
Phase Transitions and Amorphization of M2AgF4 (M = Na, K, Rb) Compounds at High Pressure

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Instrumental Methods
2.3. Computational Methods
3. Results
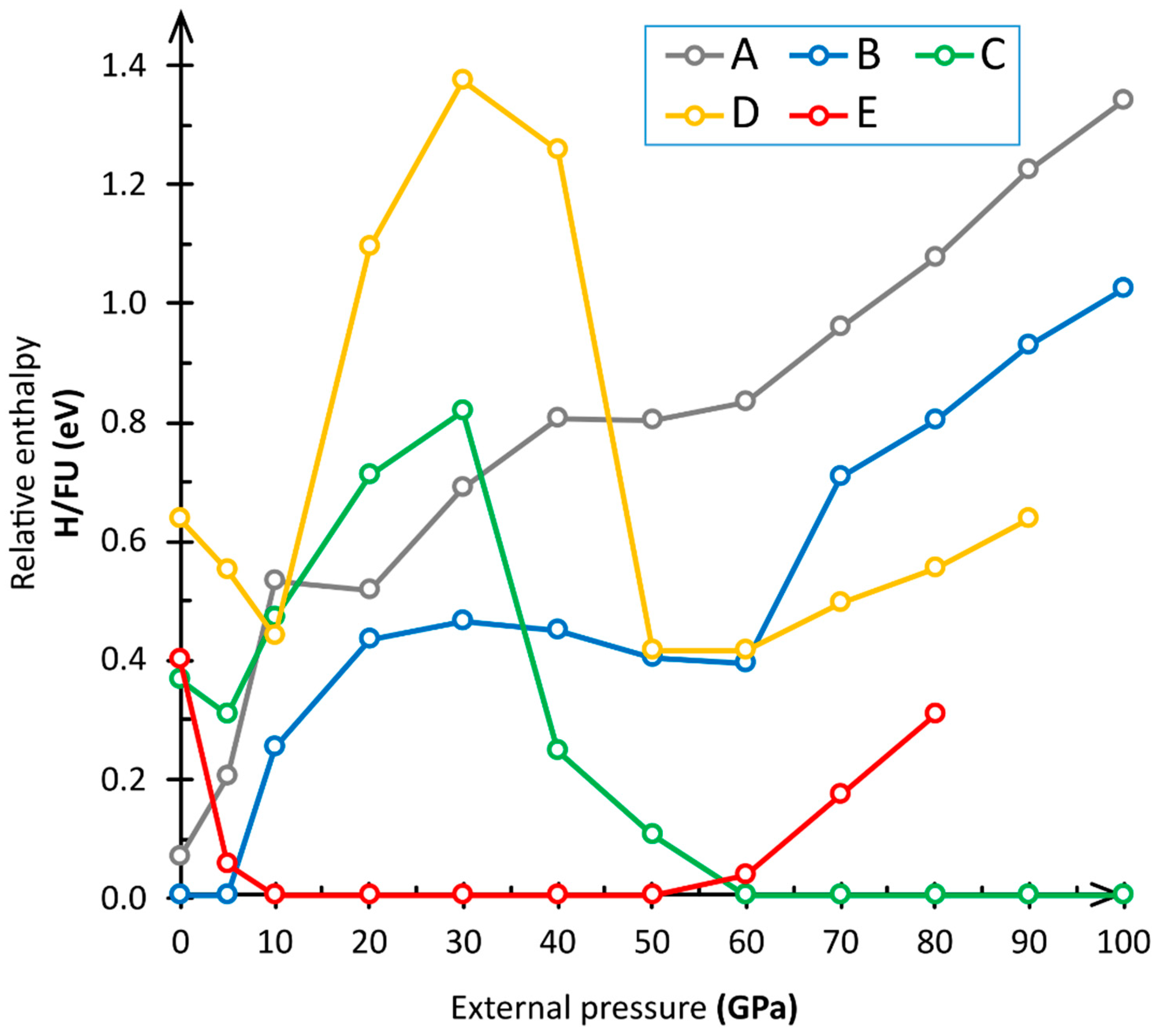
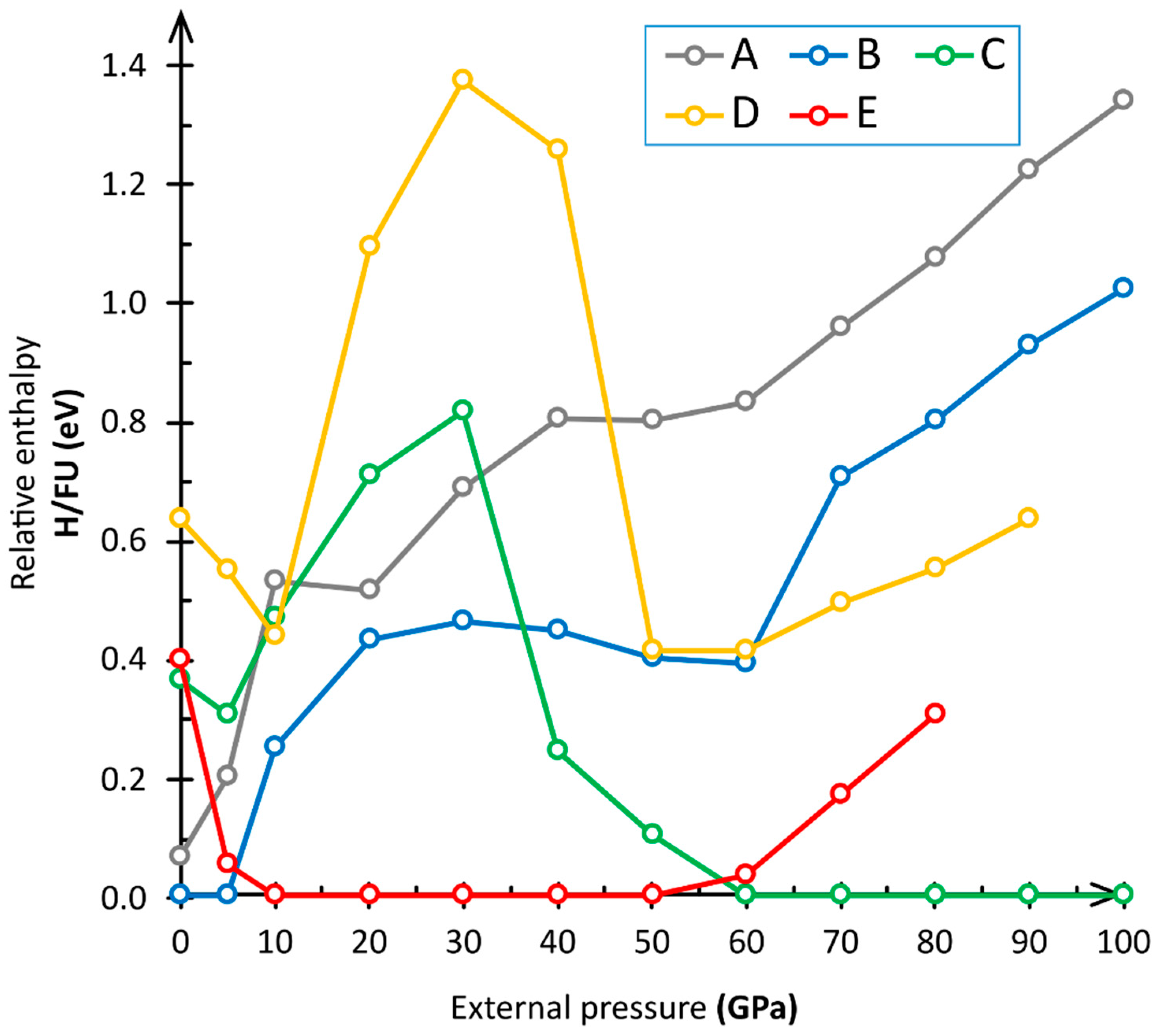
3.1. Computational Results
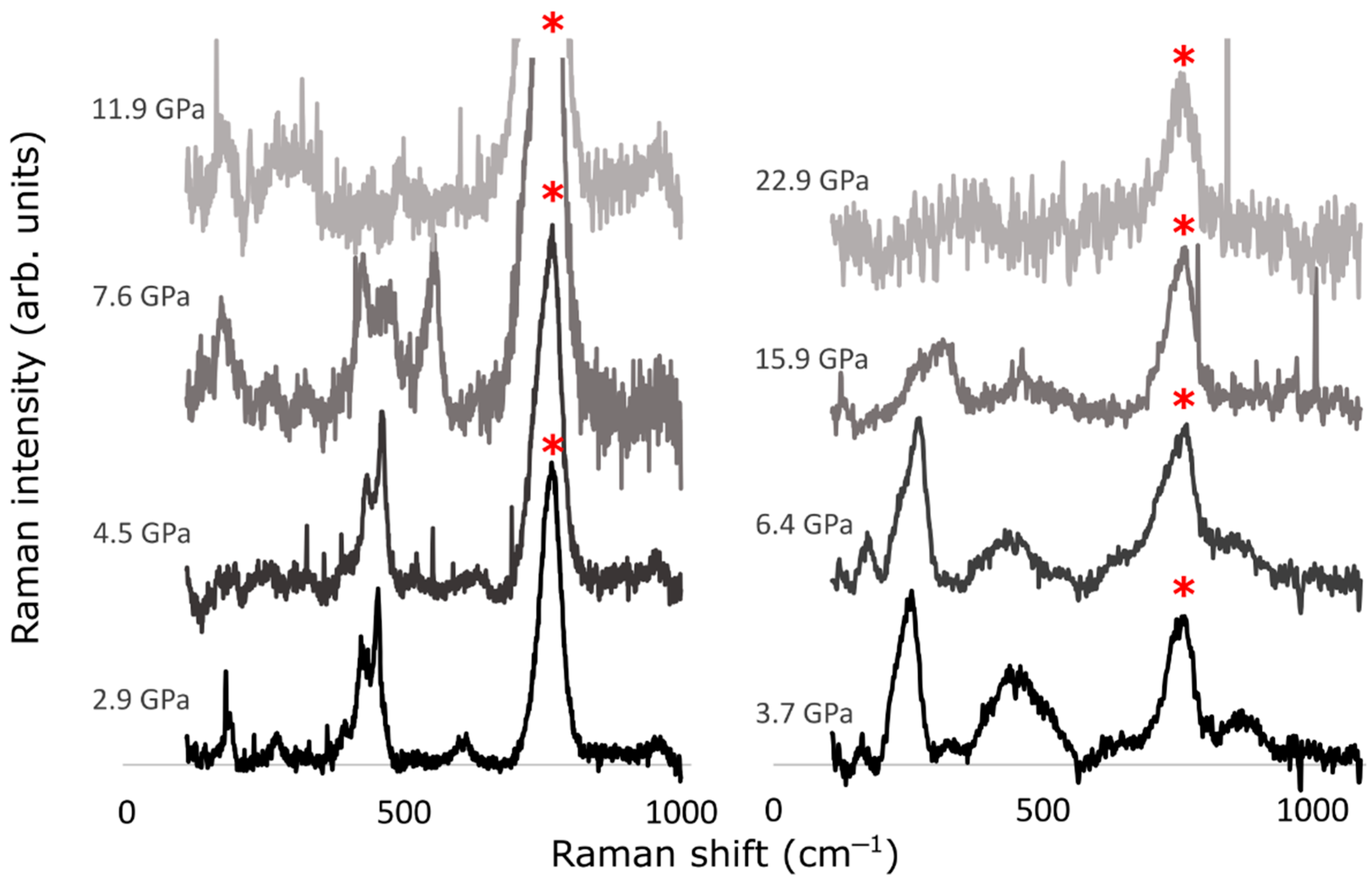
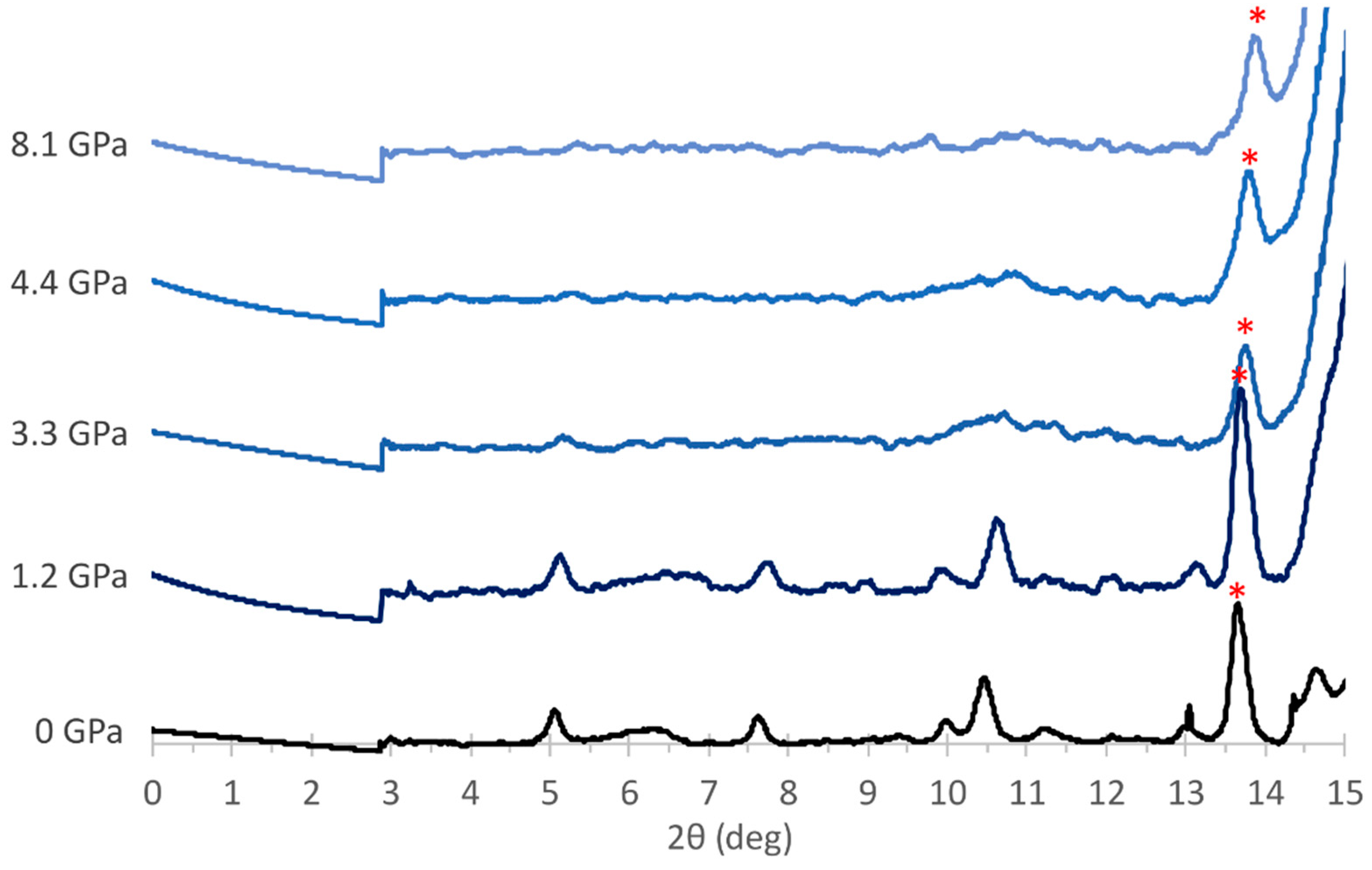
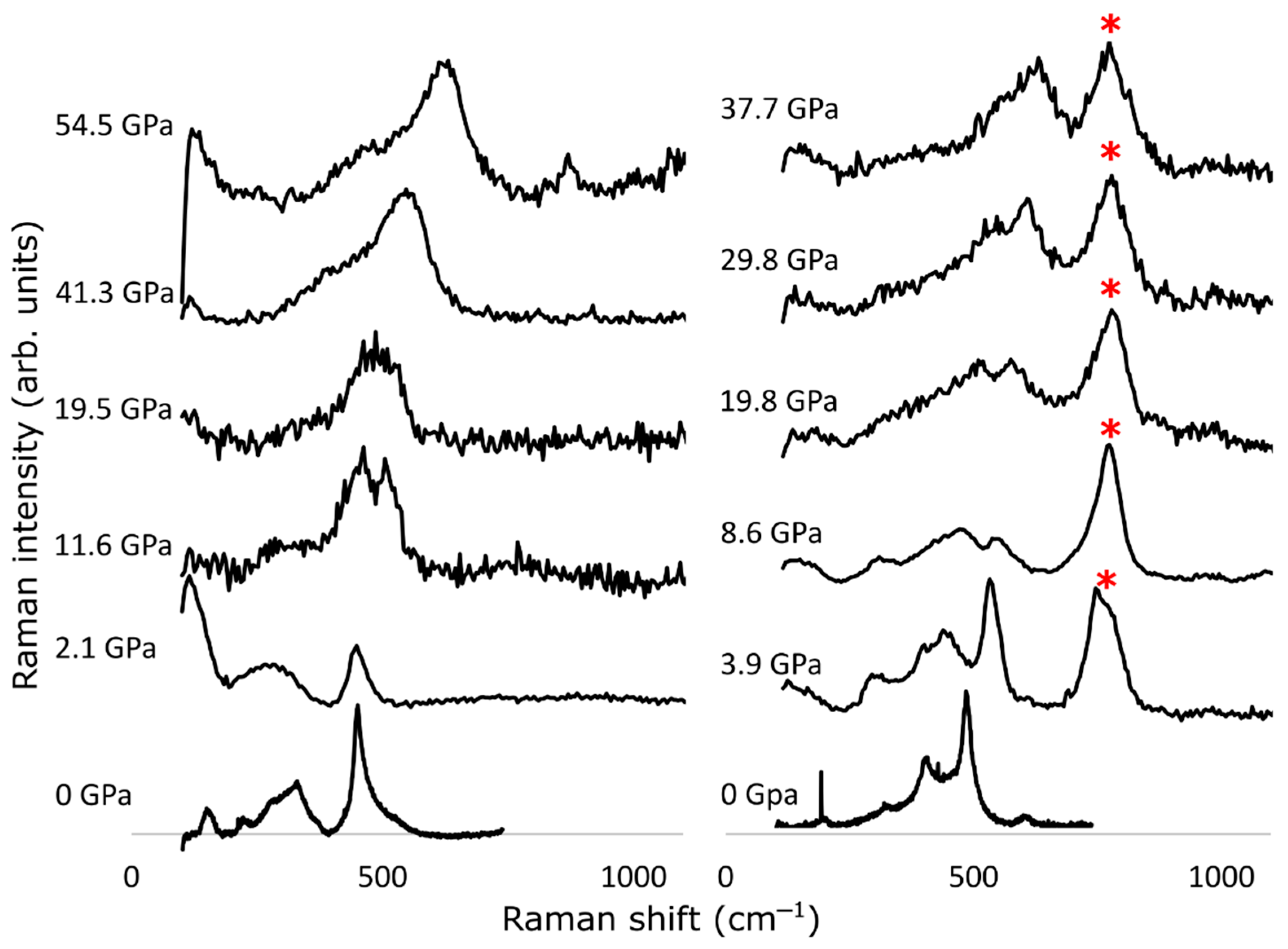
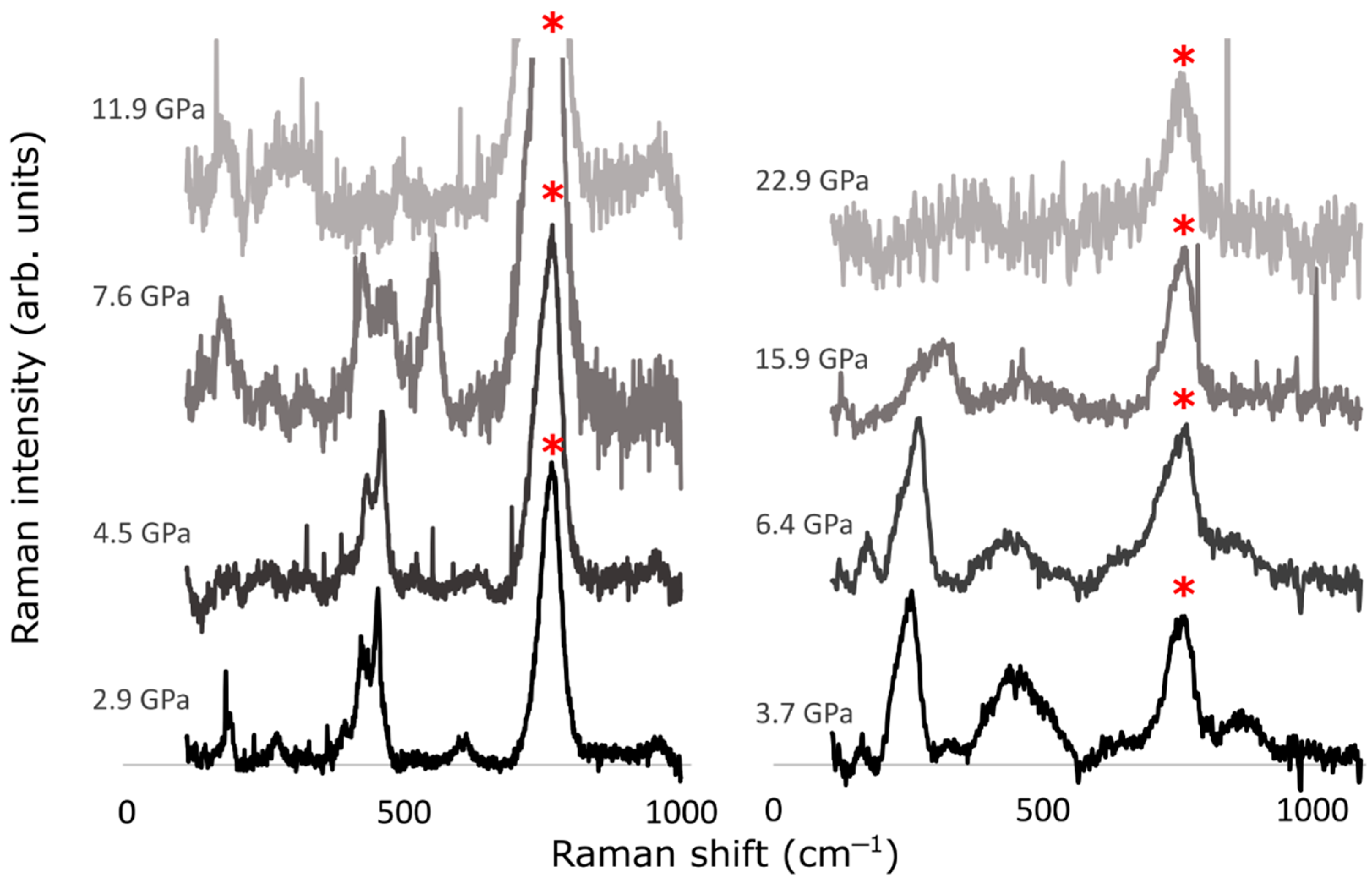
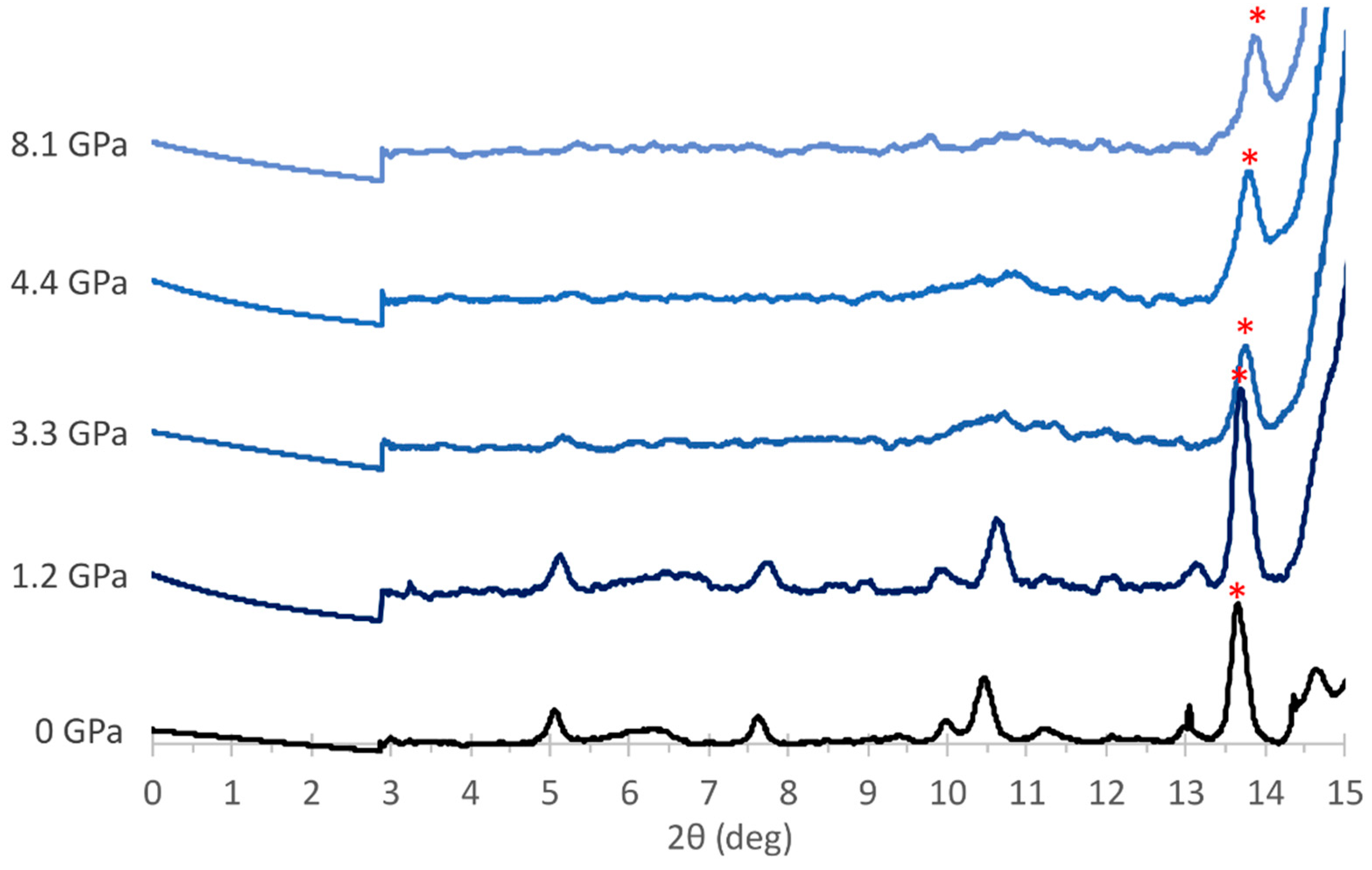
3.2. Experimental Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mazej, Z.; Goreshnik, E.; Jagličić, Z.; Gaweł, B.; Łasocha, W.; Grzybowska, D.; Jaroń, T.; Kurzydłowski, D.; Malinowski, P.; Koźminski, W.; et al. KAgF3, K2AgF4 and K3Ag2F7: Important Steps towards a Layered Antiferromagnetic Fluoroargentate(II). CrystEngComm 2009, 11, 1702–1710. [Google Scholar] [CrossRef]
- McLain, S.E.; Dolgos, M.R.; Tennant, D.A.; Turner, J.F.C.; Barnes, T.; Proffen, T.; Sales, B.C.; Bewley, R.I. Magnetic Behaviour of Layered Ag Fluorides. Nat. Mater. 2006, 5, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Grochala, W. On Similaritsies and Differences of the Electronic Structure for Cu(II)/O2-and Ag(II)/F1-Infinite Layer Compounds. Scr. Mater. 2006, 55, 811–814. [Google Scholar] [CrossRef]
- Bednorz, J.G.; Müller, K.A. Possible High Tc Superconductivity in the Ba-La-Cu-O System. Z. Phys. B Condens. Matter 1986, 64, 189–193. [Google Scholar] [CrossRef]
- Grochala, W.; Mazej, Z. Chemistry of Silver(II): A Cornucopia of Peculiarities. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2015, 373, 20140179. [Google Scholar] [CrossRef]
- Grochala, W.; Hoffmann, R. Real and Hypothetical Intermediate-Valence AgII/AgIII and AgII/AgI Fluoride Systems as Potential Superconductors. Angew. Chem. Int. Ed. 2001, 40, 2742–2781. [Google Scholar] [CrossRef]
- Mazej, Z.; Kurzydłowski, D.; Grochala, W. Unique Silver(II) Fluorides: The Emerging Electronic and Magnetic Materials. In Photonic and Electronic Properties of Fluoride Materials: Progress in Fluorine Science Series; Tressaud, A., Poeppelmeier, K.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 1, pp. 231–260. ISBN 9780128017951. [Google Scholar]
- Zhang, X.; Zhang, G.; Jia, T.; Zeng, Z.; Lin, H.Q. α-K2AgF4: Ferromagnetism Induced by the Weak Superexchange of Different eg Orbitals from the Nearest Neighbor Ag Ions. AIP Adv. 2016, 6, 055702. [Google Scholar] [CrossRef] [Green Version]
- Kurzydłowski, D.; Derzsi, M.; Mazej, Z.; Grochala, W. Crystal, Electronic, and Magnetic Structures of M2AgF4 (M = Na–Cs) Phases as Viewed from the DFT+U Method. Dalton Trans. 2016, 45, 16255–16261. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Mazej, Z.; Grochala, W. Na2AgF4: 1D Antiferromagnet with Unusually Short Ag2+⋯Ag2+ Separation. Dalton Trans. 2013, 42, 2167–2173. [Google Scholar] [CrossRef]
- Odenthal, R.H.; Paus, D.; Hoppe, R. Zur Magnetochemie Des Zweiwertigen Silbers Neue Fluoroargentate(II): Cs2AgF4, Rb2AgF4 Und K2AgF4. Z. Anorg. Allg. Chem. 1974, 407, 144–150. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Derzsi, M.; Budzianowski, A.; Jagličić, Z.; Koźmiński, W.; Mazej, Z.; Grochala, W. Polymorphism of Fluoroargentates(II): Facile Collapse of a Layered Network of α-K2AgF4 Due to the Insufficient Size of the Potassium Cation. Eur. J. Inorg. Chem. 2010, 2919–2925. [Google Scholar] [CrossRef]
- Gawraczynski, J.; Kurzydłowski, D.; Ewings, R.A.; Bandaru, S.; Gadomski, W.; Mazej, Z.; Ruani, G.; Bergenti, I.; Jaron, T.; Ozarowski, A.; et al. Silver Route to Cuprate Analogs. In Proceedings of the National Academy of Sciences of the United States of America. Proc. Natl. Acad. Sci. USA. 2019, 116, 1495–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzydłowski, D.; Mazej, Z.; Jagličić, Z.; Filinchuk, Y.; Grochala, W. Structural Transition and Unusually Strong Antiferromagnetic Superexchange Coupling in Perovskite KAgF3. Chem. Commun. 2013, 49, 6262. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Wang, Y.; Dewaele, A.; Wu, C.; Fratanduono, D.E.; Eggert, J.; Klotz, S.; Dziubek, K.F.; Loubeyre, P.; Fat’yanov, O.V.; et al. Toward an International Practical Pressure Scale: A Proposal for an IPPS Ruby Gauge (IPPS-Ruby2020). High Press. Res. 2020, 40, 299–314. [Google Scholar] [CrossRef]
- Dewaele, A.; Loubeyre, P.; Mezouar, M. Equations of State of Six Metals above 94 GPa. Phys. Rev. B 2004, 70, 094112. [Google Scholar] [CrossRef] [Green Version]
- Lonie, D.C.; Zurek, E. XtalOpt: An Open-Source Evolutionary Algorithm for Crystal Structure Prediction. Comput. Phys. Commun. 2011, 182, 372–387. [Google Scholar] [CrossRef]
- Avery, P.; Falls, Z.; Zurek, E. XtalOpt Version R11: An Open-Source Evolutionary Algorithm for Crystal Structure Prediction. Comput. Phys. Commun. 2018, 222, 418–419. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Derzsi, M.; Zurek, E.; Grochala, W. Fluorides of Silver under Large Compression. Chem. Eur. J. 2021, 27, 5536–5545. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous- Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-Functional Theory and Strong Interactions: Orbital Ordering in Mott-Hubbard Insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Ruzsinszky, A.; Csonka, G.; Vydrov, O.; Scuseria, G.; Constantin, L.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.T.; Schotte, K.-D. Structural Changes and Jahn-Teller-Effect of K2CuF4 under Pressure. J. Phys. Soc. Jpn. 2005, 74, 3221–3226. [Google Scholar] [CrossRef]
- Ishizuka, M.; Terai, M. Pressure-Induced Structural Phase Transition in the Two-Dimensional Heisenberg Ferromagnet K2CuF4. Phys. Rev. B 1998, 57, 64–67. [Google Scholar] [CrossRef]
- Kurzydłowski, D.; Jaroń, T.; Ozarowski, A.; Hill, S.; Jagličić, Z.; Filinchuk, Y.; Mazej, Z.; Grochala, W. Local and Cooperative Jahn-Teller Effect and Resultant Magnetic Properties of M2AgF4 (M = Na–Cs) Phases. Inorg. Chem. 2016, 55, 11479–11489. [Google Scholar] [CrossRef]
- McMillan, P.F. Chemistry at High Pressure. Chem. Soc. Rev. 2006, 35, 855. [Google Scholar] [CrossRef]
- Prewitt, C.T.; Downs, R.T. Chapter 9 High-Pressure Crystal Chemistry. Rev. Mineral. Geochem. 1998, 37, 283–317. [Google Scholar]
- Grochala, W.; Hoffmann, R.; Feng, J.; Ashcroft, N.W. The Chemical Imagination at Work in Very Tight Places. Angew. Chem. Int. Ed. 2007, 46, 3620–3642. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, A.; Gawraczyński, J.; Jaroń, T.; Kurzydłowski, D.; Budzianowski, A.; Mazej, Z.; Leszczyński, P.J.; Prakapenka, V.B.; Derzsi, M.; Struzhkin, V.V.; et al. High-Pressure Behavior of Silver Fluorides up to 40 GPa. Inorg. Chem. 2017, 56, 14651–14661. [Google Scholar] [CrossRef] [PubMed]
- Kroll, P. Searching Insight into the Atomistic Structure of SiCO Ceramics. J. Mater. Chem. 2010, 20, 10528–10534. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
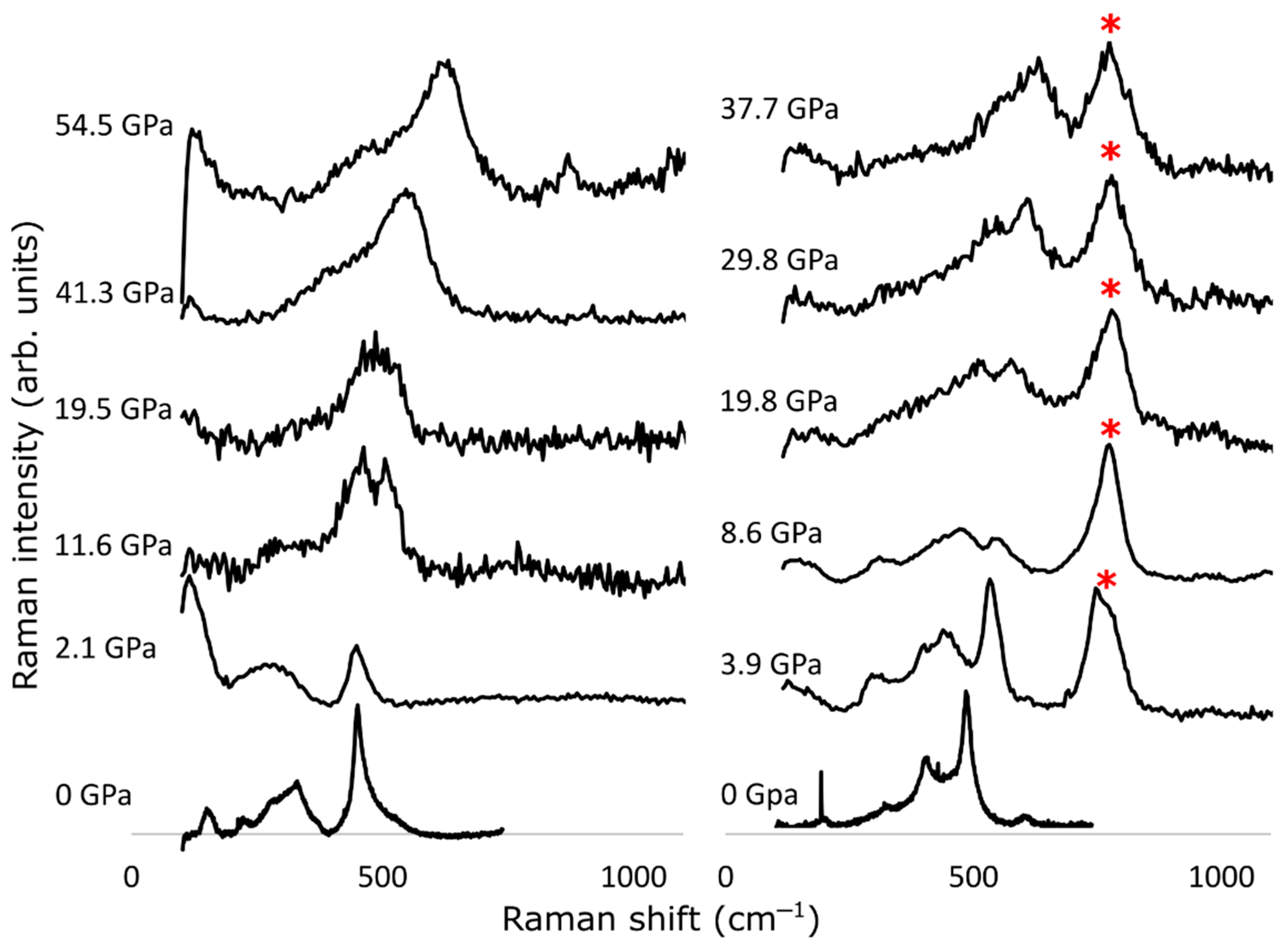{kind=link}
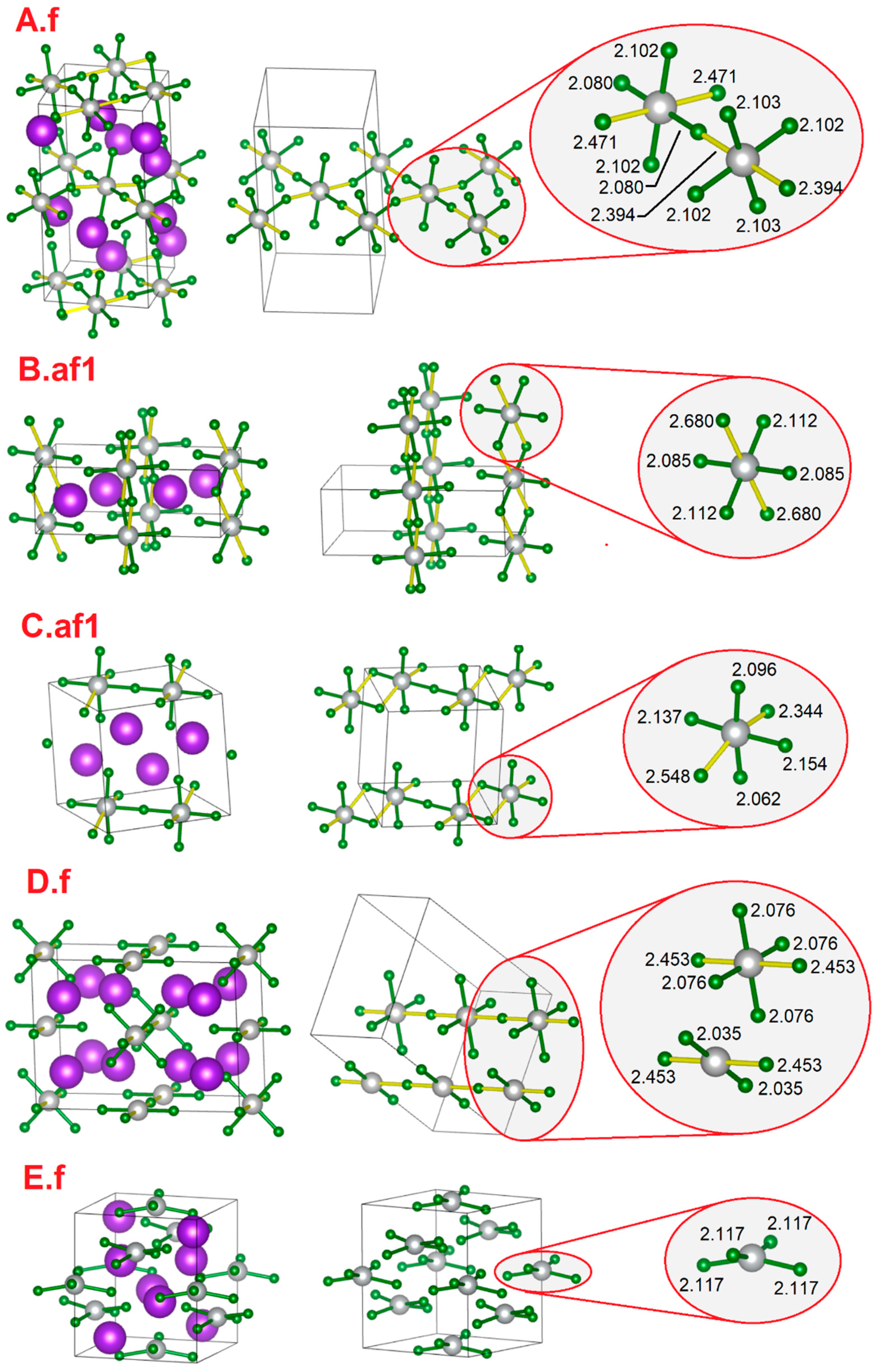
| External Pressure (GPa) | 0 | 0 | 0 | 0 | 0 | 10 | 60 |
|---|---|---|---|---|---|---|---|
| Magnetic model (SM) | A.f | B.af1 | C.af1 | D.f | E.f | E.f | C.f |
| Z | 4 | 2 & | 2 | 4 | 4 | 4 | 2 |
| Symmetry | Cmce† | P21/c | P-1 | Immm | I-42d | I-42d | P-1 |
| a [Å] | 6.33 | 3.71 | 6.03 | 11.60 | 7.30 | 6.92 | 4.64 |
| b [Å] | 6.33 | 10.22 | 6.93 | 4.91 | 7.30 | 6.92 | 5.95 |
| c [Å] | 12.48 | 6.38 | 7.08 | 8.26 | 8.20 | 7.63 | 5.95 |
| α [°] | 90 | 90 | 94.5 | 90 | 90 | 90 | 113.5 |
| β [°] | 90 | 91.9 | 112.7 | 90 | 90 | 90 | 103.7 |
| γ [°] | 90 | 90 | 114.0 | 90 | 90 | 90 | 105.6 |
| V/Z [Å3] | 125.0 | 121.0 | 119.7 | 117.4 | 109.2 | 91.3 | 66.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawraczyński, J.; Wolański, Ł.; Grzelak, A.; Mazej, Z.; Struzhkin, V.; Grochala, W. Phase Transitions and Amorphization of M2AgF4 (M = Na, K, Rb) Compounds at High Pressure. Crystals 2022, 12, 458. https://doi.org/10.3390/cryst12040458
Gawraczyński J, Wolański Ł, Grzelak A, Mazej Z, Struzhkin V, Grochala W. Phase Transitions and Amorphization of M2AgF4 (M = Na, K, Rb) Compounds at High Pressure. Crystals. 2022; 12(4):458. https://doi.org/10.3390/cryst12040458
Chicago/Turabian StyleGawraczyński, Jakub, Łukasz Wolański, Adam Grzelak, Zoran Mazej, Viktor Struzhkin, and Wojciech Grochala. 2022. "Phase Transitions and Amorphization of M2AgF4 (M = Na, K, Rb) Compounds at High Pressure" Crystals 12, no. 4: 458. https://doi.org/10.3390/cryst12040458
APA StyleGawraczyński, J., Wolański, Ł., Grzelak, A., Mazej, Z., Struzhkin, V., & Grochala, W. (2022). Phase Transitions and Amorphization of M2AgF4 (M = Na, K, Rb) Compounds at High Pressure. Crystals, 12(4), 458. https://doi.org/10.3390/cryst12040458