack propagation in Al containing Mg–Si clusters formed during natural aging. Materials 2023, 16, 883. [Google Scholar] [CrossRef] [PubMed]
- Komanduri, R.; Chandrasekaran, N.; Raff, L.M. Molecular dynamics (MD) simulation of uniaxial tensile of some single-crystal cubic metals at nanolevel. Int. J. Mech. Sci. 2001, 43, 2237–2260. [Google Scholar] [CrossRef]
- Xu, S.; Deng, X. Nanoscale void nucleation and growth and crack tip stress evolution ahead of a growing crack in a single crystal. Nanotechnology 2008, 19, 115705. [Google Scholar] [CrossRef]
- Cui, C.B.; Beom, H.G. Molecular dynamics simulations of edge cracks in copper and aluminum single crystals. Mater. Sci. Eng. A 2014, 15, 102–109. [Google Scholar] [CrossRef]
- Zhuo, X.R.; Kim, J.H.; Gyu Beom, H. Atomistic investigation of crack growth resistance in a single-crystal Al-nanoplate. J. Mater. Res. 2016, 9, 1185–1192. [Google Scholar] [CrossRef]
- Ding, J.; Wang, L.-S.; Song, K.; Liu, B.; Huang, X. Molecular dynamics simulation of crack propagation in single-crystal Aluminum plate with central cracks. J. Nanomater. 2017, 2017, 5181206. [Google Scholar] [CrossRef]
- Mikelani, M.; Panjepour, M.; Taherizadeh, A. Investigation on mechanical properties of nanofoam aluminum single crystal: Using the method of molecular dynamics simulation. Appl. Phys. A Mater. Sci. Process. 2020, 126, 921. [Google Scholar] [CrossRef]
- Ji, H.; Ren, K.; Ding, L.; Wang, T.; Li, J.-M.; Yang, J. Molecular dynamics simulation of the interaction between cracks in single crystal Aluminum. Mater. Today Commun. 2022, 30, 103020. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, Q.; Liu, R.; Yue, Z.; Tang, M.; Li, X. Molecular dynamics simulation of crack propagation behaviors at the Ni/Ni3Al grain boundary. RSC Adv. 2014, 4, 32749. [Google Scholar] [CrossRef]
- Hou, N.X.; Wen, Z.X.; Yue, Z.F. Creep behavior of single crystal superalloy specimen under temperature gradient condition. Mater. Sci. Eng. A 2009, 510–511, 42–45. [Google Scholar] [CrossRef]
- Mao, H.; Wen, Z.; Yue, Z.; Wang, B. The evolution of plasticity for nickel-base single crystal cooled blade with film cooling holes. Mater. Sci. Eng. A 2013, 587, 79–84. [Google Scholar] [CrossRef]
- Kim, J.; Suh, C.; Amanov, A.; Kim, H.; Pyun, Y. Rotary bending fatigue properties of Inconel 718 alloys by ultrasonic nanocrystal surface modification technique. J. Eng. 2015, 13, 133–137. [Google Scholar] [CrossRef]
- Yang, X.F.; He, C.Y.; Yuan, G.J.; Chen, H.; Wang, R.Z.; Jia, Y.F.; Tu, S.T. The effects of grain boundary structures on crack nucleation in nickel nanolaminsted structure: A molecular dynamics study. Comput. Mater. Sci. 2021, 186, 110019. [Google Scholar] [CrossRef]
- Mishin, Y.; Farkas, D.; Mehl, M.J.; Papaconstantopoulos, D.A. Interatomic potentials for monatomic metals from experimental data and ab initio calculations. Phys. Rev. B 1999, 59, 3393–3407. [Google Scholar] [CrossRef]
- Wu, W.-P.; Yao, Z.-Z. Molecular dynamics simulation of stress distribution and microstructure evolution ahead of a growing crack in single crystal nickel. Theor. Appl. Fract. Mech. 2012, 62, 67–75. [Google Scholar] [CrossRef]
- Sung, P.-H.; Chen, T.-C. Studies of crack growth and propagation of single-crystal nickel by molecular dynamics. Comput. Mater. Sci. 2015, 102, 151–158. [Google Scholar] [CrossRef]
- Ma, L.; Xiao, S.; Deng, H.; Hu, W. Atomistic simulation of mechanical properties and crack propagation on irradiated nickel. Comput. Mater. Sci. 2016, 120, 21–28. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S. Molecular dynamics simulation of crack propagation in nanoscale polycrystal nickel based on different strain rate. Metal 2017, 7, 432. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S.; Zhu, X.; Zhao, Y. Mechanisms of crack propagation in nanoscale single crystal, bicrystal and tricrystal nickels based on the molecular dynamics simulation. Results Phys. 2017, 7, 1722–1733. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S. Investigation on dislocation-based mechanisms of void growth and coalescence on single and nanotwinned nickels by molecular dynamics simulation. Philos. Mag. 2017, 97, 2772–2794. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S.; Zhu, X.; Zhao, Y. A molecular dynamics study of intercrystalline crack propagation in nano-nickel bicrystal films with (010) twist boundary. Eng. Fract. Mech. 2016, 168, 147–159. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S.; Zhu, X.; Zhao, Y. Influence of twist angle on crack propagation of nanoscale bicrystal nickel film based on molecular dynamics simulation. Phys. E Low-Dimens. Syst. Nanostruct. 2017, 87, 281–294. [Google Scholar] [CrossRef]
- Zhang, J.; Ghosh, S. Molecular dynamics based study and characterization of deformation mechanisms near a crack in a crystalline material. J. Mech. Phys. Solids 2013, 61, 1670–1690. [Google Scholar] [CrossRef]
- Glenn, J.; Martyna, D.J.; Tobias, M.L. Klein. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystal: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. A Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The open visualization tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Heyes, D.M. Pressure tensor of partial-charge and point-dipole lattices with bulk and surface geometries. Phys. Rev. B 1994, 49, 755–764. [Google Scholar] [CrossRef]
- Sirk, T.W.; Moore, S.; Brown, E.F. Characteristics of thermal conductivity in classical water models. J. Chem. Phys. 2013, 138, 064505. [Google Scholar] [CrossRef] [PubMed]
- Aidan, P.; Thompson, S.J.; Plimpton, W.M. General formation of pressure and stress tensor for arbitrary many-body interaction potentials under periodic boundary conditions. J. Chem. Phys. 2009, 131, 154107. [Google Scholar]
- Honeycutt, J.D.; Andersen, H.C. Andersen. Molecular dynamics study of melting and freezing of small Lennard-Jones clusters. J. Phys. Chem. 1987, 91, 4950–4963. [Google Scholar] [CrossRef]
- Faken, D.; Jónsson, H. Systematic analysis of local atomic structure combined with 3D computer graphics. Comput. Mater. Sci. 1994, 2, 279–286. [Google Scholar] [CrossRef]
- Lee, B.-J.; Shim, J.-H.; Baskes, M.I. Semiempirical atomic potentials for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, Al, and Pb based on first and second nearest-neighbor modified embedded atom method. Phys. Rev. B 2003, 68, 144112. [Google Scholar] [CrossRef]
- Ackland, G.J.; Tichy, G.; Vitek, V.; Finnis, M.W. Simple N-body potentials for the noble metals and nickel. Philos. Mag. A 1987, 56, 735–756. [Google Scholar] [CrossRef]
- Foiles, S.M.; Baskes, M.I.; Daw, M.S. Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys. Phys. Rev. B 1986, 33, 7983–7991. [Google Scholar] [CrossRef]
- Sainath, G.; Choudhary, B.K. Atomistic simulations on ductile-brittle transition in <111> BCC Fe nanowires. J. Appl. Phys. 2017, 122, 095101. [Google Scholar]
- Gordon, P.A.; Neeraj, T.; Luton, M.J.; Farkas, D. Crack-tip deformation mechanisms in α-Fe and binary Fe alloys: An atomistic study on single crystals. Metall. Mater. Trans. A 2007, 38A, 2191–2202. [Google Scholar] [CrossRef]
- Sainath, G.; Nagesha, A. Atomistic simulations of twin boundary effect on the crack growth behavior in BCC Fe. Trans. Indian Natl. Acad. Eng. 2022, 7, 433–439. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MEAM | EAM/FS | EAM | ||
|---|---|---|---|---|
| Surface energy (erg/cm2) | (100) plane | 1943 | 1444 | 1580 |
| (110) plane | 2057 | 1548 | 1730 | |
| (111) plane | 1606 | 1153 | 1450 | |
| Stacking fault energy (erg/cm2) | 125 | 33 | -- | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, X.; Liang, Y.; Gu, J.; Peng, G. The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study. Crystals 2023, 13, 585. https://doi.org/10.3390/cryst13040585
Qin X, Liang Y, Gu J, Peng G. The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study. Crystals. 2023; 13(4):585. https://doi.org/10.3390/cryst13040585
Chicago/Turabian StyleQin, Xinmao, Yilong Liang, Jiabao Gu, and Guigui Peng. 2023. "The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study" Crystals 13, no. 4: 585. https://doi.org/10.3390/cryst13040585
APA StyleQin, X., Liang, Y., Gu, J., & Peng, G. (2023). The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study. Crystals, 13(4), 585. https://doi.org/10.3390/cryst13040585