

NMR Crystallography as a Vital Tool in Assisting Crystal Structure Determination from Powder XRD Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
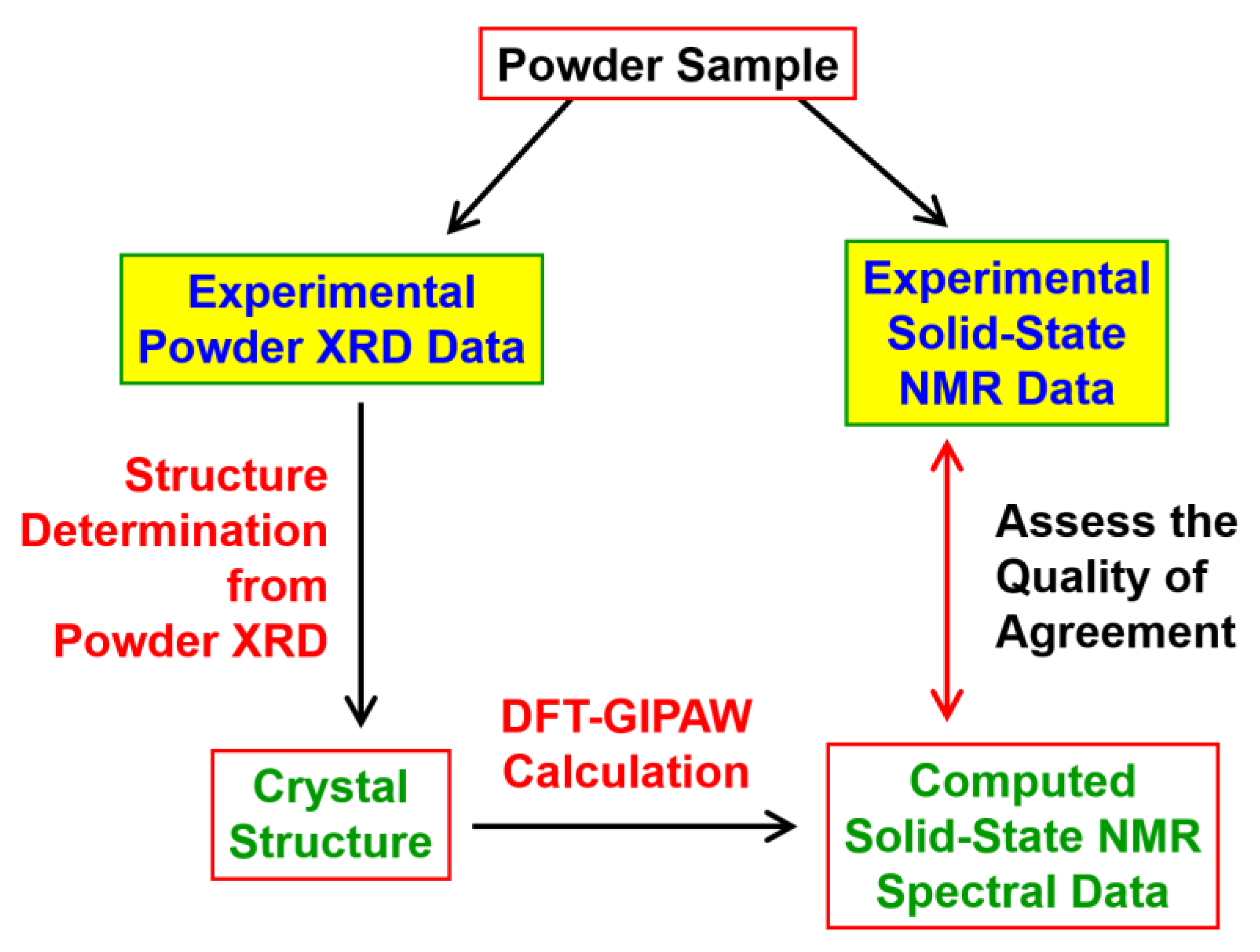
2. Overview of Opportunities to Utilize Solid-State NMR Data to Assist Structure Determination from Powder XRD Data
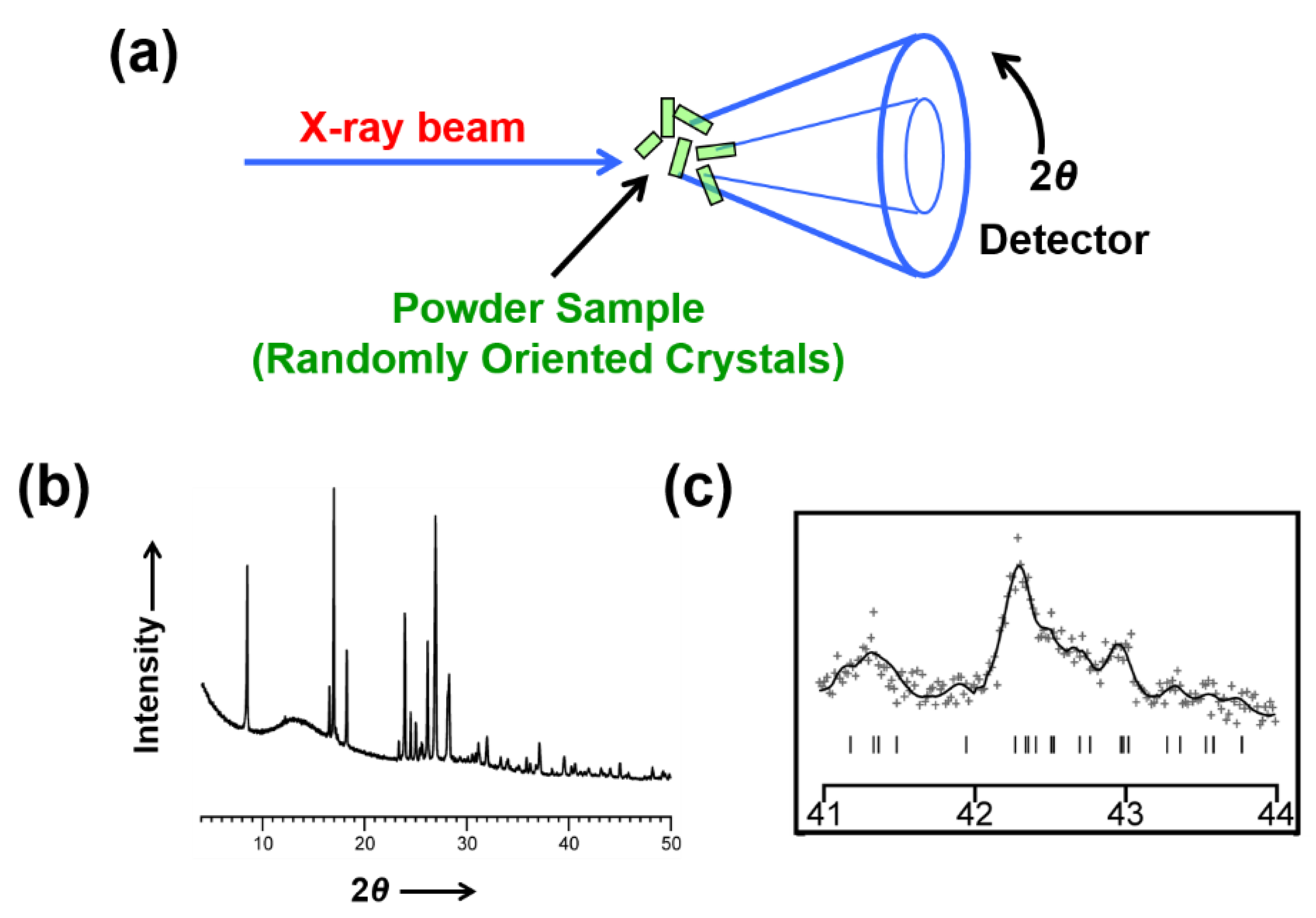
3. Overview of Structure Determination from Powder XRD Data
4. Examples of Using Solid-State NMR Data to Assist the Process of Structure Determination from Powder XRD Data
4.1. Determining the Number of Molecules in the Asymmetric Unit
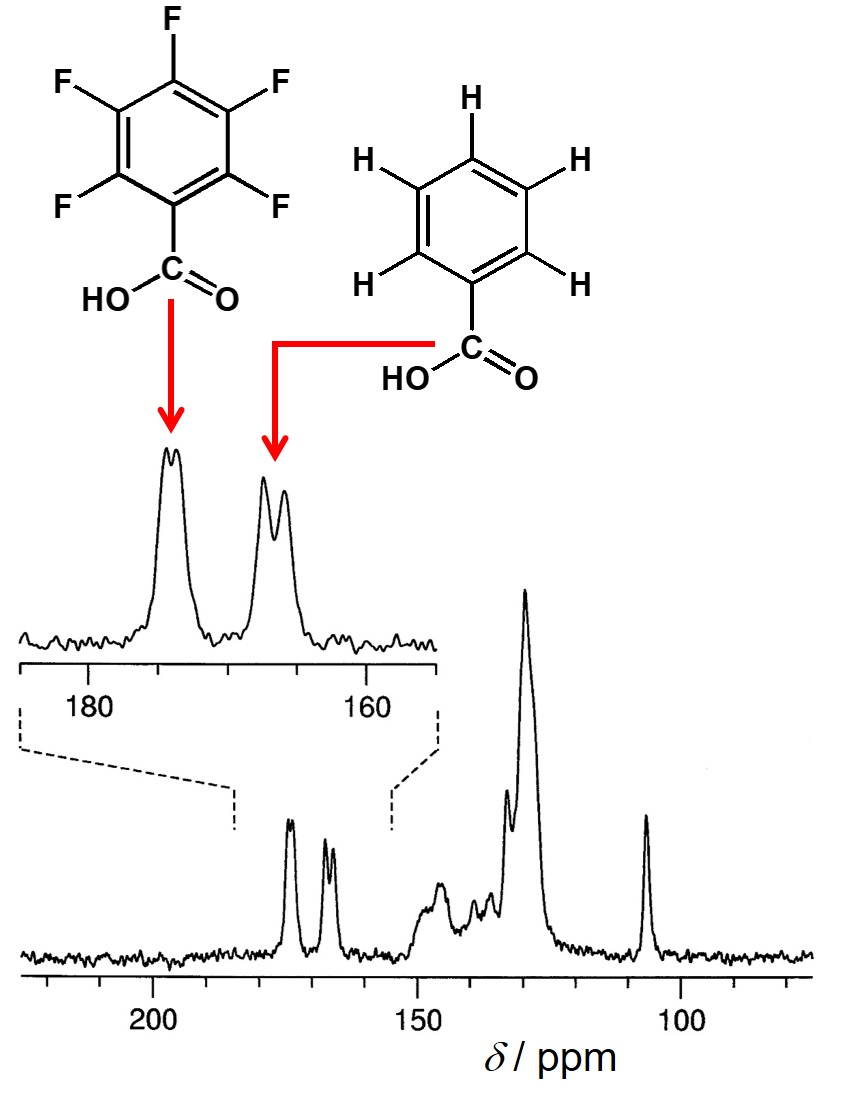
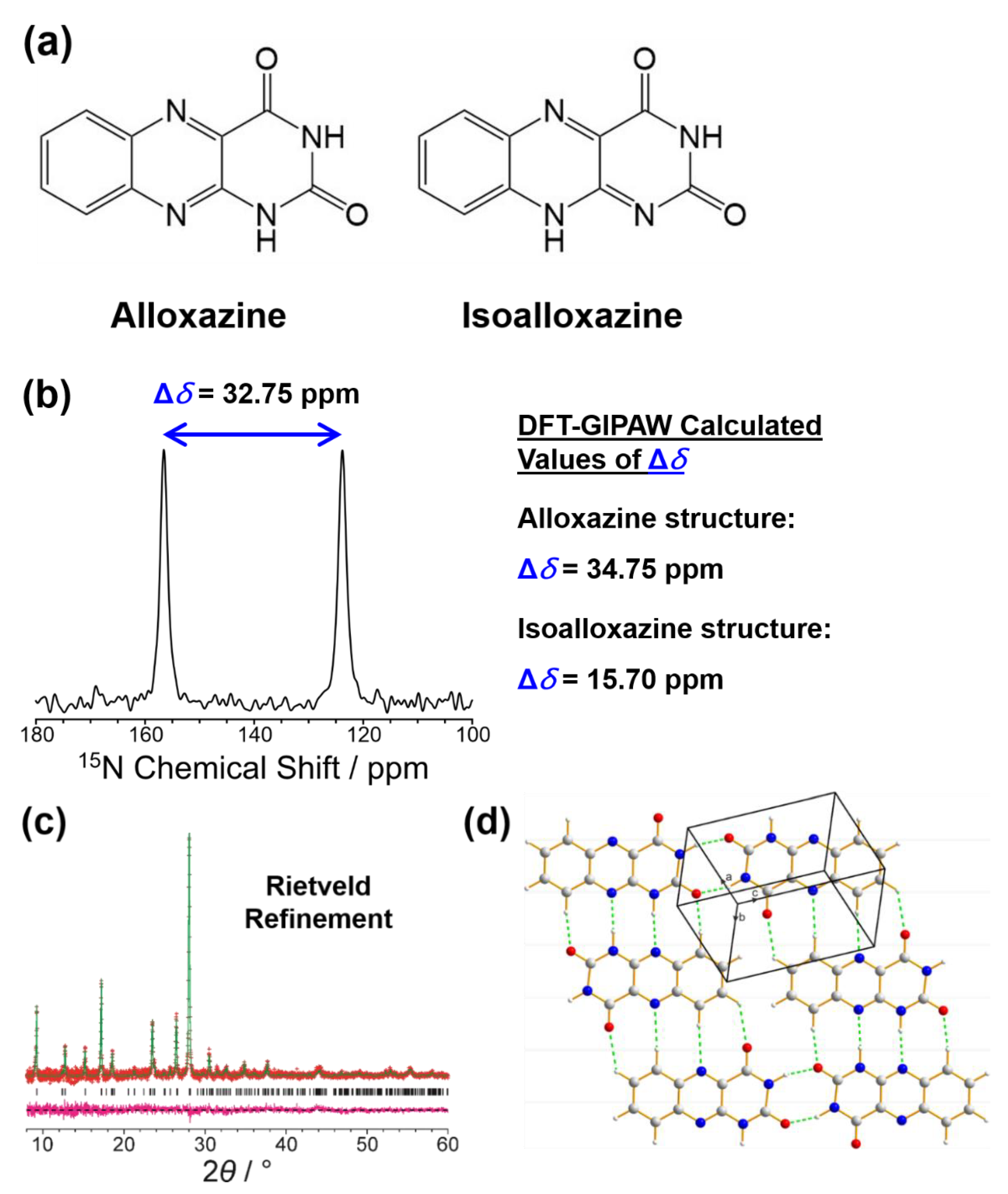
4.2. Determining the Geometric Features of the Molecule in the Crystal Structure, Focusing on Establishing the Correct Tautomeric Form
4.3. Detecting Disorder in a Crystal Structure from Solid-State NMR Data
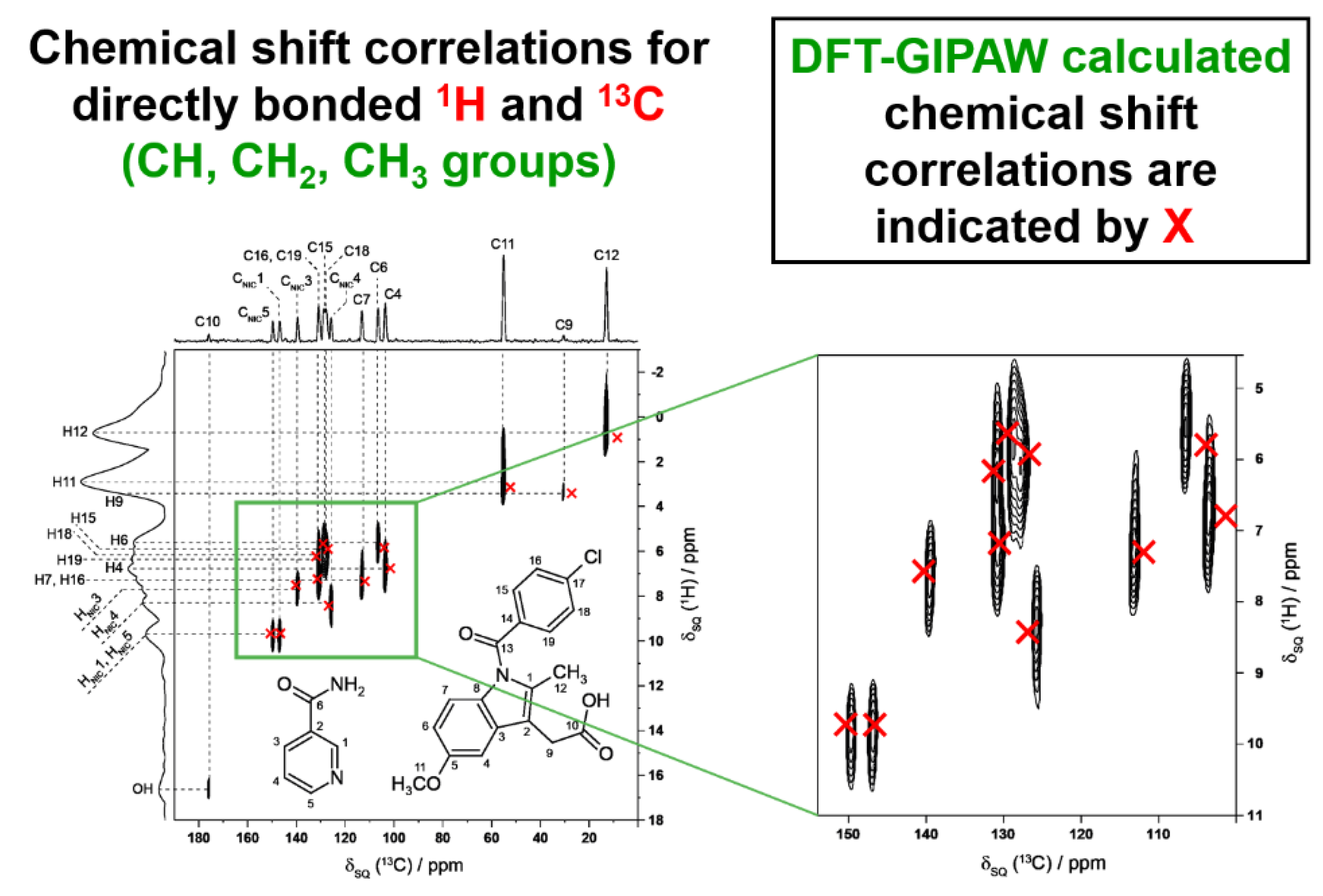
4.4. Enhancing Structure Solution Calculations Using Insights on Specific Internuclear Interactions
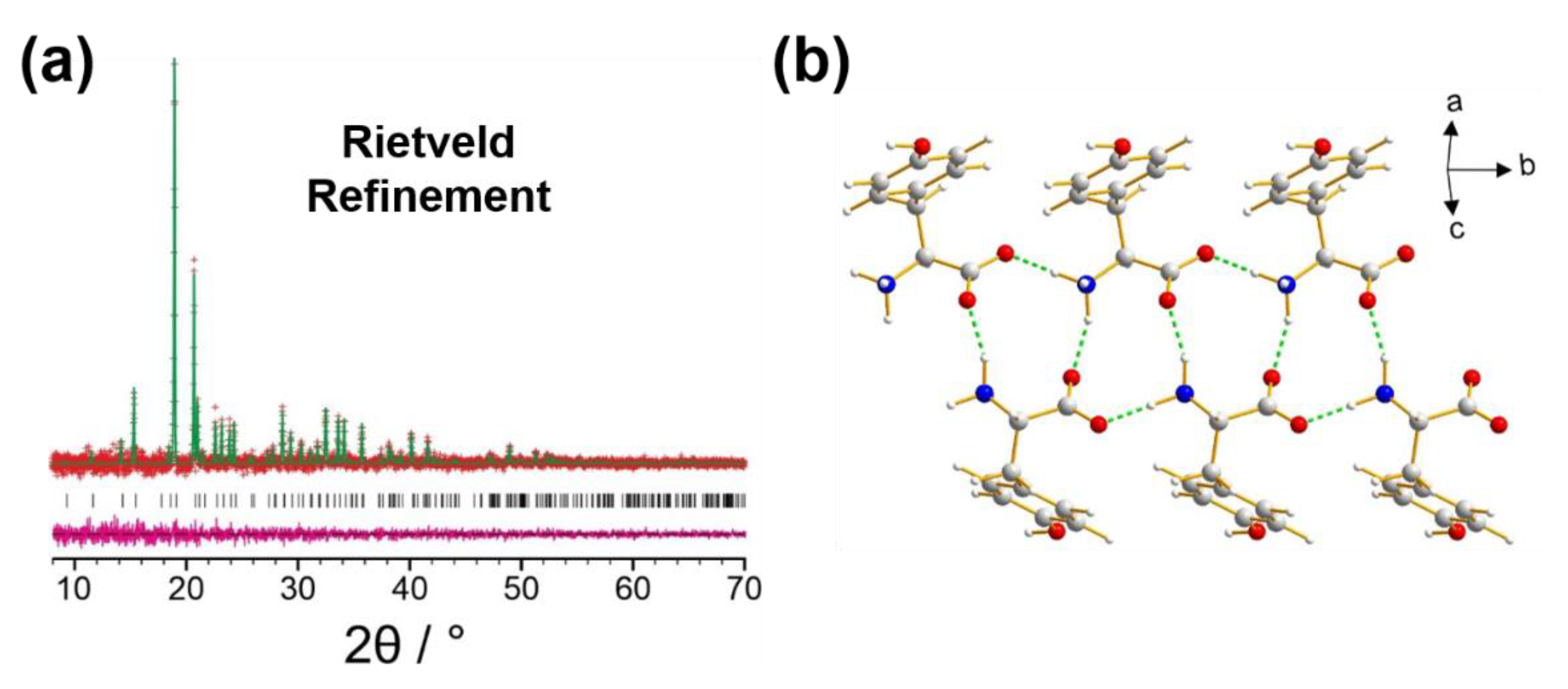
5. Examples of Using Solid-State NMR Data in Validation of the Final Crystal Structure Determined from Powder XRD Data
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Structure from Diffraction Methods; Bruce, D.W.; O’Hare, D.; Walton, R.I. (Eds.) John Wiley & Sons: Chichester, UK, 2014. [Google Scholar]
- Harris, K.D.M.; Tremayne, M. Crystal structure determination from powder diffraction data. Chem. Mater. 1996, 8, 2554–2570. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M.; Kariuki, B.M. Contemporary advances in the use of powder X-ray diffraction for structure determination. Angew. Chem. Int. Ed. 2001, 40, 1626–1651. [Google Scholar] [CrossRef]
- Chernyshev, V.V. Structure determination from powder diffraction. Russ. Chem. Bull. 2001, 50, 2273–2292. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Cheung, E.Y. How to determine structures when single crystals cannot be grown: Opportunities for structure determination of molecular materials using powder diffraction data. Chem. Soc. Rev. 2004, 33, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Tremayne, M. The impact of powder diffraction on the structural characterization of organic crystalline materials. Phil. Trans. R. Soc. A 2004, 362, 2691–2707. [Google Scholar] [CrossRef]
- Černý, R.; Favré-Nicolin, V. Direct space methods of structure determination from powder diffraction: Principles, guidelines and perspectives. Z. Kristallogr. 2007, 222, 105–113. [Google Scholar] [CrossRef]
- Tsue, H.; Horiguchi, M.; Tamura, R.; Fujii, K.; Uekusa, H. Crystal structure solution of organic compounds from X-ray powder diffraction data. J. Synth. Org. Chem. Jpn. 2007, 65, 1203–1212. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K. Structure determination from powder diffraction data. Acta Crystallogr. Sect. A 2008, 64, 52–64. [Google Scholar] [CrossRef]
- Powder Diffraction: Theory and Practice; Dinnebier, R.E.; Billinge, S.J.L. (Eds.) RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Harris, K.D.M. Powder diffraction crystallography of molecular solids. Top. Curr. Chem. 2012, 315, 133–178. [Google Scholar]
- Martí-Rujas, J. Structural elucidation of microcrystalline MOFs from powder X-ray diffraction. Dalton Trans. 2020, 49, 13897–13916. [Google Scholar] [CrossRef]
- Egami, T.; Billinge, S.J.L. Underneath the Bragg Peaks: Structural Analysis of Complex Materials; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Young, C.A.; Goodwin, A.L. Applications of pair distribution function methods to contemporary problems in materials chemistry. J. Mater. Chem. 2011, 21, 6464–6476. [Google Scholar] [CrossRef]
- Billinge, S.J.L. The rise of the X-ray atomic pair distribution function method: A series of fortunate events. Phil. Trans. R. Soc. A 2019, 377, 20180413. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K. NMR Crystallography: The use of chemical shifts. Solid State Sci. 2004, 6, 1025–1037. [Google Scholar] [CrossRef]
- Senker, J.; Seyfarth, L.; Voll, J. Determination of rotational symmetry elements in NMR Crystallography. Solid State Sci. 2004, 6, 1039–1052. [Google Scholar] [CrossRef]
- Taulelle, F. NMR Crystallography: Crystallochemical formula and space group selection. Solid State Sci. 2004, 6, 1053–1057. [Google Scholar] [CrossRef]
- Elena, B.; Pintacuda, G.; Mifsud, N.; Emsley, L. Molecular structure determination in powders by NMR Crystallography from proton spin diffusion. J. Am. Chem. Soc. 2006, 128, 9555–9560. [Google Scholar] [CrossRef]
- NMR Crystallography; Harris, R.K.; Wasylishen, R.E.; Duer, M.J. (Eds.) John Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- Martineau, C.; Senker, J.; Taulelle, F. NMR Crystallography. Ann. Rep. NMR Spectrosc. 2014, 82, 1–57. [Google Scholar]
- Ashbrook, S.E.; McKay, D. Combining solid-state NMR spectroscopy with first-principles calculations – a guide to NMR Crystallography. Chem. Commun. 2016, 52, 7186–7204. [Google Scholar] [CrossRef]
- Bryce, D.L. NMR Crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef]
- Hodgkinson, P. NMR Crystallography of molecular organics. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118–119, 10–53. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef]
- Harris, R.K.; Hodgkinson, P.; Pickard, C.J.; Yates, J.R.; Zorin, V. Chemical shift computations on a crystallographic basis: Some reflections and comments. Magn. Reson. Chem. 2007, 45, S174–S186. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azaïs, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-principles calculation of NMR parameters using the gauge including projector augmented wave method: A chemist’s point of view. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Pan, Z.; Cheung, E.Y.; Harris, K.D.M.; Constable, E.C.; Housecroft, C.E. A case study in direct-space structure determination from powder X-ray diffraction data: Finding the hydrate structure of an organic molecule with significant conformational flexibility. Cryst. Growth Des. 2005, 5, 2084–2090. [Google Scholar] [CrossRef]
- Filip, X.; Borodi, G.; Filip, C. Testing the limits of sensitivity in a solid-state structural investigation by combined X-ray powder diffraction, solid-state NMR, and molecular modelling. Phys. Chem. Chem. Phys. 2011, 13, 17978–17986. [Google Scholar] [CrossRef]
- Dudenko, D.V.; Williams, P.A.; Hughes, C.E.; Antzutkin, O.N.; Velaga, S.P.; Brown, S.P.; Harris, K.D.M. Exploiting the synergy of powder X-ray diffraction and solid-state NMR spectroscopy in structure determination of organic molecular solids. J. Phys. Chem. C 2013, 117, 12258–12265. [Google Scholar] [CrossRef]
- Li, P.; Chu, Y.; Wang, L.; Wenslow, R.M.; Yu, K.; Zhang, H.; Deng, Z. Structure determination of the theophylline-nicotinamide cocrystal: A combined powder XRD, 1D solid-state NMR, and theoretical calculation study. CrystEngComm 2014, 16, 3141–3147. [Google Scholar] [CrossRef]
- Li, X.; Bond, A.D.; Johansson, K.E.; Van de Streek, J. Distinguishing tautomerism in the crystal structure of (Z)-N-(5-ethyl-2,3-dihydro-1,3,4-thiadiazol-2-ylidene)-4-methylbenzene sulfonamide using DFT-D calculations and 13C solid-state NMR. Acta Crystallogr. Sect. C 2014, 70, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.N.M.; Cook, D.S.; Iuga, D.; Walton, R.I.; Marsh, A.; Brown, S.P. An NMR Crystallography study of the hemihydrate of 2′,3′-O-isopropylidineguanosine. Solid State Nucl. Magn. Reson. 2015, 65, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardo, M.; Santos, S.M.; Babaryk, A.A.; López, C.; Alkorta, I.; Elguero, J.; Claramunt, R.M.; Mafra, L. Diazole-based powdered cocrystal featuring a helical hydrogen-bonded network: Structure determination from PXRD, solid-state NMR and computer modeling. Solid State Nucl. Magn. Reson. 2015, 65, 49–63. [Google Scholar] [CrossRef]
- Watts, A.E.; Maruyoshi, K.; Hughes, C.E.; Brown, S.P.; Harris, K.D.M. Combining the advantages of powder X-ray diffraction and NMR Crystallography in structure determination of the pharmaceutical material cimetidine hydrochloride. Cryst. Growth Des. 2016, 16, 1798–1804. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M.; Lightfoot, P.; Bruce, P.G. Crystal structure determination from powder diffraction data by Monte Carlo methods. J. Am. Chem. Soc. 1994, 116, 3543–3547. [Google Scholar] [CrossRef]
- Kariuki, B.M.; Serrano-González, H.; Johnston, R.L.; Harris, K.D.M. The application of a genetic algorithm for solving crystal structures from powder diffraction data. Chem. Phys. Lett. 1997, 280, 189–195. [Google Scholar] [CrossRef]
- Kariuki, B.M.; Psallidas, K.; Harris, K.D.M.; Johnston, R.L.; Lancaster, R.W.; Staniforth, S.E.; Cooper, S.M. Structure determination of a steroid directly from powder diffraction data. Chem. Commun. 1999, 1677–1678. [Google Scholar] [CrossRef]
- Tedesco, E.; Turner, G.W.; Harris, K.D.M.; Johnston, R.L.; Kariuki, B.M. Structure determination of an oligopeptide directly from powder diffraction data. Angew. Chem. Int. Ed. 2000, 39, 4488–4491. [Google Scholar] [CrossRef]
- Courvoisier, E.; Williams, P.A.; Lim, G.K.; Hughes, C.E.; Harris, K.D.M. The crystal structure of l-arginine. Chem. Commun. 2012, 48, 2761–2763. [Google Scholar] [CrossRef]
- Williams, P.A.; Hughes, C.E.; Lim, G.K.; Kariuki, B.M.; Harris, K.D.M. Discovery of a new system exhibiting abundant polymorphism: m-aminobenzoic acid. Cryst. Growth. Des. 2012, 12, 3104–3113. [Google Scholar] [CrossRef]
- Ma, X.; Lim, G.K.; Harris, K.D.M.; Apperley, D.C.; Horton, P.N.; Hursthouse, M.B.; James, S.L. Efficient, scalable and solvent-free mechanochemical synthesis of the OLED material Alq3 (q = 8-hydroxyquinolinate). Cryst. Growth. Des. 2012, 12, 5869–5872. [Google Scholar] [CrossRef]
- Martí-Rujas, J.; Meazza, L.; Lim, G.K.; Terraneo, G.; Pilati, T.; Harris, K.D.M.; Metrangolo, P.; Resnati, G. An adaptable and dynamically porous organic salt traps unique tetrahalide dianions. Angew. Chem. Int. Ed. 2013, 52, 13444–13448. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Hughes, C.E.; Harris, K.D.M. l-Lysine: Exploiting powder X-ray diffraction to complete the set of crystal structures of the 20 directly-encoded proteinogenic amino acids. Angew. Chem. Int. Ed. 2015, 54, 3973–3977. [Google Scholar] [CrossRef]
- Al Rahal, O.; Hughes, C.E.; Williams, P.A.; Logsdail, A.J.; Diskin-Posner, Y.; Harris, K.D.M. Polymorphism of l-tryptophan. Angew. Chem. Int. Ed. 2019, 58, 18788–18792. [Google Scholar] [CrossRef]
- Brekalo, I.; Yuan, W.; Mottillo, C.; Lu, Y.; Zhang, Y.; Casaban, J.; Holman, K.T.; James, S.L.; Duarte, F.; Williams, P.A.; et al. Manometric real-time studies of the mechanochemical synthesis of zeolitic imidazolate frameworks. Chem. Sci. 2020, 11, 2141–2147. [Google Scholar] [CrossRef]
- Sun, T.; Hughes, C.E.; Guo, L.; Wei, L.; Harris, K.D.M.; Zhang, Y.; Ma, Y. Direct-space structure determination of covalent organic frameworks from 3D electron diffraction data. Angew. Chem. Int. Ed. 2020, 59, 22638–22644. [Google Scholar] [CrossRef]
- Al Rahal, O.; Williams, P.A.; Hughes, C.E.; Kariuki, B.M.; Harris, K.D.M. Structure determination of multicomponent crystalline phases of (S)-ibuprofen and l-proline from powder X-ray diffraction data, augmented by complementary experimental and computational techniques. Cryst. Growth Des. 2021, 21, 2498–2507. [Google Scholar] [CrossRef]
- Albesa-Jové, D.; Kariuki, B.M.; Kitchin, S.J.; Grice, L.; Cheung, E.Y.; Harris, K.D.M. Challenges in direct-space structure determination from powder diffraction data: A molecular material with four independent molecules in the asymmetric unit. ChemPhysChem 2004, 5, 414–418. [Google Scholar] [CrossRef]
- Pan, Z.; Xu, M.; Cheung, E.Y.; Harris, K.D.M.; Constable, E.C.; Housecroft, C.E. Understanding structural properties of a dendrimeric material directly from powder X-ray diffraction data. J. Phys. Chem. B 2006, 110, 11620–11623. [Google Scholar] [CrossRef]
- MacLean, E.J.; Tremayne, M.; Kariuki, B.M.; Harris, K.D.M.; Iqbal, A.F.M.; Hao, Z. Structural understanding of a polymorphic system by structure solution and refinement from powder X-ray diffraction data: The α and β phases of the latent pigment DPP-Boc. J. Chem. Soc. Perkin Trans. 2 2000, 1513–1519. [Google Scholar] [CrossRef]
- Tremayne, M.; Kariuki, B.M.; Harris, K.D.M. Structure determination of a complex organic solid from X-ray powder diffraction data by a generalized Monte Carlo method: The crystal structure of red fluorescein. Angew. Chem. Int. Ed. 1997, 36, 770–772. [Google Scholar] [CrossRef]
- Smalley, C.J.H.; Logsdail, A.J.; Hughes, C.E.; Iuga, D.; Young, M.T.; Harris, K.D.M. Solid-state structural properties of alloxazine determined from powder XRD data in conjunction with DFT-D calculations and solid-state NMR spectroscopy: Unraveling the tautomeric identity and pathways for tautomeric interconversion. Cryst. Growth Des. 2022, 22, 524–534. [Google Scholar] [CrossRef] [PubMed]
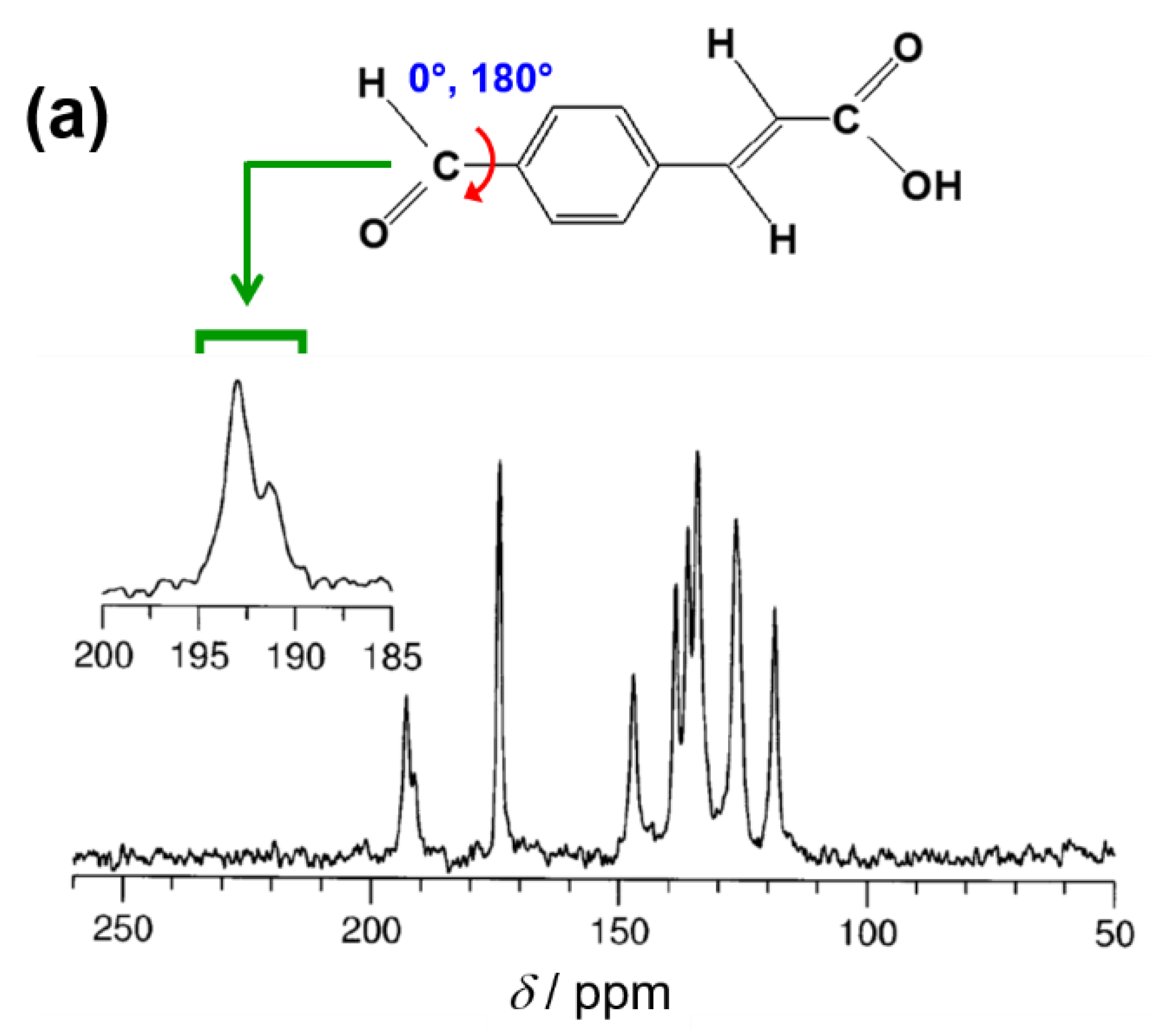
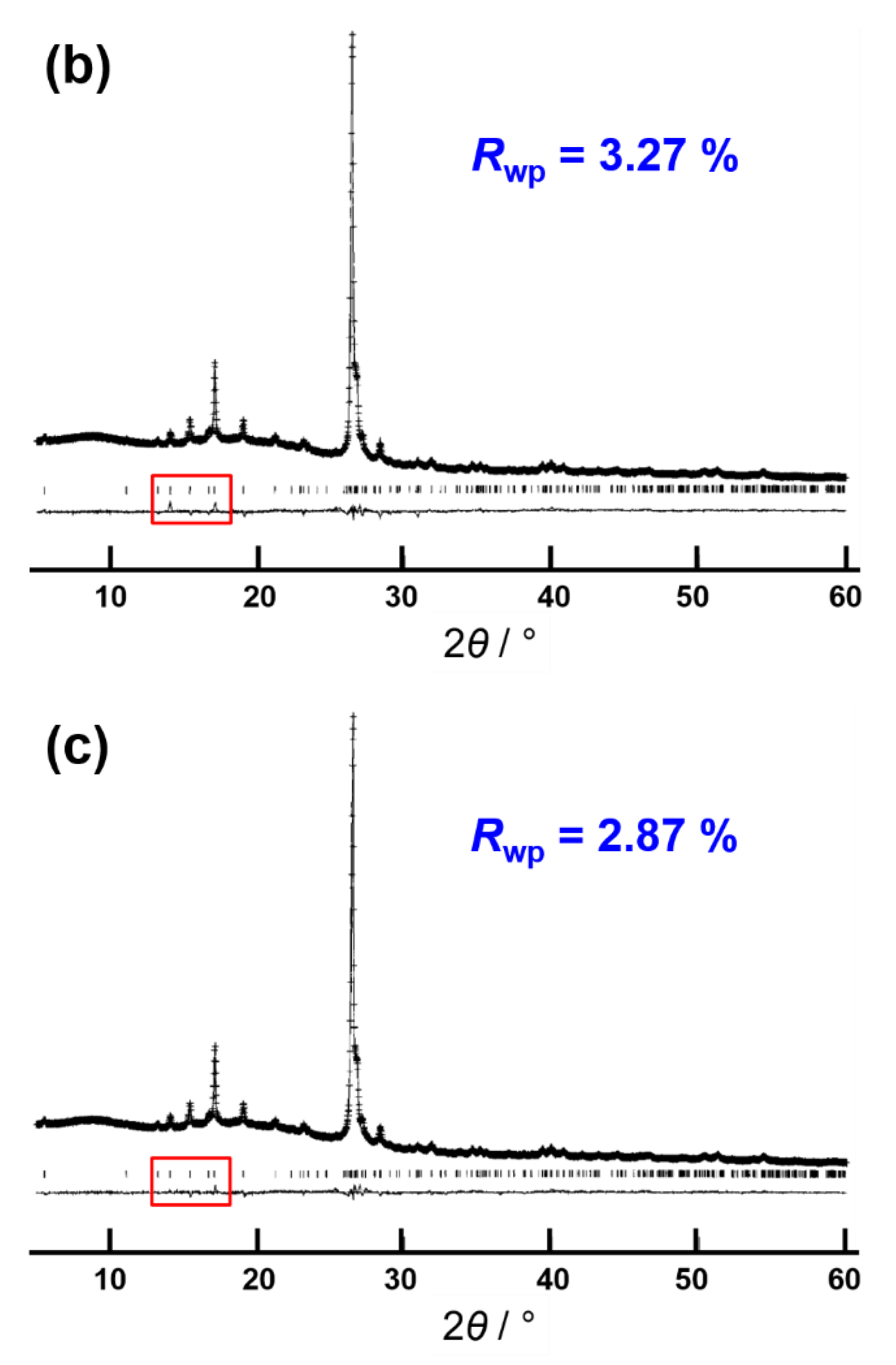
- Meejoo, S.; Kariuki, B.M.; Kitchin, S.J.; Cheung, E.Y.; Albesa-Jové, D.; Harris, K.D.M. Structural aspects of the β-polymorph of (E)-4-formylcinnamic acid: Structure determination directly from powder diffraction data and elucidation of structural disorder from solid-state NMR. Helv. Chim. Acta 2003, 86, 1467–1477. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Thomas, J.M. Probing polymorphism and reactivity in the organic solid state using 13C NMR spectroscopy: Studies of p-formyl-trans-cinnamic acid. J. Solid State Chem. 1991, 94, 197–205. [Google Scholar] [CrossRef]
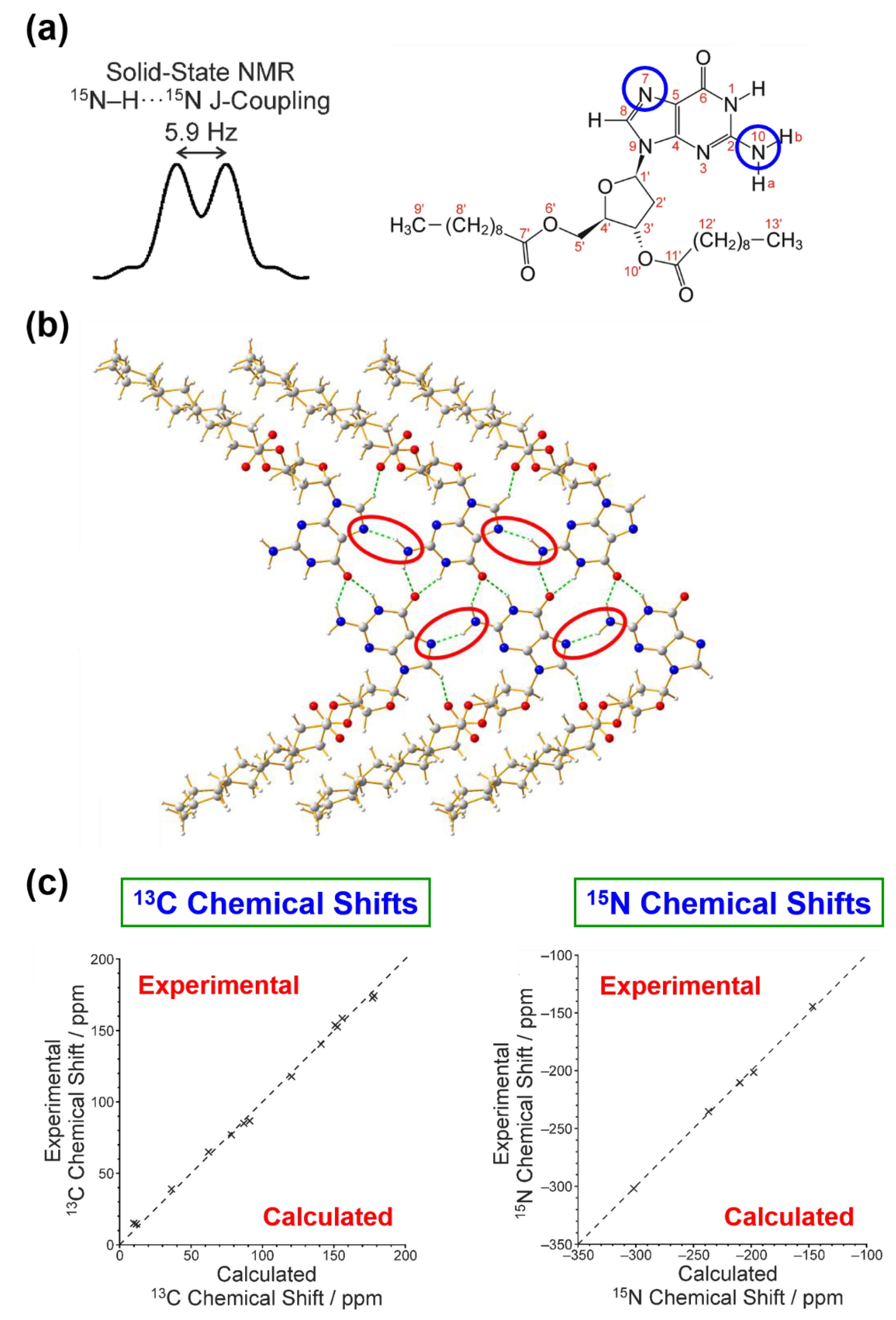
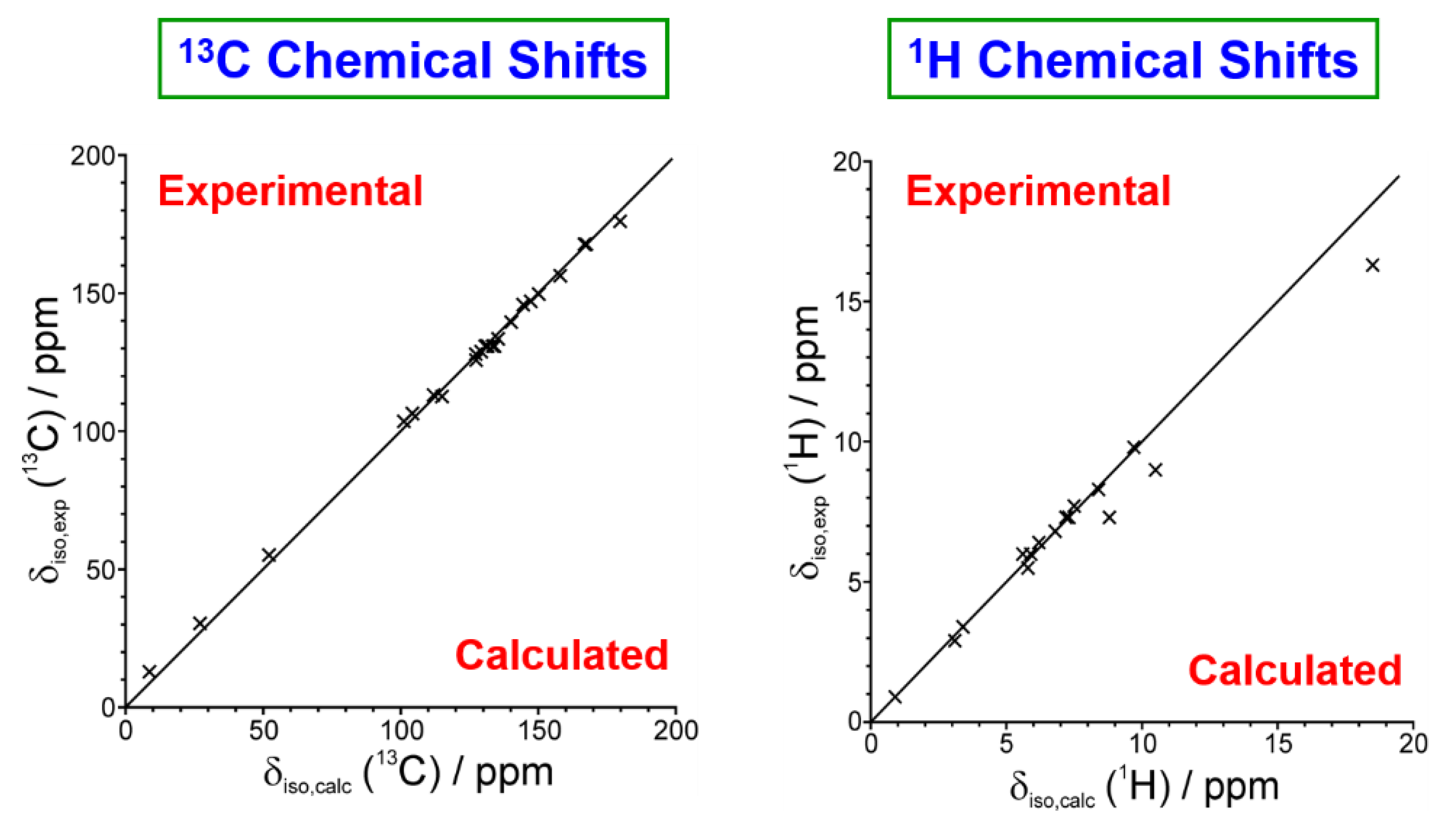
- Hughes, C.E.; Reddy, G.N.M.; Masiero, S.; Brown, S.P.; Williams, P.A.; Harris, K.D.M. Determination of a complex crystal structure in the absence of single crystals: Analysis of powder X-ray diffraction data, guided by solid-state NMR and periodic DFT calculations, reveals a new 2′-deoxyguanosine structural motif. Chem. Sci. 2017, 8, 3971–3979. [Google Scholar] [CrossRef]
- Dingley, A.J.; Grzesiek, S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Gindin, V.A.; Golubev, N.S.; Dunger, A.; Reibke, R.; Kirpekar, S.; Malkina, O.L.; Limbach, H.-H. Nuclear magnetic resonance of hydrogen bonded clusters between F− and (HF)n: Experiment and theory. Ber. Bunsenges. Phys. Chem. 1998, 102, 422–428. [Google Scholar] [CrossRef]
- Pham, T.N.; Masiero, S.; Gottarelli, G.; Brown, S.P. Identification by 15N refocused INADEQUATE MAS NMR of intermolecular hydrogen bonding that directs the self-assembly of modified DNA bases. J. Am. Chem. Soc. 2005, 127, 16018–16019. [Google Scholar] [CrossRef]
- Pham, T.N.; Griffin, J.M.; Masiero, S.; Lena, S.; Gottarelli, G.; Hodgkinson, P.; Filipe, C.; Brown, S.P. Quantifying hydrogen-bonding strength: The measurement of 2hJNN couplings in self-assembled guanosines by solid-state 15N spin-echo MAS NMR. Phys. Chem. Chem. Phys. 2007, 9, 3416–3423. [Google Scholar] [CrossRef]
- Webber, A.L.; Masiero, S.; Pieraccini, S.; Burley, J.C.; Tatton, A.S.; Iuga, D.; Pham, T.N.; Spada, G.P.; Brown, S.P. Identifying guanosine self assembly at natural isotopic abundance by high-resolution 1H and 13C solid-state NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 19777–19795. [Google Scholar] [CrossRef] [PubMed]
- Alhalaweh, A.; Velaga, S.P. Formation of cocrystals from stoichiometric solutions of incongruently saturating systems by spray drying. Cryst. Growth Des. 2010, 10, 3302–3305. [Google Scholar] [CrossRef]
- Maruyoshi, K.; Iuga, D.; Antzutkin, O.N.; Alhalaweh, A.; Velaga, S.P.; Brown, S.P. Identifying the intermolecular hydrogen-bonding supramolecular synthons in an indomethacin-nicotinamide cocrystal by solid-state NMR. Chem. Commun. 2012, 48, 10844–10846. [Google Scholar] [CrossRef] [PubMed]
- Pickard, C.J.; Salager, E.; Pintacuda, G.; Elena, B.; Emsley, L. Resolving structures from powders by NMR Crystallography using combined proton spin diffusion and plane wave DFT calculations. J. Am. Chem. Soc. 2007, 129, 8932–8933. [Google Scholar]
- Webber, A.L.; Elena, B.; Griffin, J.M.; Yates, J.R.; Pham, T.N.; Mauri, F.; Pickard, C.J.; Gil, A.M.; Stein, R.; Lesage, A.; et al. Complete 1H resonance assignment of β-maltose from 1H–1H DQ-SQ CRAMPS and 1H (DQ-DUMBO)–13C SQ refocused INEPT 2D solid-state NMR spectra and first principles GIPAW calculations. Phys. Chem. Chem. Phys. 2010, 12, 6970–6983. [Google Scholar] [CrossRef]
- Brown, S.P.; Zhu, X.X.; Saalwachter, K.; Spiess, H.W. An investigation of the hydrogen-bonding structure in bilirubin by 1H double-quantum magic-angle spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 2001, 123, 4275–4285. [Google Scholar] [CrossRef]
- Mafra, L.; Santos, S.M.; Siegel, R.; Alves, I.; Paz, F.A.A.; Dudenko, D.; Spiess, H.W. Packing interactions in hydrated and anhydrous forms of the antibiotic ciprofloxacin: A solid-state NMR, X-ray diffraction, and computer simulation study. J. Am. Chem. Soc. 2012, 134, 71–74. [Google Scholar] [CrossRef]
- Dumez, J.N.; Pickard, C.J. Calculation of NMR chemical shifts in organic solids: Accounting for motional effects. J. Chem. Phys. 2009, 130, 104701. [Google Scholar] [CrossRef]
- de Gortari, I.; Portella, G.; Salvatella, X.; Bajaj, V.S.; van der Wel, P.C.A.; Yates, J.R.; Segall, M.D.; Pickard, C.J.; Payne, M.C.; Vendruscolo, M. Time averaging of NMR chemical shifts in the MLF peptide in the solid state. J. Am. Chem. Soc. 2010, 132, 5993–6000. [Google Scholar] [CrossRef]
- Smalley, C.J.H.; Hoskyns, H.E.; Hughes, C.E.; Johnstone, D.N.; Willhammar, T.; Young, M.T.; Pickard, C.J.; Logsdail, A.J.; Midgley, P.A.; Harris, K.D.M. A structure determination protocol based on combined analysis of 3D-ED data, powder XRD data, solid-state NMR data and DFT-D calculations reveals the structure of a new polymorph of l-tyrosine. Chem. Sci. 2022, 13, 5277–5288. [Google Scholar] [CrossRef]
- Dawson, D.M.; Moran, R.F.; Sneddon, S.; Ashbrook, S.E. Is the 31P chemical shift anisotropy of aluminophosphates a useful parameter for NMR Crystallography? Magn. Reson. Chem. 2019, 57, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Burgess, K.M.N.; Bryce, D.L. On the crystal structure of the vaterite polymorph of CaCO3: A calcium-43 solid-state NMR and computational assessment. Solid State Nucl. Magn. Reson. 2015, 65, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.T.; Vojvodin, C.S.; Schurko, R.W. Dispersion-corrected DFT methods for applications in nuclear magnetic resonance crystallography. J. Phys. Chem. A 2020, 124, 10312–10323. [Google Scholar] [CrossRef] [PubMed]
- Chierotti, M.R.; Gobetto, R. NMR Crystallography: The use of dipolar interactions in polymorph and co-crystal investigation. CrystEngComm 2013, 15, 8599–8612. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Mollica, G.; Juramy, M.; Hughes, C.E.; Williams, P.A.; Ziarelli, F.; Viel, S.; Thureau, P.; Harris, K.D.M. Insights into the crystallization and structural evolution of glycine dihydrate by in-situ solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 6619–6623. [Google Scholar] [CrossRef]
- Thureau, P.; Sturniolo, S.; Zilka, M.; Ziarelli, F.; Viel, S.; Yates, J.R.; Mollica, G. Reducing the computational cost of NMR Crystallography of organic powders at natural isotopic abundance with the help of 13C-13C dipolar couplings. Magn. Reson. Chem. 2019, 57, 256–264. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pislak, D.M. A review on combination of ab initio molecular dynamics and NMR parameters calculations. Int. J. Mol. Sci. 2021, 22, 4378. [Google Scholar] [CrossRef]
- Iftimie, R.; Minary, P.; Tuckerman, M.E. Ab initio molecular dynamics: Concepts, recent developments, and future trends. Proc. Natl. Acad. Sci. USA 2005, 102, 6654–6659. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harris, K.D.M. NMR Crystallography as a Vital Tool in Assisting Crystal Structure Determination from Powder XRD Data. Crystals 2022, 12, 1277. https://doi.org/10.3390/cryst12091277
Harris KDM. NMR Crystallography as a Vital Tool in Assisting Crystal Structure Determination from Powder XRD Data. Crystals. 2022; 12(9):1277. https://doi.org/10.3390/cryst12091277
Chicago/Turabian StyleHarris, Kenneth D. M. 2022. "NMR Crystallography as a Vital Tool in Assisting Crystal Structure Determination from Powder XRD Data" Crystals 12, no. 9: 1277. https://doi.org/10.3390/cryst12091277
APA StyleHarris, K. D. M. (2022). NMR Crystallography as a Vital Tool in Assisting Crystal Structure Determination from Powder XRD Data. Crystals, 12(9), 1277. https://doi.org/10.3390/cryst12091277