
Structural Characterization of Alzheimer DNA Promoter Sequences from the Amyloid Precursor Gene in the Presence of Thioflavin T and Analogs

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
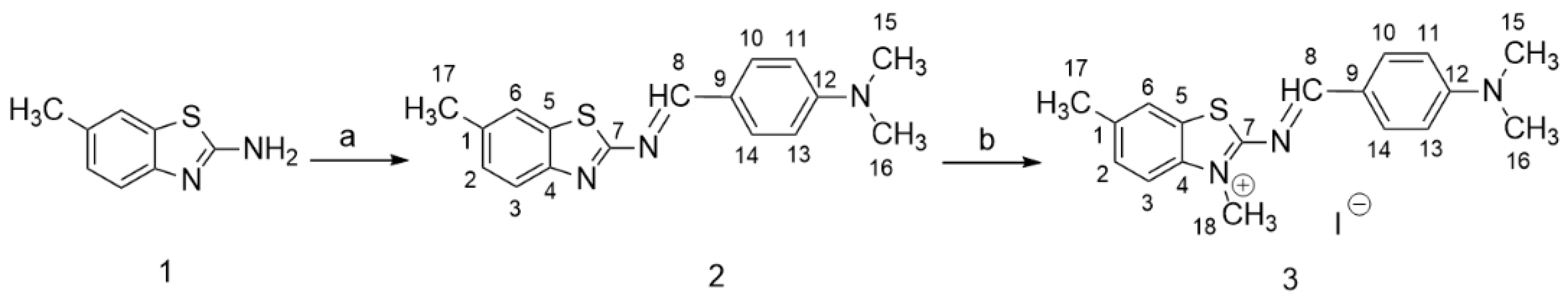
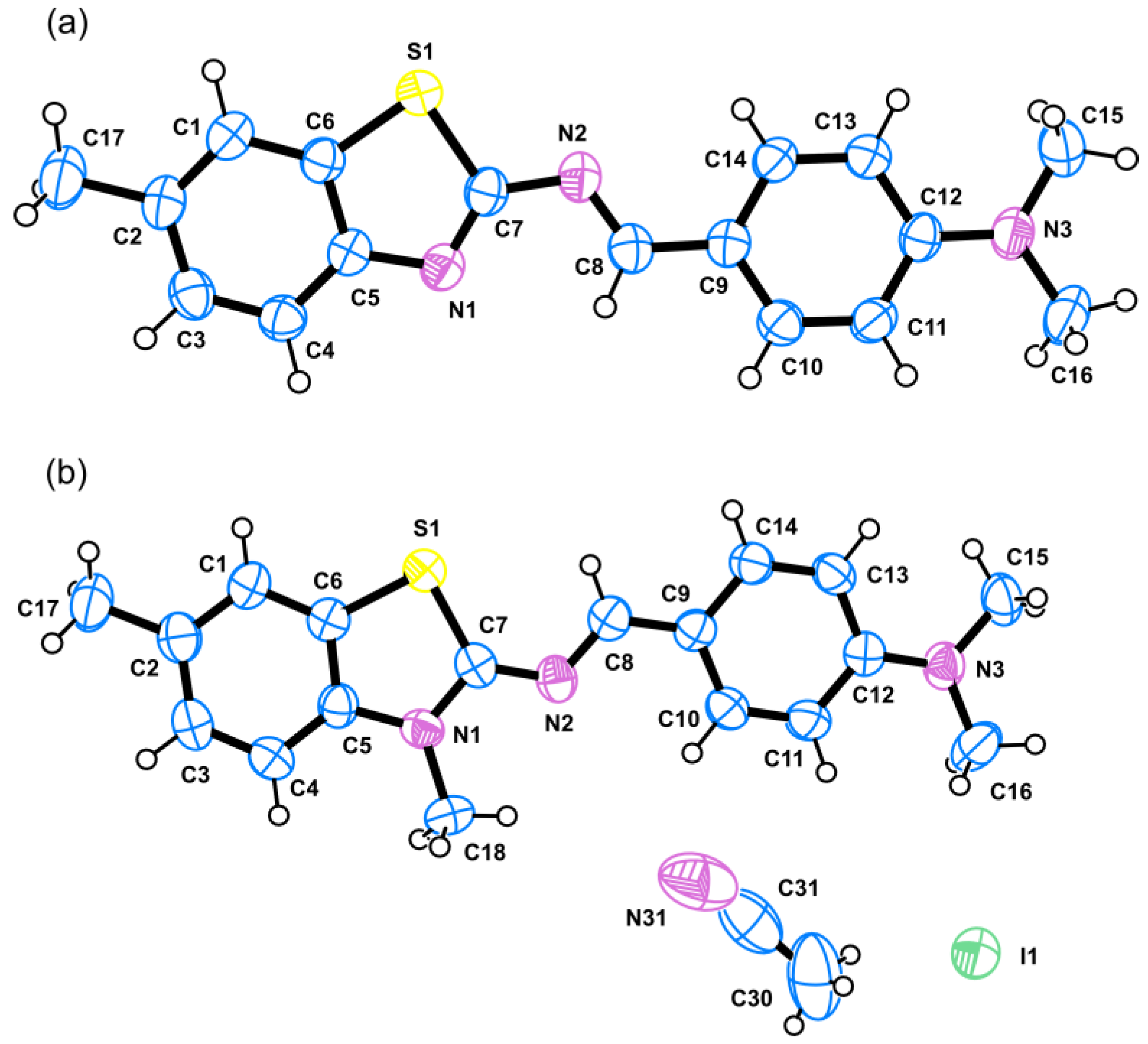
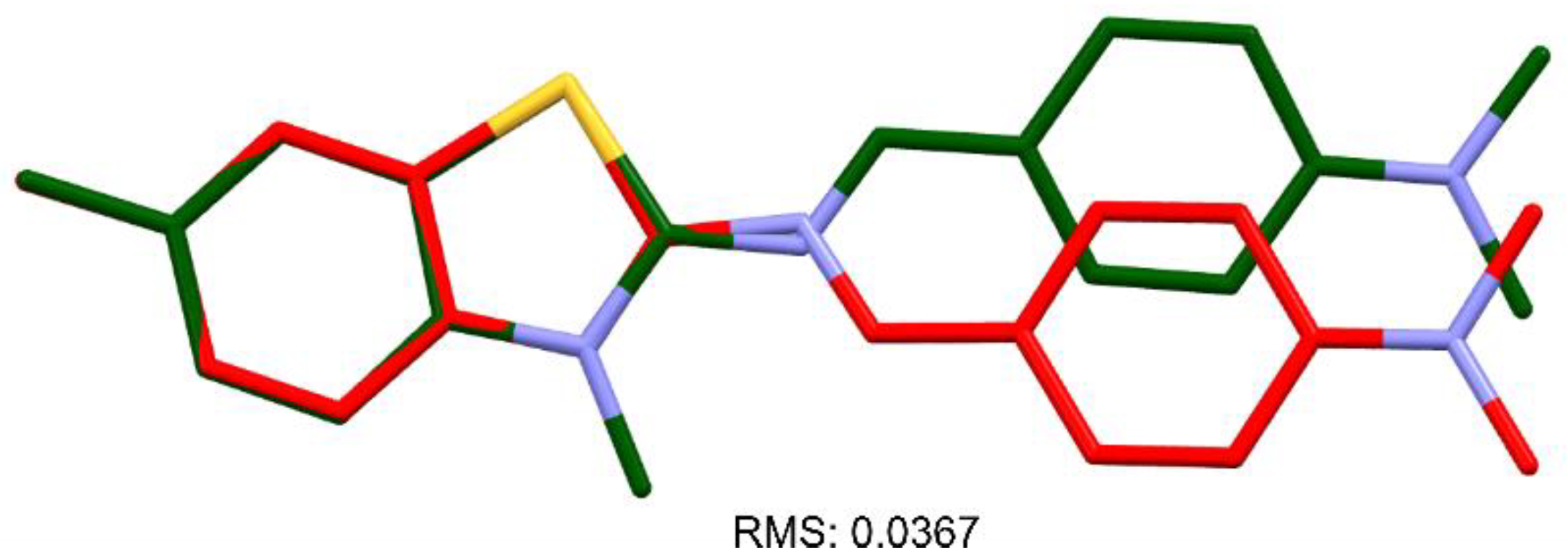
2.2. Synthesis of New Analogues of Thioflavin T Derivatives
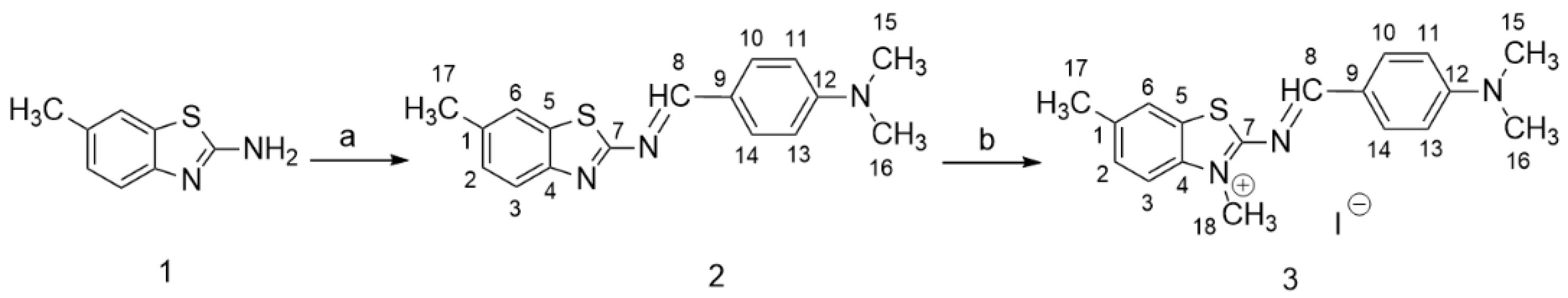
2.2.1. N,N-Dimethyl-4-(((6-methylbenzo[d]thiazol-2-yl)imino)methyl)aniline (2)
2.2.2. 2-((4-(Dimethylamino)benzylidene)amino)-3,6-dimethylbenzo[d]thiazol-3-ium iodide (3)
2.2.3. Nuclear Magnetic Resonance Spectroscopy (NMR Spectroscopy)
2.3. Powder X-ray Diffraction (PXRD)
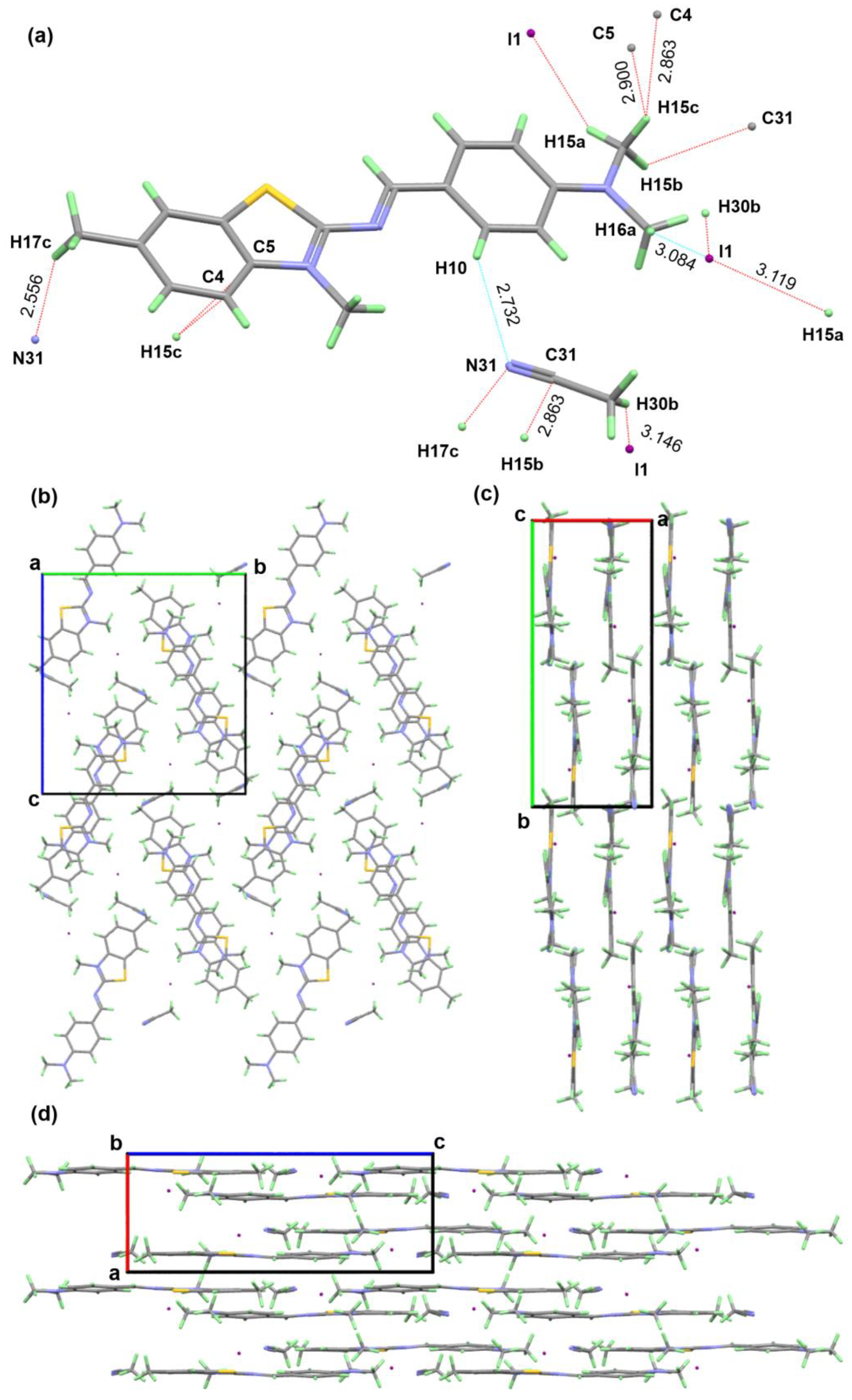
2.4. Single Crystal X-ray Diffraction (SCXRD)
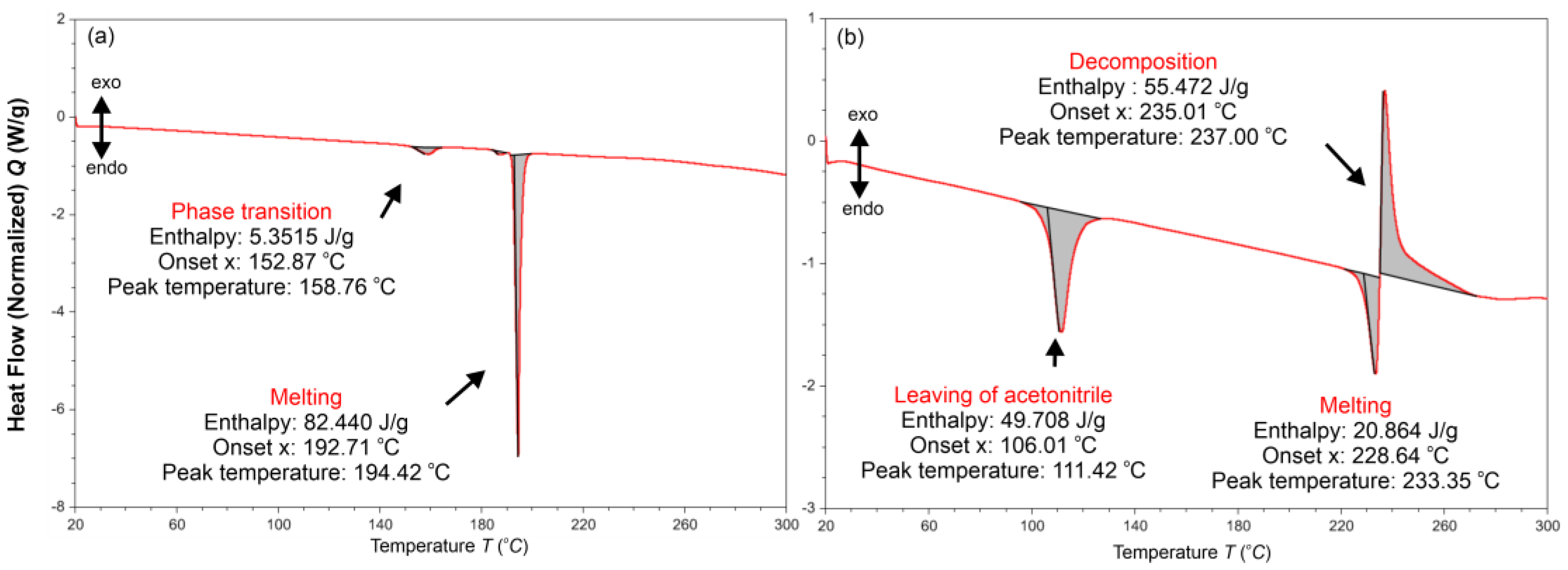
2.5. Thermal Analysis (DSC)
2.6. Fluorescent Intercalator Displacement (FID) Assay
2.7. Samples Crystallization
2.8. Macromolecules Data Collection and Processing
2.9. Macromolecules Structure Solution and Refinement
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The early events that initiate β-amyloid aggregation in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef]
- Stanciu, G.; Luca, A.; Rusu, R.; Bild, V.; Beschea Chiriac, S.; Solcan, C.; Bild, W.; Ababei, D. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, P.; Patel, B.; Makwana, V.; Jadhav, H.R.; Kiefel, M.; Davey, A.; Reekie, T.A.; Rudrawar, S.; Kassiou, M. Peptides, peptidomimetics, and carbohydrate–peptide conjugates as amyloidogenic aggregation inhibitors for Alzheimer’s disease. ACS Chem. Neurosci. 2018, 9, 1530–1551. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondareff, W.; Mountjoy, C.; Roth, M.; Hauser, D. Neurofibrillary degeneration and neuronal loss in Alzheimer’s disease. Neurobiol. Aging 1989, 10, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Association, A. Early signs and symptoms of Alzheimer’s. Alzheimer’s Dement. 2019, 1–88. [Google Scholar]
- Souza, L.C.d.; Sarazin, M.; Teixeira Júnior, A.L.; Caramelli, P.; Santos, A.E.d.; Dubois, B. Biomarcadores da doença de Alzheimer. Arq. Neuro-Psiquiatr. 2014, 72, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Maezawa, I.; Hong, H.S.; Liu, R.; Wu, C.Y.; Cheng, R.H.; Kung, M.P.; Kung, H.F.; Lam, K.S.; Oddo, S.; LaFerla, F.M. Congo red and thioflavin-T analogs detect Aβ oligomers. J. Neurochem. 2008, 104, 457–468. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Bocchetta, M.; Chételat, G.; Rabinovici, G.D.; De Leon, M.J.; Kaye, J.; Reiman, E.M.; Scheltens, P.; Barkhof, F.; Black, S.E. Imaging markers for Alzheimer disease: Which vs how. Neurology 2013, 81, 487–500. [Google Scholar] [CrossRef]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils—Current status. J. Chem. Biol. 2010, 3, 1–18. [Google Scholar] [CrossRef]
- Jamali, M.; Mohajer, S.; Sheikhlary, S.; Ara, M.H.M. Z-scan optical method complements the Thioflavin T assay for investigation of anti-Alzheimer’s impact of polyphenols. Photodiagnosis Photodyn. Ther. 2022, 39, 102914. [Google Scholar] [CrossRef]
- Biancardi, A.; Biver, T.; Mennucci, B. Fluorescent dyes in the context of DNA-binding: The case of Thioflavin T. Int. J. Quantum Chem. 2017, 117, e25349. [Google Scholar] [CrossRef]
- Wang, H.; Peng, P.; Liu, S.; Li, T. Thioflavin T behaves as an efficient fluorescent ligand for label-free ATP aptasensor. Anal. Bioanal. Chem. 2016, 408, 7927–7934. [Google Scholar] [CrossRef]
- Gu, P.; Lu, Y.; Li, S.; Ma, C. A Label-Free Fluorescence Aptasensor Based on G-Quadruplex/Thioflavin T Complex for the Detection of Trypsin. Molecules 2022, 27, 6093. [Google Scholar] [CrossRef]
- Liu, L.; Shao, Y.; Peng, J.; Liu, H.; Zhang, L. Selective recognition of ds-DNA cavities by a molecular rotor: Switched fluorescence of thioflavin T. Mol. BioSystems 2013, 9, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine III, H. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Salbaum, J.M.; Weidemann, A.; Lemaire, H.; Masters, C.; Beyreuther, K. The promoter of Alzheimer’s disease amyloid A4 precursor gene. EMBO J. 1988, 7, 2807–2813. [Google Scholar] [CrossRef]
- Chernak, J.M. Structural features of the 5′ upstream regulatory region of the gene encoding rat amyloid precursor protein. Gene 1993, 133, 255–260. [Google Scholar] [CrossRef]
- Brohede, J.; Rinde, M.; Winblad, B.; Graff, C. A DNA methylation study of the amyloid precursor protein gene in several brain regions from patients with familial Alzheimer disease. J. Neurogenet. 2010, 24, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, W. Adenosine Triphosphate Binding Cassette Subfamily C Member 1 (ABCC1) Modulates Amyloid Precursor Protein (APP) Processing: A Potential Therapeutic Target for the Treatment of Alzheimer’s Disease. Ph.D. Thesis, Arizona State University, Tempe, AZ, USA, 2021. [Google Scholar]
- Rebecchi, I.M.M.; Rodrigues, A.C.; Arazi, S.S.; Genvigir, F.D.V.; Willrich, M.A.V.; Hirata, M.H.; Soares, S.A.; Bertolami, M.C.; Faludi, A.A.; Bernik, M.M.S. ABCB1 and ABCC1 expression in peripheral mononuclear cells is influenced by gene polymorphisms and atorvastatin treatment. Biochem. Pharmacol. 2009, 77, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Morales, S.; Guijarro, F.G.; Garcia Ruano, J.L.; Cid, M.B. A general aminocatalytic method for the synthesis of aldimines. J. Am. Chem. Soc. 2014, 136, 1082–1089. [Google Scholar] [CrossRef]
- Bruker AXS. APEX3, SAINT-Plus, XPREP; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Faruggia, L. ORTEP-3 V2. 02 for Windows; University of Glasgow: Glasgow, Scotland, 2008. [Google Scholar]
- Luft, J.R.; Detitta, G.T. HANGMAN: A macromolecular hanging-drop vapor-diffusion technique. J. Appl. Crystallogr. 1992, 25, 324–325. [Google Scholar] [CrossRef]
- Krohn, M.; Bracke, A.; Avchalumov, Y.; Schumacher, T.; Hofrichter, J.; Paarmann, K.; Fröhlich, C.; Lange, C.; Brüning, T.; von Bohlen und Halbach, O. Accumulation of murine amyloid-β mimics early Alzheimer’s disease. Brain 2015, 138, 2370–2382. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Brüning, T.; Plath, A.-S.; Alfen, F. Cerebral amyloid-β proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Jepsen, W.M.; De Both, M.; Siniard, A.L.; Ramsey, K.; Piras, I.S.; Naymik, M.; Henderson, A.; Huentelman, M.J. Adenosine triphosphate binding cassette subfamily C member 1 (ABCC1) overexpression reduces APP processing and increases alpha-versus beta-secretase activity, in vitro. Biol. Open 2021, 10, bio054627. [Google Scholar] [CrossRef]
- Karthik, S.; Thirugnanasambandam, A.; Mandal, P.K.; Gautham, N. Crystal structure of d (CCGGGGTACCCCGG) 2 at 1.4 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 32–41. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Lu, X.-J.; Olson, W.K. Web 3DNA—A web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009, 37, W240–W246. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Lin, N.; Wu, X.; Cai, Z.; Shi, J.; Tong, B.; Dong, Y. The fluorescence properties of 4′-Methoxychalcone derivates modified by substituents and investigation of lysosomal imaging. Dye. Pigment. 2022, 199, 110091. [Google Scholar] [CrossRef]
- Azum, N.; Taib, L.A.; Al Angari, Y.M.; Asiri, A.M.; Denti, M.; Zhao, W.; Usta, H.; Facchetti, A. π-Conjugated donor-acceptor small molecule thin-films on gold electrodes for reducing the metal work-function. Thin Solid Films 2016, 616, 320–327. [Google Scholar] [CrossRef]
- Suyambulingam, J.K.; Karvembu, R.; Bhuvanesh, N.S.; Enoch, I.V.M.V.; Selvakumar, P.M.; Premnath, D.; Subramanian, C.; Mayakrishnan, P.; Kim, S.-H.; Chung, I.-M. Synthesis, structure, biological/chemosensor evaluation and molecular docking studies of aminobenzothiazole Schiff bases. J. Adhes. Sci. Technol. 2020, 34, 2590–2612. [Google Scholar] [CrossRef]
- Hazra, D.K.; Mukherjee, M.; Mukherjee, A.K. Ab initio powder structure analysis and theoretical study of two thiazole derivatives. J. Mol. Struct. 2013, 1039, 153–159. [Google Scholar] [CrossRef]
- Russell, W.; Newman, C.; Williamson, D. A simple cytochemical technique for demonstration of DNA in cells infected with mycoplasmas and viruses. Nature 1975, 253, 461–462. [Google Scholar] [CrossRef]
- Waring, M. Complex formation between ethidium bromide and nucleic acids. J. Mol. Biol. 1965, 13, 269–282. [Google Scholar] [CrossRef]
- Rahman, Y.; Afrin, S.; Husain, M.A.; Sarwar, T.; Ali, A.; Tabish, M. Unravelling the interaction of pirenzepine, a gastrointestinal disorder drug, with calf thymus DNA: An in vitro and molecular modelling study. Arch. Biochem. Biophys. 2017, 625, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Sarwar, T.; Husain, M.A.; Ishqi, H.M.; Tabish, M. Studying non-covalent drug–DNA interactions. Arch. Biochem. Biophys. 2015, 576, 49–60. [Google Scholar] [CrossRef]
- Malicka, J.; Gryczynski, I.; Lakowicz, J.R. DNA hybridization assays using metal-enhanced fluorescence. Biochem. Biophys. Res. Commun. 2003, 306, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Afrin, S.; Rahman, Y.; Sarwar, T.; Husain, M.A.; Ali, A.; Tabish, M. Molecular spectroscopic and thermodynamic studies on the interaction of anti-platelet drug ticlopidine with calf thymus DNA. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 186, 66–75. [Google Scholar] [CrossRef] [PubMed]
- González-Ruiz, V.; Olives, A.I.; Martín, M.A.; Ribelles, P.; Ramos, M.T.; Menéndez, J.C. An overview of analytical techniques employed to evidence drug-DNA interactions. Applications to the design of genosensors. Biomed. Eng. Trends Res. Technol. 2011, 32, 215–219. [Google Scholar]
- Cao, Y.; He, X.-w. Studies of interaction between safranine T and double helix DNA by spectral methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1998, 54, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Olson, W.K.; Lu, X.-J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res. 2019, 47, W26–W34. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; Tanaka, Y.; Fukunaga, J.-i.; Fujiwara, K.; Chiba, M.; Iibuchi, H.; Tanaka, T.; Nakamura, Y.; Kawai, G.; Kozu, T. Solution structure of a DNA mimicking motif of an RNA aptamer against transcription factor AML1 Runt domain. J. Biochem. 2013, 154, 513–519. [Google Scholar] [CrossRef] [PubMed]

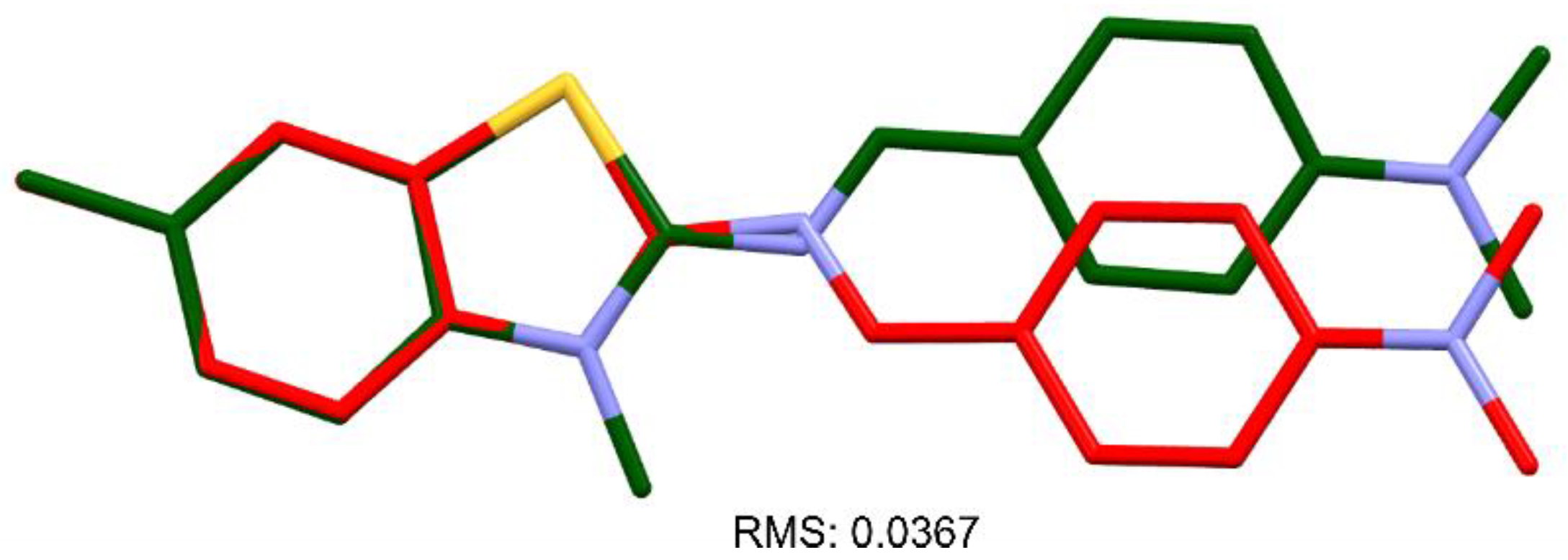

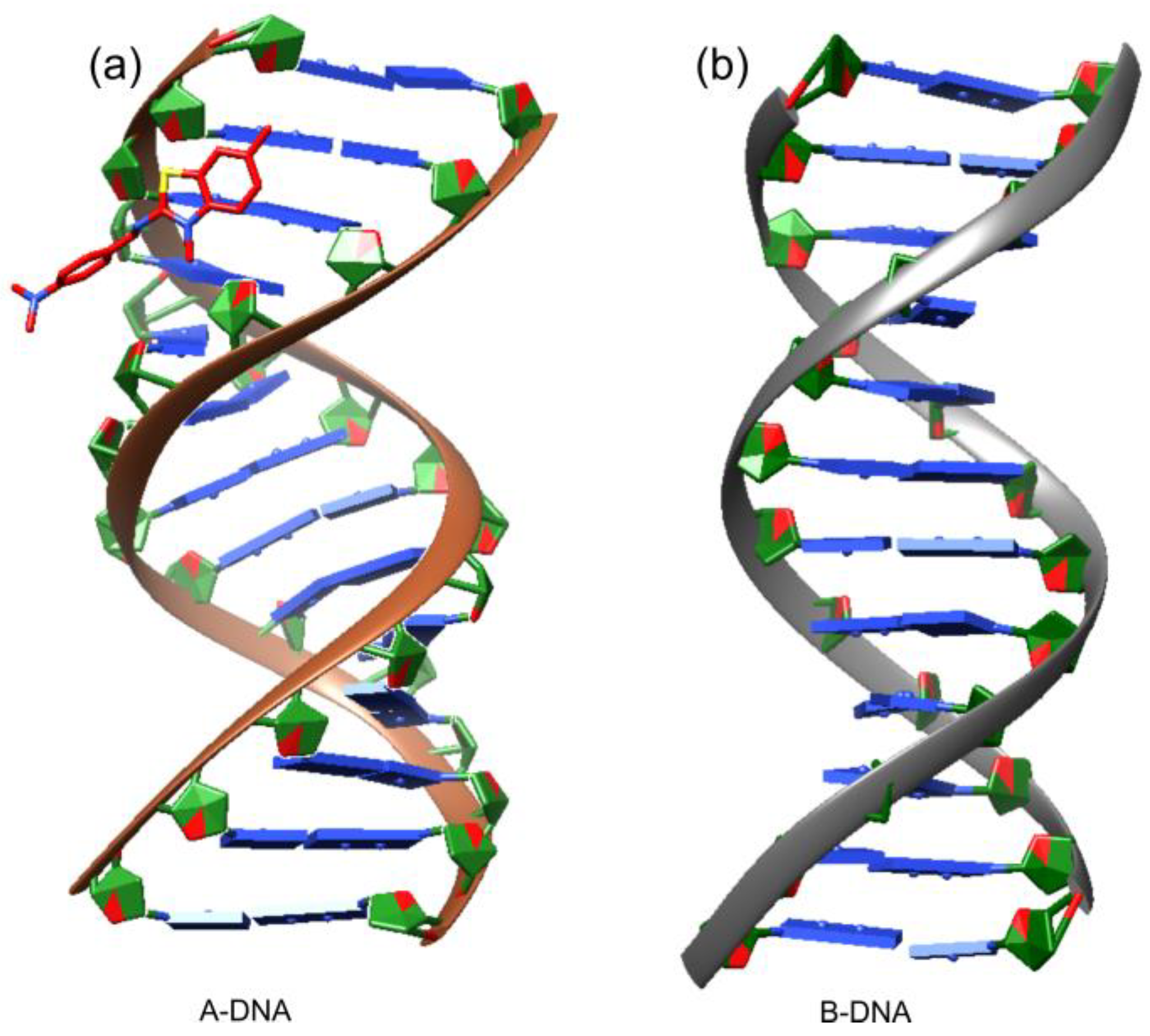
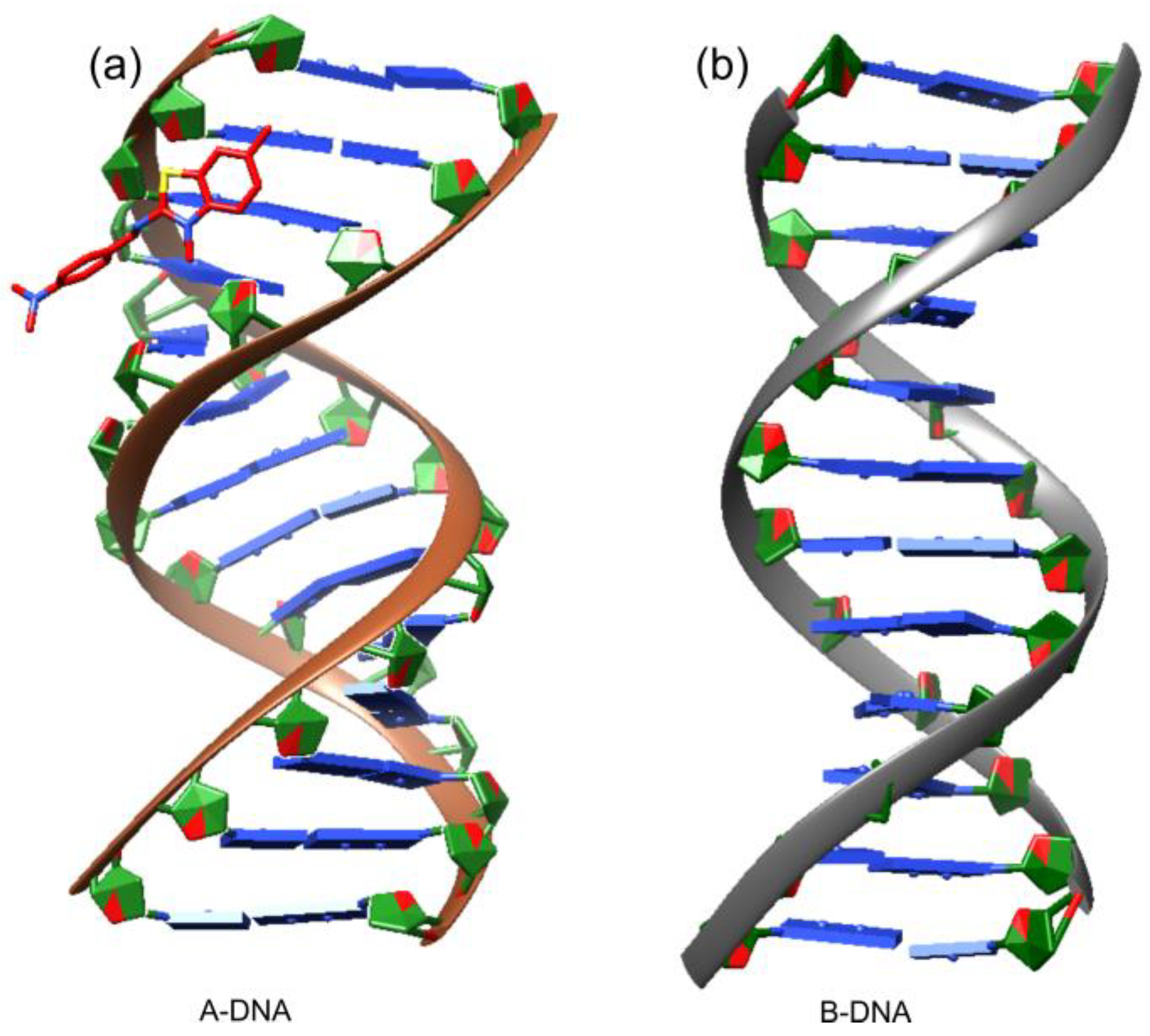
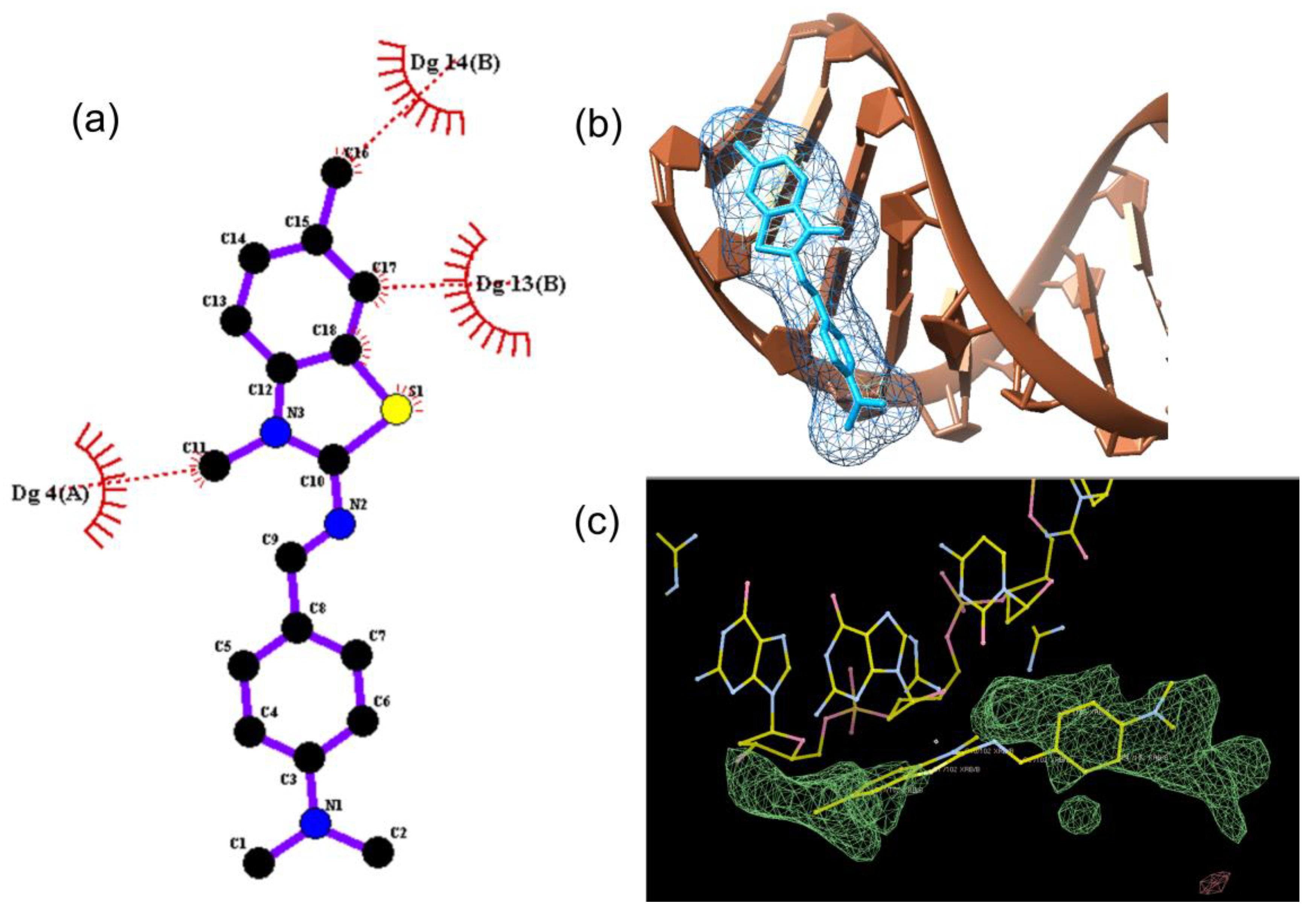
 for DNA nucleosides and with
for DNA nucleosides and with  for ligand XRB, (b) detailed visualization of XRB density generated with UCSF Chimera [42] and (c) positive omit electron density (green) of the XRB using Coot [40] and Refmac [39].
for DNA nucleosides and with for ligand XRB, (b) detailed visualization of XRB density generated with UCSF Chimera [42] and (c) positive omit electron density (green) of the XRB using Coot [40] and Refmac [39].
for ligand XRB, (b) detailed visualization of XRB density generated with UCSF Chimera [42] and (c) positive omit electron density (green) of the XRB using Coot [40] and Refmac [39].
for DNA nucleosides and with for ligand XRB, (b) detailed visualization of XRB density generated with UCSF Chimera [42] and (c) positive omit electron density (green) of the XRB using Coot [40] and Refmac [39].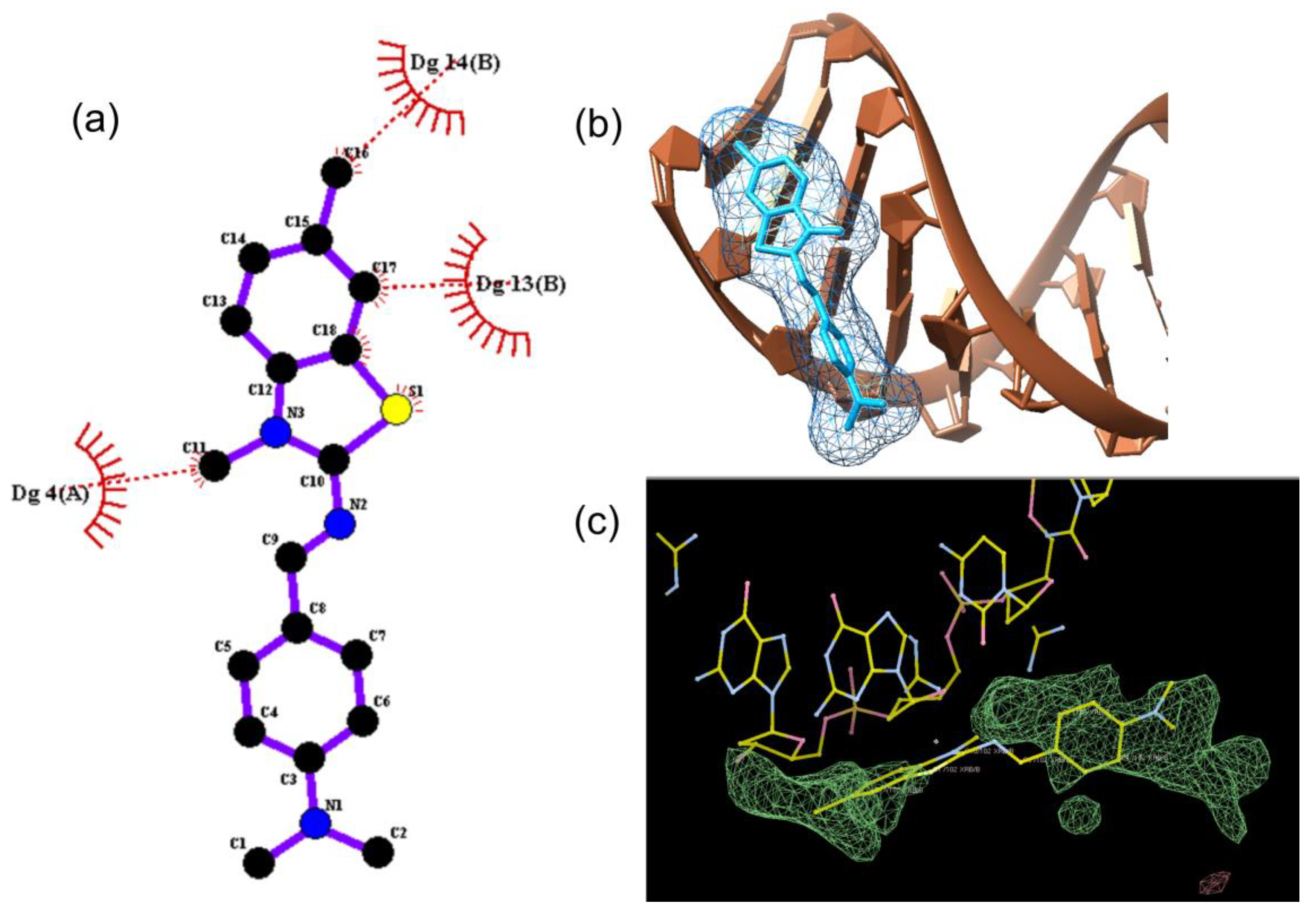

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 2 | 3 |
|---|---|---|
| Empirical formula | C17H17N3S | C20H23IN4S |
| Formula weight | 295.39 | 478.38 |
| Temperature | 290 K | 290 K |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21/c | P212121 |
| a/Å | 16.1220 (7) | 7.0148 (9) |
| b/Å | 6.1860 (3) | 16.778 (2) |
| c/Å | 16.5215 (7) | 18.144 (2) |
| α/° | 90 | 90 |
| β/° | 114.5490 (10) | 90 |
| γ/° | 90 | 90 |
| Volume/Å3 | 1498.76 (12) | 2135.5 (5) |
| Z | 4 | 4 |
| ρcalcg/cm3 | 1.309 | 1.488 |
| μ/mm−1 | 0.213 | 1.608 |
| F(000) | 624.0 | 960.0 |
| Crystal size/mm3 | 0.25 × 0.25 × 0.2 | 0.3 × 0.05 × 0.05 |
| Radiation | MoKα λ = 0.71073 | MoKα λ = 0.71073 |
| 2Θ range for data collection/° | 5.556 to 52.742 | 4.49 to 52.736 |
| Reflections collected/independent | 40281/3028 | 76891/4377 |
| Rint/Rsigma | 0.0372/0.0160 | 0.1356/0.0459 |
| Data/restraints/parameters | 3028/0/225 | 4377/0/240 |
| Goodness-of-fit on F2 | 1.104 | 1.071 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0490 wR2 = 0.1375 | R1 = 0.0391 wR2 = 0.0748 |
| Final R indexes [all data] | R1 = 0.0529 wR2 = 0.1401 | R1 = 0.0753 wR2 = 0.0885 |
| Largest diff. peak/hole/e Å−3 | 0.30/−0.21 | 0.28/−0.59 |
| CCDC number | 2211388 | 2198995 |
| PDB Code | 8ASK | 8ASH |
|---|---|---|
| space group | H3 | P41212 |
| cell dimensions | ||
| a, b, c, Å | 64.49, 64.49, 46.19 | 41.76, 41.76, 88.96 |
| α, β, γ, º | 90, 90, 120 | 90, 90, 90 |
| independent molecules | 2 | 2 |
| diffraction data | ||
| wavelength, Å | 0.99 | 0.99 |
| resolution, Å | 2.96 | 1.84 |
| reflections | 2057 | 7383 |
| completeness, % | 96.95 | 98.8 |
| I/σ(I) | 50.43 | 50.8 |
| redundancy | 10.4 (8.5) | 9.2 (8.9) |
| Rmerge % | 4.24 (23) | 4.9 (29) |
| Refinement | ||
| reflections used | 1399 | 6934 |
| resolution, Å | 2.955 | 1.84 |
| R (Rfree) % | 12.5 (23.0) | 14.1 (19.3) |
| no. of atoms | 492 | 590 |
| DNA | 492 | 568 |
| XRB | n/a | 22 |
| average B fatcor, Å2 | 91 | 43 |
| r.m.s.d. | ||
| bond lengths, Å | 0.006 | 0.014 |
| bond angles, ∘ | 1.196 | 1.730 |
| Pair | Shear | Stretch | Stagger | Buckle | Propeller | Opening | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8ASH | 5WV7 | 8ASH | 5WV7 | 8ASH | 5WV7 | 8ASH | 5WV7 | 8ASH | 5WV7 | 8ASH | 5WV7 | ||
| 1 | C-G | 0.21 | 0.24 | −0.03 | −0.07 | −0.22 | −0.13 | −8.32 | −8.49 | 6.57 | 5.97 | 1.09 | 1.72 |
| 2 | C-G | 0.11 | 0.12 | −0.13 | −0.14 | 0.04 | 0.07 | 1.4 | 1.25 | 1.38 | 1.01 | −1.18 | −0.87 |
| 3 | G_C | −0.24 | −0.25 | −0.14 | −0.12 | −0.16 | −0.17 | 1.78 | 1.6 | 0.41 | 1.01 | −2.18 | −1.65 |
| 4 | G_C | −0.25 | −0.24 | −0.13 | −0.14 | −0.18 | −0.15 | −4.85 | −4.95 | −2.39 | −2.63 | −0.8 | −1.46 |
| 5 | G_C | −0.17 | −0.17 | −0.17 | −0.17 | −0.23 | −0.25 | −6.03 | −6.5 | −11.59 | −11.09 | −1.8 | −1.73 |
| 6 | G_C | −0.14 | −0.19 | −0.07 | −0.07 | −0.2 | −0.18 | −11.16 | −11.92 | −14.34 | −13.99 | 3.3 | 2.95 |
| 7 | T-A | 0.05 | 0 | −0.07 | −0.09 | 0.13 | 0.15 | 1.44 | 0.88 | 0.2 | 1.03 | 4.15 | 4.47 |
| 8 | A-T | 0.1 | 0.16 | −0.1 | −0.11 | 0.04 | 0.03 | 10.37 | 11.26 | −6.34 | −5.41 | 0.16 | 1 |
| 9 | C-G | 0.27 | 0.3 | −0.12 | −0.08 | −0.37 | −0.39 | 14.35 | 14.94 | −11.38 | −11.71 | 2.27 | 2.33 |
| 10 | C-G | 0.22 | 0.21 | −0.19 | −0.19 | 0.11 | 0.1 | −5.08 | −4.85 | −8.15 | −7.95 | −3.12 | −3.46 |
| 11 | C-G | 0.14 | 0.17 | −0.26 | −0.25 | −0.08 | −0.08 | 2.75 | 3.25 | −2.54 | −2.38 | −2 | −2.14 |
| 12 | C-G | 0.11 | 0.09 | −0.08 | −0.09 | −0.11 | −0.14 | 4.13 | 4.63 | −1.66 | −1.23 | −0.34 | −0.18 |
| 13 | G-C | −0.15 | −0.15 | −0.17 | −0.15 | 0 | −0.01 | −0.7 | −1.13 | −1.32 | −1.58 | −0.86 | −0.57 |
| 14 | G-C | −0.24 | −0.24 | −0.12 | −0.13 | −0.2 | −0.15 | −3.55 | −2.36 | −0.94 | −0.59 | 0.33 | −0.03 |
| Pair | Shear | Stretch | Stagger | Buckle | Propeller | Opening | |
|---|---|---|---|---|---|---|---|
| 1 | G-C | 0.22 | −0.11 | 0.06 | 0.29 | −18.04 | −3.79 |
| 2 | C-G | 0.07 | 0.05 | 0.13 | 1.64 | −6.19 | 3.2 |
| 3 | C-G | 0.14 | 0.13 | 0.32 | −9.14 | −15.53 | 4.9 |
| 4 | C-G | −0.33 | −0.2 | 0.52 | −7.57 | −12.39 | 0.76 |
| 5 | A-T | 0.01 | −0.38 | 0.17 | 5.51 | −13.33 | −3.51 |
| 6 | C-G | 0.25 | −0.12 | 0.05 | −0.79 | −15.92 | −2.19 |
| 7 | C-G | 0.32 | −0.04 | −0.15 | 2.16 | −10.89 | 1.66 |
| 8 | A-T | −0.15 | −0.12 | −0.08 | 0.21 | −11.77 | 0.75 |
| 9 | C-G | 0.11 | −0.13 | 0 | 6.54 | −14.52 | 1.39 |
| 10 | G-C | −0.02 | −0.05 | 0.31 | 8.07 | −8.33 | 1.25 |
| 11 | G-C | 0.01 | −0.28 | 0.43 | 3.41 | −7.86 | −3.06 |
| 12 | C-G | −0.04 | 0.04 | 0.7 | −9.52 | −12.05 | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbirkova-Dimitrova, H.; Rusew, R.; Kuvandjiev, N.; Heroux, A.; Doukov, T.; Shivachev, B.L. Structural Characterization of Alzheimer DNA Promoter Sequences from the Amyloid Precursor Gene in the Presence of Thioflavin T and Analogs. Crystals 2022, 12, 1717. https://doi.org/10.3390/cryst12121717
Sbirkova-Dimitrova H, Rusew R, Kuvandjiev N, Heroux A, Doukov T, Shivachev BL. Structural Characterization of Alzheimer DNA Promoter Sequences from the Amyloid Precursor Gene in the Presence of Thioflavin T and Analogs. Crystals. 2022; 12(12):1717. https://doi.org/10.3390/cryst12121717
Chicago/Turabian StyleSbirkova-Dimitrova, Hristina, Rusi Rusew, Nikola Kuvandjiev, Annie Heroux, Tzanko Doukov, and Boris L. Shivachev. 2022. "Structural Characterization of Alzheimer DNA Promoter Sequences from the Amyloid Precursor Gene in the Presence of Thioflavin T and Analogs" Crystals 12, no. 12: 1717. https://doi.org/10.3390/cryst12121717
APA StyleSbirkova-Dimitrova, H., Rusew, R., Kuvandjiev, N., Heroux, A., Doukov, T., & Shivachev, B. L. (2022). Structural Characterization of Alzheimer DNA Promoter Sequences from the Amyloid Precursor Gene in the Presence of Thioflavin T and Analogs. Crystals, 12(12), 1717. https://doi.org/10.3390/cryst12121717