Abstract
Multicomponent pharmaceutical materials offer new opportunities to address drug physicochemical issues and to obtain improved drug formulation, especially on oral administration drugs. This work reports three new multicomponent pharmaceutical crystals of the non-steroidal anti-inflammatory drug diclofenac and the nucleobases adenine, cytosine, and isocytosine. They have been synthesized by mechanochemical methods and been characterized in-depth in solid-state by powder and single crystal X-ray diffraction, as well as other techniques such as thermal analyses and infrared spectroscopy. Stability and solubility tests were also performed on these materials. This work aimed to evaluate the physicochemical properties of these solid forms, which revealed thermal stability improvement. Dissociation of the new phases was observed in water, though. This fact is consistent with the reported observed layered structures and BFDH morphology calculations.
1. Introduction
Diclofenac (DIC), a phenylacetic derivative non-steroidal anti-inflammatory drug (NSAID), is widely used in human and veterinary practice for the treatment of acute and chronic pain as well as in inflammatory and degenerative rheumatic diseases [1,2]. Diclofenac exerts its action through the inhibition of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) enzymes, which inhibit the synthesis of prostaglandins [3]. According to the Biopharmaceutics Classification System (BCS), DIC is a class II drug with low solubility and high permeability [4]. Due to its low solubility (0.9 ± 0.1 μg/mL) [5], achieving its minimum effective concentration requires a higher dosage in the formulation. However, the side effects of DIC have shown dosage-dependency; these include gastrointestinal damage and bleeding, nausea, hepatotoxicity, or renal failure. Moreover, when DIC is administrated orally, its low solubility increases the residence time in the stomach and the contact with the gastric mucosa, increasing the risk of gastric damage [6]. However, poor solubility is a major drawback not only for DIC but also for other Active Pharmaceutical Ingredients (APIs). For that reason, significant efforts have been made by both the industry and academia to develop new methodologies to enhance the physicochemical properties of APIs. Pharmaceutical multicomponent solid forms have gained much interest in the last decade due to their great potential to overcome drug performance limitations [7]. These solid forms are crystalline materials composed of two or more components. At least one must be an API, and the other, called cocrystal former or coformer, must be pharmaceutically acceptable, which means to be recognized as a safe molecule. Both components are in a stoichiometric ratio and interact through non-covalent interactions, mainly hydrogen bonds. These non-covalent interactions guide the organization of the molecules in the crystalline structure and allow the modulation of the physicochemical properties without covalent alterations of the API, whose activity and efficacy remain intact [8]. The literature reports pharmaceutical salts and cocrystals of DIC with amide (isonicotinamide [9]), amine (metformin [10], L-proline [11]), and xanthine (theophylline [12]), as well as pyridine-based coformers [13].
Nucleobases, the main component of nucleic acids, have attracted interest from the crystal engineering point of view because they can establish different hydrogen bond patterns [14]. This ability has been explored previously to form cocrystals and salts through NH···O=C hydrogen bond motifs [15]. Amine-carbonyl synthon has a remarkable key role in the transfer of genetic information and nucleic acid-protein recognition [16]. Moreover, nucleobase-derived drugs exhibit different biological roles, including anti-viral, antibacterial, and antitumoral activities [17,18].
This work reports the synthesis and physicochemical characterization of new multicomponent forms with diclofenac and a nucleobase: adenine, cytosine, and isocytosine (Scheme 1). The single crystal structure of all solid forms is thoroughly described, providing valuable insights into the structural differences that drive their physicochemical properties, mainly stability and solubility.
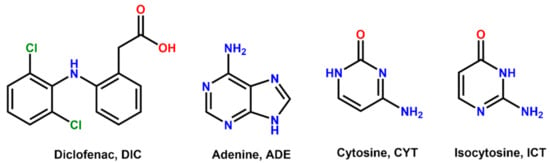
Scheme 1.
Chemical formula of diclofenac (DIC), adenine (ADE), cytosine (CYT), and isocytosine (ICT).
2. Materials and Methods
2.1. Materials
Sodium diclofenac (DICNa), adenine, cytosine, and isocytosine were commercially available from Sigma-Aldrich (purity > 98%, Sigma-Aldrich, St. Louis, MO, USA). All solvents were also purchased from Sigma-Aldrich and were used as received.
Synthesis of Diclofenac Acid Form
Diclofenac acid form (DIC) was obtained from hydrolysis of DICNa. For this purpose, 5 mol of DICNa (1.590 g) were dissolved in 30 mL of ultrapure water (Milli-Q, Millipore, Burlington, MA, USA) at 40 °C. HCl 1 M was added dropwise to the solution until no more diclofenac was precipitating. The product was filtrated and washed three times with cold deionized water and let dry at 35 °C for 24 h. Powder X-ray diffraction (PXRD) was used to confirm the purity of DIC.
2.2. Coformer Selection
A search of the Cambridge Structural Database (CSD) [19] was conducted to identify complementary functional groups with the potential for molecular recognition with DIC. A virtual cocrystal screening was performed afterwards using COSMOQuick software [20] (COSMOlogic, Germany, Version 1.4), calculating the excess enthalpy (Hex) of mixing between DIC and selected coformers from an internal library.
2.3. General Procedure for Mechanochemical Synthesis
Mechanochemical experiments were carried out via liquid-assisted grinding (LAG) using a Retsh MM200 ball mill (Retsch, Haan, Germany) operating for 30 min at a 25 Hz frequency using methanol as a solvent.
Synthesis of DIC–ADE: A mixture of DIC (74.1 mg, 0.25 mmol) and ADE (33.80 mg, 0.25 mmol) in a 1:1 stoichiometric ratio was placed in a 10 mL stainless-steel jar along with 100 μL of methanol and two stainless-steel balls of 5 mm diameter.
Synthesis of DIC–CYT: A mixture of DIC (74.1 mg, 0.25 mmol) and CYT (22.20 mg, 0.25 mmol) in a 1:1 stoichiometric ratio was placed in a 10 mL stainless-steel jar along with 100 μL of methanol and two stainless-steel balls of 5 mm diameter.
Synthesis of DIC–ICT: A mixture of DIC (74.1 mg, 0.25 mmol) and ICT (22.20 mg, 0.25 mmol) in a 1:1 stoichiometric ratio was placed in a 10 mL stainless-steel jar along with 100 μL of methanol and two stainless-steel balls of 5 mm diameter.
2.4. Powder X-ray Diffraction (PXRD)
PXRD data were collected using a Bruker D8 Advance Series II Vαrio diffractometer (Bruker-AXS, Karlsruhe, Germany) equipped with a LYNXEYE detector and Cu-Kα1 radiation (1.5406 Å). Diffraction patterns were collected over a 2θ range of 5–60° using a continuous step size of 0.02° and a total acquisition time of 30 min.
2.5. Preparation of Single Crystals
Single crystals were grown from saturated solutions (methanol) of the polycrystalline material obtained from LAG synthesis. Suitable single crystals for X-ray diffraction studies were grown by slow solvent evaporation at room temperature for two days.
2.6. Single-Crystal X-ray Diffraction (SCXRD)
Measured crystals were prepared under inert conditions immersed in perfluoropolyether as protecting oil for manipulation. Suitable crystals were mounted on MiTeGen Micromounts™, and these samples were used for data collection. Data for DIC–ADE, DIC–CYT, and DIC–ICT were collected with a Bruker D8 Venture diffractometer with graphite monochromated Cu-Kα radiation (λ = 1.54178 Å). The data were processed with APEX4 suite [21]. The structures were solved by intrinsic phasing using the ShelXT program [22], which revealed the position of all non-hydrogen atoms. These atoms were refined on F2 by a full-matrix least-squares procedure using an anisotropic displacement parameter [23]. All hydrogen atoms were located in difference Fourier maps and included as fixed contributions riding on attached atoms with isotropic thermal displacement parameters 1.2 or 1.5 times those of the respective atom. The Olex2 software was used as a graphical interface [24]. Intermolecular interactions were calculated using PLATON [25]. Molecular graphics were generated using Mercury [26,27]. The crystallographic data for the reported structures were deposited with the Cambridge Crystallographic Data Center as supplementary publication no. CCDC 2180776-2180778. Additional crystal data are shown in Table 1. Copies of the data can be obtained free of charge at https://www.ccdc.cam.ac.uk/structures/.

Table 1.
Crystallographic data and structure refinement of DIC polymorphs and new solid forms.
2.7. Thermal Analysis
Simultaneous thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed using a Mettler Toledo TGA/DSC1 thermal analyzer (Mettler Toledo, Columbus, OH, USA). Samples (3–5 mg) were placed into sealed aluminium pans and heated in a stream of nitrogen (100 mL min−1) from 25 to 400 °C at a heating rate of 10 °C min−1.
2.8. Infrared Spectroscopy
Fourier-transform infrared (FT–IR) spectroscopic measurements were performed on a Bruker Tensor 27 FT–IR instrument (Bruker Corporation, Billerica, MA, USA) equipped with a single-reflection diamond crystal platinum ATR unit and OPUS data collection program. The scanning range was from 4000 to 400 cm−1 with a resolution of 4 cm−1.
2.9. Stability Test
Stability in aqueous solution was evaluated through slurry experiments. Excess of powder samples of each phase was added to 1 mL of water and stirred for 24 h in sealed vials. The solids were collected, filtered, and dried for further analysis by PXRD.
Stability at accelerated ageing conditions was also studied: 200 mg of solid was placed in watch glasses and left at 40 °C in 75% relative humidity using a Memmert HPP110 climate chamber (Memmert, Schwabach, Germany). The samples were subjected to the above-accelerated stability conditions for two months. PXRD was used to monitor the stability of the solid forms.
2.10. Solubility Test
Solubility studies were performed using the Crystal16 equipment (Technobis Crystallization Systems, Alkmaar, The Netherlands) in water. The equipment comprised four individually controlled reactors, each with a working volume of 1 mL, allowing the measurement of cloud and clear points based on the turbidity of 16 aliquots of 1 mL of solution in parallel and automatically. Each solution was heated at 0.3 °C/min from 20 to 90 °C with a magnetic stirring rate of 700 rpm, held at this temperature for 10 min and then cooled to 20 °C at 0.3 °C/min. The dissolution temperature for each compound was measured using different amounts of solid, and the solubility data of the pure components were fitted to a quadratic equation [29] using the CrystalClear software (Technobis Crystallization Systems, Alkmaar, The Netherlands).
3. Results and Discussion
3.1. Coformer Selection
Before the experimental trials were conducted, a virtual cocrystal screening was performed to improve the success ratio. A survey on the Cambridge Structural Database (CSD version 5.43, update from June 2022) based on DIC resulted in 70 hits. After excluding the three reported polymorphs [28,30,31] and metal complexes [32,33,34,35,36,37,38,39,40,41], the dataset contained 28 hits corresponding to multicomponent forms (salts, cocrystals, hydrates, and solvates). Only in one hit, the dimer DIC–DIC, observed in the monoclinic DIC polymorphs [28,30], was maintained; meanwhile, the remaining hits exhibited common structural features for DIC salts, COO−···amine and COO−··· ammonium synthons, or cocrystals: COOH···N(pyridine) and COOH···N(imidazole) synthons. According to the above-mentioned, our main prerequisite for the coformer selection was having the above-referred N-groups and being a safe molecule. From our library of coformers, two groups of molecules fulfil these criteria: amino acids and nucleobases. COSMOQuick software was used to validate our selection. Table 2 shows calculations from a list of candidates to form multicomponent crystals with DIC. The list includes our reported coformers and other coformer molecule involved in the formation of cocrystals/salts reported in the survey. Compounds with negative Hex values show an increased probability of forming cocrystals.
Table 2.
Ranking of potential DIC coformers used in this work, based on COSMOQuick calculations.
3.2. Mechanochemical Synthesis
Liquid-Assisted Grinding is a versatile and efficient methodology widely used to obtain pharmaceutical multicomponent solid forms [42]. A screening through LAG was conducted with DIC and all coformers listed in Table 2. Unfortunately, despite the promising results obtained by the COSMOQuick analysis, only those LAG synthesis with the coformers ADE, CYT, and ICT were successful and achieved new phases whilst the other coformers yielded only physical mixtures of the two components. The product of these reactions was characterized by PXRD and compared with the X-ray powder pattern of the parent components (Table S1, Figure S1). Only experiments using ADE, CYT, and ICT as coformers provided a new PXRD pattern and were used for subsequent characterization (Figure 1). After the screening procedure, the work was focused on the search for fine-tuning conditions to obtain multicomponent materials of DIC with ADE, CYT, and ICT. Neat grinding experiments resulted in physical mixtures of the components (Figure S1). Neat grinding approach only led to physical mixtures (Figure S2). It is reported that this synthesis technique sometimes yields products with low crystallinity, partial reactions, or not even a reaction at all [43]. However, it is well known that adding small amounts of liquids accelerate the reaction, which essentially drove us to the idea of using LAG synthesis instead. LAG experiments were then performed using methanol and different stoichiometries (1:1, 1:2, and 2:1) (Figure S3). A new common pattern was observed in the three stoichiometries. However, in the 1:2 and 2:1 ratios, there were also peaks corresponding to the coformer and DIC, respectively. Only the 1:1 ratio provided unique different PXRD patterns. Comparing these patterns with those simulated from the crystal structures confirmed the monophasic nature of the bulk solids (Figure S4).
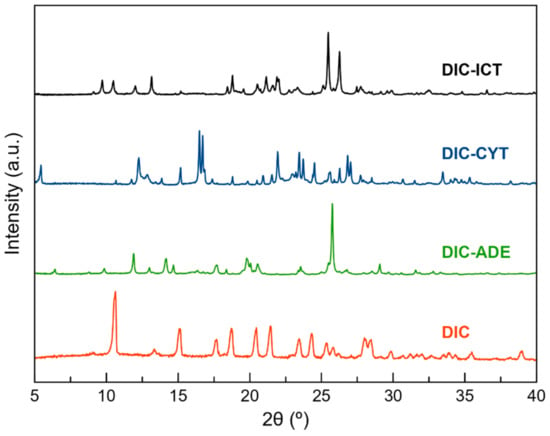
Figure 1.
PXRD patterns of DIC, DIC–ADE, DIC–CYT, and DIC–ICT obtained by Liquid-Assisted Grinding (LAG) with methanol in a 1:1 ratio.
3.3. Structural Analysis of Multicomponent Forms
DIC–ADE cocrystal crystallized in the triclinic P-1 space group. The asymmetric unit is composed of DIC and ADE in a 1:1 stoichiometric ratio, where ADE adopts its most stable and expected 9H tautomeric form. DIC exhibits its aromatic ring twisted out (dihedral angle: 71.92°) and is stabilized by an intramolecular N–H(amine)⋯O(carbonyl) hydrogen bond (Figure 2a). Centrosymmetric H-bonded adenine dimers (N29-H29···N23#2 2.00 Å, 166.6°; #2: -x + 3,-y + 2,-z) aggregate through the Hoogsteen edge (N26-H26B···N27#1: 2.10 Å, 162.7°; #1:-x + 2,-y + 1,-z), creating infinite zig-zag chains. DIC molecules connect to the chain structure by H-bonding interactions through the Watson–Crick edge (O2-H2···N21: 1.84 Å, 169°; N26-H26A···O1: 2.10 Å, 169°), resulting in an infinite tape structure (Figure 2b). C–H···F hydrogen bonds reinforce this structure and also connect adjacent tapes. Finally, the 3D structure is accomplished by piling these tapes through C=O···π and C-H···π interactions among DIC and aromatic rings from the adenine (Figure 2c).
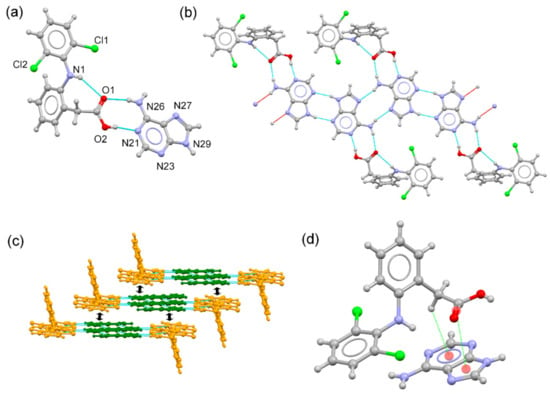
Figure 2.
(a) Asymmetric unit of the DIC–ADE cocrystal. (b) Fragment of the tape structure generated by H-bonding interactions. (c) Detailed view of the crystal packing of the DIC–ADE cocrystal. Orange: DIC molecules, green: ADE molecules. (d) C=O···π and C-H···π interactions in the DIC–ADE cocrystal.
DIC–CYT crystallized as a molecular salt in the orthorhombic Pca21 spacegroup. The asymmetric unit consisted of two symmetry-independent molecules of diclofenac anion and two symmetry-independent molecules of cytosinium cation in a 1:1 stoichiometric ratio (Figure 3a). Cytosinium over hemicytosinium duplex formation was observed in agreement with the cutoff pKa value for acids reported by Sun et al. [44] (pKa value for DIC: 4.15). The analysis of the C–O bond distances of the carboxylate group of DIC supports the salt formation [45]. In the DIC–CYT system, C–O distances were indicative of a deprotonated acid, as expected for a salt with ΔDC–O values of 0.001 Å and 0.002 Å for both DIC anions, respectively, according to the ΔDC–O values observed in salts (typically less than 0.03 Å). As in DIC–ADE, the two aromatic rings of diclofenac are bent out of plane, with dihedral angles of 81.78 and 84.06°. In the crystal, DIC− and CYT–H+ form an alternating layered structure where cytosinium molecules are associated through single-point N–H···O bonds, graph set , generating CYT–H+···CYT–H+ chains running along the a-axis [Figure 3b]. DIC− layers are reinforced by C–H···F hydrogen bonds. The two-point 2-amino-pyridinium–carboxylate synthon (N4A–H4AA···O1A, 1.85 Å, 178°, N2A–H2A+···O2A, 1.92 Å, 176.3°, and N4B–H4BA···O1B, 1.84 Å, 178.4°, N2B–H2B+···O2B, 1.92 Å, 176.5°) associates the DIC− and CYT–H+ layers, generating the supramolecular 3D structure.
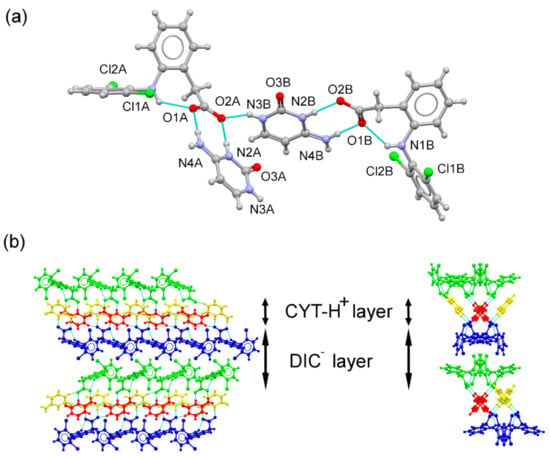
Figure 3.
(a) Asymmetric unit of the DIC–CYT molecular salt. (b) Detailed view of the packing arrangement of DIC− anions (blue and green) and CYT–H+ cations (red and yellow) in the DIC–CYT crystal structure (viewed along the b and a axes), showing an alternating layered structure.
DIC–ICT crystallized in the triclinic P1 spacegroup. Both diclofenac and isocytosine components are present in their neutral and ionic forms, resulting in a hybrid solid with a cocrystal and a salt in the asymmetric unit (Figure 4a). In the DIC–ICT system, C–O distances confirmed the presence of carboxylate and carboxylic groups, as expected for ΔDC–O values observed for the two symmetry-independent diclofenac molecules (0.080 Å and 0.006 Å for neutral diclofenac and diclofenac anion, respectively). Isocytosine and isocytosinium molecules formed a dimeric structure through H-bonds involving the 2-amino-pyridinium–carbonyl synthon (graph set motif ). These dimers connect with adjacent dimers using the amine-carbonyl synthon (graph set motif ), generating a chain structure. The two-point 2-amino-pyridine–carboxylic (N4B–H4BA···O1B, 2.02 Å, 166.5°, and O2B–H2B···N2B#1, 1.80 Å, 171.7°; #1: x + 1, y, z) and 2-amino-pyridinium–carboxylate (N4A–H4AB···O1A, 1.82 Å, 158.1°, and N2A–H2A+···O2A, 1.99 Å, 174.0°) synthons connect components to build up a ribbon structure (Figure 4b). C–H···π interactions (C13B-H13D···Cg; H···Cg distance: 2.97 Å; C-H···Cg angle: 118°; Cg = C7B-C8B-C9B-C10B-C11B-C12B) associate these ribbons to form a layered structure. Finally, weak C–H···F hydrogen bonds connect these layers to create the 3D structure.
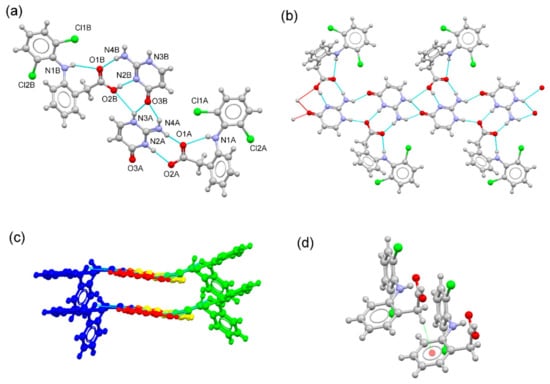
Figure 4.
(a) Asymmetric unit of the DIC–ICT multicomponent form. (b) Fragment of the ribbon structure generated by H-bonding interactions between DIC components and a chain of –ICT–H+···ICT– dimmers. (c) Detailed view of the packing arrangement of ribbons structures containing DIC (blue), DIC− (green), ICT (yellow), and ICT–H+ (red), building up a layered structure. (d) Detailed view of the C–H···π interaction between DIC molecules.
3.4. Thermal Analysis
DSC was used to evaluate the thermal behaviour and to determine the melting point of the new DIC phases. Figure 5 shows the melting point of DIC, as well as the DSC traces of DIC–ADE, DIC–CYT, and DIC–ICT. Each plot shows a well-defined endothermic event that corresponds with the melting point of the material. A single endothermic transition indicates the absence of solvation or hydration phenomena and also demonstrates the stability of the phase until the melting point. Above the melting point, some endothermic events are also observed, corresponding to the degradation of the samples. The multicomponent materials display a melting point that falls in a region between the melting point of DIC (179 °C) and the coformer (ADE: 360 °C; CYT: 320–325 °C; ICT:248–254 °C). This feature has already been described by other researchers [46]. A higher melting point was obtained through multicomponent crystallization, resulting in better thermal stability, probably due to stronger intermolecular interactions between DIC and nucleobases. TGA showed no weight loss until melting, suggesting that the new DIC phases were not hydrated or solvated. The occurrence of mass loss was observed after melting points, which was attributed to the degradation of cocrystals (Figure S5).
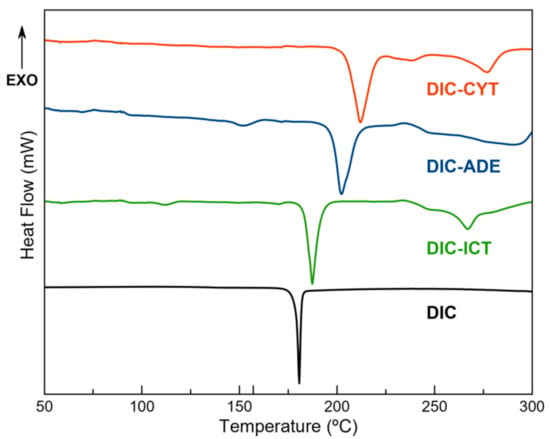
Figure 5.
Differential scanning calorimetry (DSC) traces of DIC and multicomponent compounds DIC–ADE, DIC–CYT, and DIC–ICT.
3.5. Fourier Transform Infrared (FT–IR) Spectroscopy
Due to its simplicity and reduced consumption of time and samples, FT–IR spectroscopy is a widely used technique for detecting new multicomponent materials [47]. Functional groups exhibit defined bands in the IR spectrum, and intermolecular interactions, such as hydrogen bonds, induce changes in the position of these bands. Hence, the study of the shifts can detect the formation of a cocrystal or a salt and gives information about the groups involved in the interaction [48].
Figure 6 shows the FT–IR spectra of DIC and DIC multicomponent materials. The DIC spectrum has a characteristic band at 3322 cm−1, ascribed to the stretching mode of –NH. In DIC–ADE, DIC–CYT, and DIC–ICT, this band is shifted to 3326, 3300, and 3298 cm−1, respectively. Another characteristic band of DIC is the C=O stretching vibration that appears at 1961 cm−1. This band is shifted to 1671 (DIC–ADE), 1693 (DIC–CYT), and 1678 cm−1 (DIC–ICT). FT–IR data support the information observed in SCXRD, where –COOH and –NH groups from DIC and coformers, respectively, drive the formation of the crystalline structures.
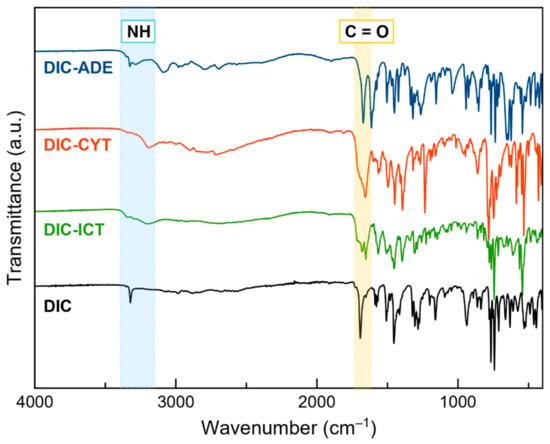
Figure 6.
Fourier transform infrared (FT–IR) spectra of DIC, DIC–ADE, DIC–CYT, and DIC–ICT.
3.6. Stability Studies
Thermodynamic stability in an aqueous solution was evaluated by placing an excess of the sample in a vial and stirring in deionized water at 25 °C. After 24 h, the product was filtered, dried at room temperature, and characterized by PXRD. Powder patterns showed high stability for the pure DIC and DIC–ADE cocrystal. However, DIC–CYT and DIC–ICT phases could not remain stable for more than 3 h and 30 min, respectively (Figure 7 and Figure S7). Multicomponent DIC phases were also stored at accelerating ageing conditions (40 °C, 75% RH). At the determined time, samples were characterized by PXRD to assess their stability. DIC was included in the experiment for better comparison with the new materials. Under these conditions, it was observed that all samples remained stable for two months (Figure 8).
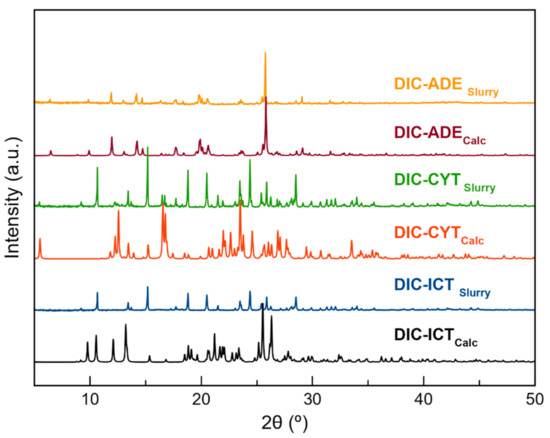
Figure 7.
PXRD diagrams corresponding to the stability of DIC–ADE, DIC–CYT, and DIC–ICT in aqueous slurry experiments at 24 h.
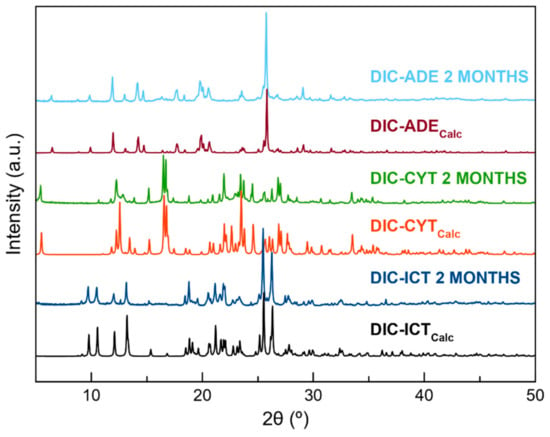
Figure 8.
PXRD diagrams corresponding to the stability of DIC–ADE, DIC–CYT, and DIC–ICT in accelerated ageing conditions (40 °C, 75% RH) at two months.
3.7. Solubility Studies
As observed in the previous section, only the DIC–ADE cocrystal was thermodynamically stable in a water solution at room temperature. Initial attempts to determine solubility by the shake-flask method [49] were not possible due to the overlap of UV absorption maxima of both the API and coformer (Figure S8). Evaluation of solubility was then performed by a polythermal method using the Crystal16 equipment. Results showed an improvement in the solubility of DIC–ADE (0.993 mg/mL, Figure 9) compared with the reported solubility of DIC (0.9 μg/mL) [5]. Although the difference in solubility between DIC and the DIC–ADE cocrystal was significant, the amount of solubility improvement is not significant compared with the solubility of the sodium salt (16.18 mg/mL) [50]. The layered structure observed in the DIC–ADE cocrystal directly impacts the solubility improvement of DIC. Different studies have reported the effect of a layer structure consisting of high-solubility molecules on the solubility of multicomponent solid forms [51,52,53]. Although the dimeric DIC structure is disrupted and a better solubility is obtained in DIC–ADE, the intercalated layers composed of low water-soluble ADE molecules do not confer enough solubility improvement themselves in comparison with other multicomponent DIC solids.
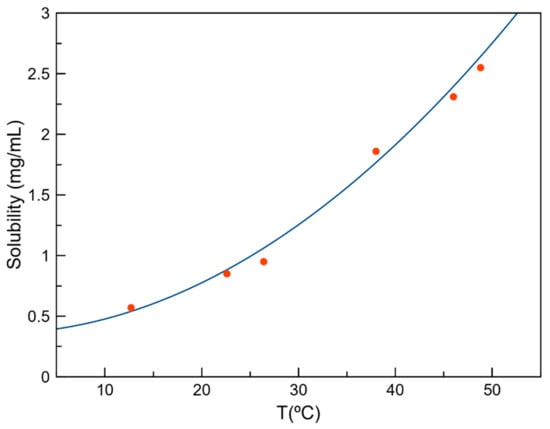
Figure 9.
Solubility curve for DIC–ADE in water as a function of concentration and temperature.
A potential risk observed in the use of multicomponent systems is their tendency to experience unexpected dissociation in contact with water or with high relative humidity (RH), which leads to a return to the respective free API and coformer [54,55] and denies the solubility advantage achieved by multicomponent solid formation. To rationalize the dissociation observed for DIC–CYT and DIC–ICT solids, crystal morphologies of the three reported multicomponent DIC forms were computed using the Bravais–Friedel–Donnay–Harker (BFDH) method included in the visualization software package Mercury [27]. As described previously, all the crystal structures consist of alternate layers of type DIC···coformer···DIC··· and these supramolecular arrangements seem responsible for the enhanced properties. Figure 10 shows the predicted morphologies for the reported multicomponent solids. Notably, the facets with the largest surface, following the order: DIC–ICT (57.2%) > DIC–CYT (50%) > DIC–ADE (24.2%), contain hydrogen bond donor and acceptor groups that potentially could interact with water during the dissolution process. Water solubility depends not only on the groups exposed on the surface of a crystal but on other different factors, including density, coformer solubility, or lattice energy [56]. We cannot argue that the high polarity of the crystal surfaces could impact the solubility performance by itself, but it does affect the dissolution of the reported solids as evidenced by the rapid dissociation observed for DIC–CYT and DIC–ICT during the slurry experiments in water. Dissolution is carried out at higher rate in the DIC–CYT and DIC–ICT species. As expected, dissolution in these species is favored by extensive surface exposure. On the other hand, dissolution of the DIC–ADE phase occurs at a slower rate, evidenced by the apparent stability at 24 h.
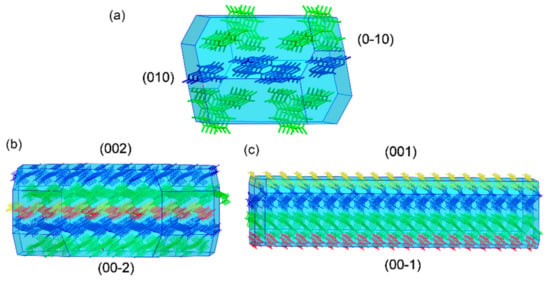
Figure 10.
BFDH-predicted morphologies of (a) DIC–ADE I (green: DIC, blue: ADE); (b) DIC–CYT (blue and green: DIC, yellow and red: CYT), and (c) DIC–ICT (DIC: blue and green: ICT: red and yellow), showing the largest faces.
4. Conclusions
In conclusion, we have described three new pharmaceutical multicomponent crystals containing DIC and nucleobases (ADE, CYT, and ICT) as coformers. Expected heterosynthons assist formation of the new solid forms, disrupting the robust acid:acid dimmer synthon observed in reported DIC polymorphs. All solids consist of alternated layered structures connected by hydrogen bonds. This supramolecular organization confers good thermal stability, and good stability under accelerated ageing conditions and seems to have an important role in the dissolution properties of the solids. Relevant insights are inferred from the BFDH calculations where CYT and ICT, containing solids, possess a large crystal surface that expose hydrogen donor and acceptor groups, which interact with the water molecules of the bulk solvent.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst12081038/s1, Figure S1. PXRD patterns of the LAG screening experiments with the coformers of Table 2; Figure S2. PXRD patterns of DIC—ADE, DIC—CYT, and DIC—ICT after neat grinding; Figure S3. PXRD patterns of DIC—ADE, DIC—CYT, and DIC—ICT after LAG in methanol using different stoichiometries; Figure S4. Experimental PXRD pattern of DIC—ADE, DIC—CYT, and DIC—ICT, compared with DIC, coformers, and the corresponding calculated powder patterns; Figure S5. TGA traces of DIC–ADE (top), DIC—CYT (middle), and DIC–ICT (bottom); Figure S6. PXRD patterns of DIC–ADE (top), DIC—CYT (middle), and DIC–ICT (bottom) with respect to the stability under accelerated ageing conditions (40 °C, 75% RH) at different time intervals; Figure S7. PXRD patterns of DIC–ADE (top), DIC—CYT (middle), and DIC–ICT (bottom) after the stability slurry assay (at 25 °C, during 24 h, in water); Figure S8. Overlapping UV spectra of diclofenac (DIC) and nucleobase coformers (ADE: adenine, CYT: cytosine, ICT: isocytosine); Figure S9. ORTEP representation showing the asymmetric unit of DIC—ADE with an atom numbering scheme (thermal ellipsoids are plotted with the 50% probability level); Figure S10. ORTEP representation showing the asymmetric unit of DIC—CYT with atom numbering scheme (thermal ellipsoids are plotted with the 50% probability level); Figure S11. ORTEP representation showing the asymmetric unit of DIC—ICT with atom numbering scheme (thermal ellipsoids are plotted with the 50% probability level); Table S1. Results of the LAG experiments between DIC and selected coformers; Table S2. Hydrogen bonds for DIC—ADE [Å and deg.]; Table S3. Hydrogen bonds for DIC—CYT [Å and deg.]; Table S4. Hydrogen bonds for DIC—ICT [Å and deg.].
Author Contributions
Conceptualization and methodology, D.C.-L.; formal analysis and investigation, C.A.-P., F.J.A.-M., H.M.B.-R., J.N.-G., A.D.-M.; writing—original draft preparation, D.C.-L., F.J.A.-M.; writing—review and editing, D.C.-L.; funding acquisition, A.D.-M. and D.C.-L.; supervision, D.C.-L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Spanish Agencia Estatal de Investigación of the Ministerio de Ciencia, Innovación y Universidades (MICIU) and co-funded with FEDER, UE, project no. PGC2018-102047-B-I00 (MCIU/AEI/FEDER, UE) and project B-FQM-478-UGR20 (FEDER-Universidad de Granada, Spain).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
F.J.A.-M. acknowledges an FPI grant (ref. PRE2019-088832).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Davies, N.M.; Andersen, K.E. Clinical Pharmacokinetics of Diclofenac. Clin. Pharmacokinet. 2012, 33, 184–213. [Google Scholar] [CrossRef] [PubMed]
- Brogden, R.N.; Heel, R.C.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Diclofenac Sodium: A Review of Its Pharmacological Properties and Therapeutic Use in Rheumatic Diseases and Pain of Varying Origin. Drugs 2012, 20, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.J. Diclofenac: An Update on Its Mechanism of Action and Safety Profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Stuart, M.; Box, K. Chasing Equilibrium: Measuring the Intrinsic Solubility of Weak Acids and Bases. Anal. Chem. 2005, 77, 983–990. [Google Scholar] [CrossRef]
- Gomaa, S. Adverse Effects Induced by Diclofenac, Ibuprofen, and Paracetamol Toxicity on Immunological and Biochemical Parameters in Swiss Albino Mice. J. Basic Appl. Zool. 2018, 79, 5. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.J.; Steed, J.W. Pharmaceutical Cocrystals, Salts and Multicomponent Systems; Intermolecular Interactions and Property Based Design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Bolla, G.; Nangia, A. Pharmaceutical Cocrystals: Walking the Talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef]
- Báthori, N.B.; Lemmerer, A.; Venter, G.A.; Bourne, S.A.; Caira, M.R. Pharmaceutical Co-Crystals with Isonicotinamide-Vitamin B3, Clofibric Acid, and Diclofenac-and Two Isonicotinamide Hydrates. Cryst. Growth Des. 2011, 11, 75–87. [Google Scholar] [CrossRef]
- Feng, W.Q.; Wang, L.Y.; Gao, J.; Zhao, M.Y.; Li, Y.T.; Wu, Z.Y.; Yan, C.W. Solid State and Solubility Study of a Potential Anticancer Drug-Drug Molecular Salt of Diclofenac and Metformin. J. Mol. Struct. 2021, 1234, 130166. [Google Scholar] [CrossRef]
- Nugrahani, I.; Utami, D.; Ibrahim, S.; Nugraha, Y.P.; Uekusa, H. Zwitterionic Cocrystal of Diclofenac and L-Proline: Structure Determination, Solubility, Kinetics of Cocrystallization, and Stability Study. Eur. J. Pharm. Sci. 2018, 117, 168–176. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Manin, A.N.; Manin, N.G.; Kuzmina, L.G.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical Cocrystals of Diflunisal and Diclofenac with Theophylline. Mol. Pharm. 2014, 11, 3707–3715. [Google Scholar] [CrossRef]
- Goswami, P.K.; Kumar, V.; Ramanan, A. Multicomponent Solids of Diclofenac with Pyridine Based Coformers. J. Mol. Struct. 2020, 1210, 128066. [Google Scholar] [CrossRef]
- Sivakova, S.; Rowan, S.J. Nucleobases as Supramolecular Motifs. Chem. Soc. Rev. 2005, 34, 9–21. [Google Scholar] [CrossRef]
- Koch, E.S.; McKenna, K.A.; Kim, H.J.; Young, V.G.; Swift, J.A. Thymine Cocrystals Based on DNA-Inspired Binding Motifs. CrystEngComm 2017, 19, 5679–5685. [Google Scholar] [CrossRef]
- Sarai, A.; Kono, H. Protein-DNA Recognition Patterns and Predictions. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 379–398. [Google Scholar] [CrossRef]
- Xia, Y.; Qu, F.; Peng, L. Triazole Nucleoside Derivatives Bearing Aryl Functionalities on the Nucleobases Show Antiviral and Anticancer Activity. Mini-Rev. Med. Chem. 2010, 10, 806–821. [Google Scholar] [CrossRef]
- Sun, R.; Wang, L. Inhibition of Mycoplasma Pneumoniae Growth by FDA-Approved Anticancer and Antiviral Nucleoside and Nucleobase Analogs. BMC Microbiol. 2013, 13, 184. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.H. The Cambridge Structural Database: A Quarter of a Million Crystal Structures and Rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Loschen, C.; Klamt, A. Solubility Prediction, Solvate and Cocrystal Screening as Tools for Rational Crystal Engineering. J. Pharm. Pharmacol. 2015, 67, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Bruker APEX4, APEX4 V2021.1 2021; Bruker-AXS: Madison, WI, USA, 2021.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Castellari, C.; Ottani, S. Two Monoclinic Forms of Diclofenac Acid. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1997, 53, 794–797. [Google Scholar] [CrossRef]
- Horst, J.H.T.; Deij, M.A.; Cains, P.W. Discovering New Co-Crystals. Cryst. Growth Des. 2009, 9, 1531–1537. [Google Scholar] [CrossRef]
- Moser, P.; Sallmann, A.; Wiesenberg, I. Synthesis and Quantitative Structure-Activity Relationships of Diclofenac Analogues. J. Med. Chem. 1990, 33, 2358–2368. [Google Scholar] [CrossRef]
- Jaiboon, N.; Yos-In, K.; Ruangchaithaweesuk, S.; Chaichit, N.; Thutivoranath, R.; Siritaedmukul, K.; Hannongbua, S. New Orthorhombic Form of 2-[(2,6-Dichlorophenyl)Amino]Benzeneacetic Acid (Diclofenac Acid). Anal. Sci. 2001, 17, 1465–1466. [Google Scholar] [CrossRef] [Green Version]
- Hamamci Alisir, S.; Dege, N. Crystal Structure of a Mixed-Ligand Silver(I) Complex of the Non-Steroidal Anti-Inflammatory Drug Diclofenac and Pyrimidine. Acta Crystallogr. Sect. E Crystallogr. Commun. 2016, 72, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Caglar, S.; Dilek, E.; Caglar, B.; Adiguzel, E.; Temel, E.; Buyukgungor, O.; Tabak, A. New Metal Complexes with Diclofenac Containing 2-Pyridineethanol or 2-Pyridinepropanol: Synthesis, Structural, Spectroscopic, Thermal Properties, Catechol Oxidase and Carbonic Anhydrase Activities. J. Coord. Chem. 2016, 69, 3321–3335. [Google Scholar] [CrossRef]
- García-García, A.; Méndez-Arriaga, J.M.; Martín-Escolano, R.; Cepeda, J.; Gómez-Ruiz, S.; Salinas-Castillo, A.; Seco, J.M.; Sánchez-Moreno, M.; Choquesillo-Lazarte, D.; Ruiz-Muelle, A.B.; et al. In Vitro Evaluation of Leishmanicidal Properties of a New Family of Monodimensional Coordination Polymers Based on Diclofenac Ligand. Polyhedron 2020, 184, 114570. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, R.P.; Bala, R.; Kariuki, B.M. Second Sphere Coordination in Oxoanion Binding: Synthesis, Spectroscopic Characterisation and Crystal Structures of Trans-[Bis(Ethylenediamine)Dinitrocobalt(III)] Diclofenac and Chlorate. J. Mol. Struct. 2007, 826, 177–184. [Google Scholar] [CrossRef]
- Biswas, P.; Dastidar, P. Anchoring Drugs to a Zinc(II) Coordination Polymer Network: Exploiting Structural Rationale toward the Design of Metallogels for Drug-Delivery Applications. Inorg. Chem. 2021, 60, 3218–3231. [Google Scholar] [CrossRef]
- Lu, C.; Laws, K.; Eskandari, A.; Suntharalingam, K. A Reactive Oxygen Species-Generating, Cyclooxygenase-2 Inhibiting, Cancer Stem Cell-Potent Tetranuclear Copper(II) Cluster. Dalton Trans. 2017, 46, 12785–12789. [Google Scholar] [CrossRef] [Green Version]
- Sayen, S.; Carlier, A.; Tarpin, M.; Guillon, E. A Novel Copper(II) Mononuclear Complex with the Non-Steroidal Anti-Inflammatory Drug Diclofenac: Structural Characterization and Biological Activity. J. Inorg. Biochem. 2013, 120, 39–43. [Google Scholar] [CrossRef]
- Lee, S.; Kapustin, E.A.; Yaghi, O.M. Coordinative Alignment of Molecules in Chiral Metal-Organic Frameworks. Science 2016, 353, 808–811. [Google Scholar] [CrossRef]
- Paul, M.; Sarkar, K.; Deb, J.; Dastidar, P. Hand-Ground Nanoscale ZnII-Based Coordination Polymers Derived from NSAIDs: Cell Migration Inhibition of Human Breast Cancer Cells. Chem.—A Eur. J. 2017, 23, 5736–5747. [Google Scholar] [CrossRef]
- Dilek, E.; Caglar, S.; Erdogan, K.; Caglar, B.; Sahin, O. Synthesis and Characterization of Four Novel Palladium(II) and Platinum(II) Complexes with 1-(2-Aminoethyl)Pyrrolidine, Diclofenac and Mefenamic Acid: In Vitro Effect of These Complexes on Human Serum Paraoxanase1 Activity. J. Biochem. Mol. Toxicol. 2018, 32, e22043. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical Preparation of Co-Crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef]
- Karki, S.; Friščić, T.; Jones, W.; Motherwell, W.D.S. Screening for Pharmaceutical Cocrystal Hydrates via Neat and Liquid-Assisted Grinding. Mol. Pharm. 2007, 4, 347–354. [Google Scholar] [CrossRef]
- Perumalla, S.R.; Pedireddi, V.R.; Sun, C.C. Protonation of Cytosine: Cytosinium vs. Hemicytosinium Duplexes. Cryst. Growth Des. 2013, 13, 429–432. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The Salt-Cocrystal Continuum: The Influence of Crystal Structure on Ionization State. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Perlovich, G. Melting Points of One- and Two-Component Molecular Crystals as Effective Characteristics for Rational Design of Pharmaceutical Systems. Acta Cryst. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 696–706. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Chakraborty, S.; Ganguly, S.; Desiraju, G.R. Synthon Identification in Co-Crystals and Polymorphs with IR Spectroscopy. Primary Amides as a Case Study. CrystEngComm 2013, 15, 4640–4654. [Google Scholar] [CrossRef]
- Heinz, A.; Strachan, C.J.; Gordon, K.C.; Rades, T. Analysis of Solid-State Transformations of Pharmaceutical Compounds Using Vibrational Spectroscopy. J. Pharm. Pharmacol. 2009, 61, 971–988. [Google Scholar] [CrossRef]
- Glomme, A.; März, J.; Dressman, J.B. Comparison of a Miniaturized Shake-Flask Solubility Method with Automated Potentiometric Acid/Base Titrations and Calculated Solubilities. J. Pharm. Sci. 2005, 94, 1–16. [Google Scholar] [CrossRef]
- Nugrahani, I.; Kumalasari, R.A.; Auli, W.N.; Horikawa, A.; Uekusa, H. Salt Cocrystal of Diclofenac Sodium-l-Proline: Structural, Pseudopolymorphism, and Pharmaceutics Performance Study. Pharmaceutics 2020, 12, 690. [Google Scholar] [CrossRef]
- Putra, O.D.; Umeda, D.; Nugraha, Y.P.; Furuishi, T.; Nagase, H.; Fukuzawa, K.; Uekusa, H.; Yonemochi, E. Solubility Improvement of Epalrestat by Layered Structure Formation: Via Cocrystallization. CrystEngComm 2017, 19, 2614–2622. [Google Scholar] [CrossRef]
- Putra, O.D.; Umeda, D.; Fujita, E.; Haraguchi, T.; Uchida, T.; Yonemochi, E.; Uekusa, H. Solubility Improvement of Benexate through Salt Formation Using Artificial Sweetener. Pharmaceutics 2018, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Sanphui, P.; Rajput, L. Tuning Solubility and Stability of Hydrochloro-Thiazide Co-Crystals. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 81–90. [Google Scholar] [CrossRef]
- McNamara, D.P.; Childs, S.L.; Giordano, J.; Iarriccio, A.; Cassidy, J.; Shet, M.S.; Mannion, R.; O’Donnell, E.; Park, A. Use of a Glutaric Acid Cocrystal to Improve Oral Bioavailability of a Low Solubility API. Pharm. Res. 2006, 23, 1888–1897. [Google Scholar] [CrossRef]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Smolenskaya, V.N.; Stahly, B.C.; Stahly, G.P. Crystal Engineering Approach to Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef] [Green Version]
- Sathisaran, I.; Dalvi, S. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).