
Refinement of RNA Structures Using Amber Force Fields


Abstract
:1. Introduction
2. Materials and Methods
2.1. Structures Examined
2.2. Refinements Using Phenix
2.3. Refinements Using 3D-RISM
3. Results
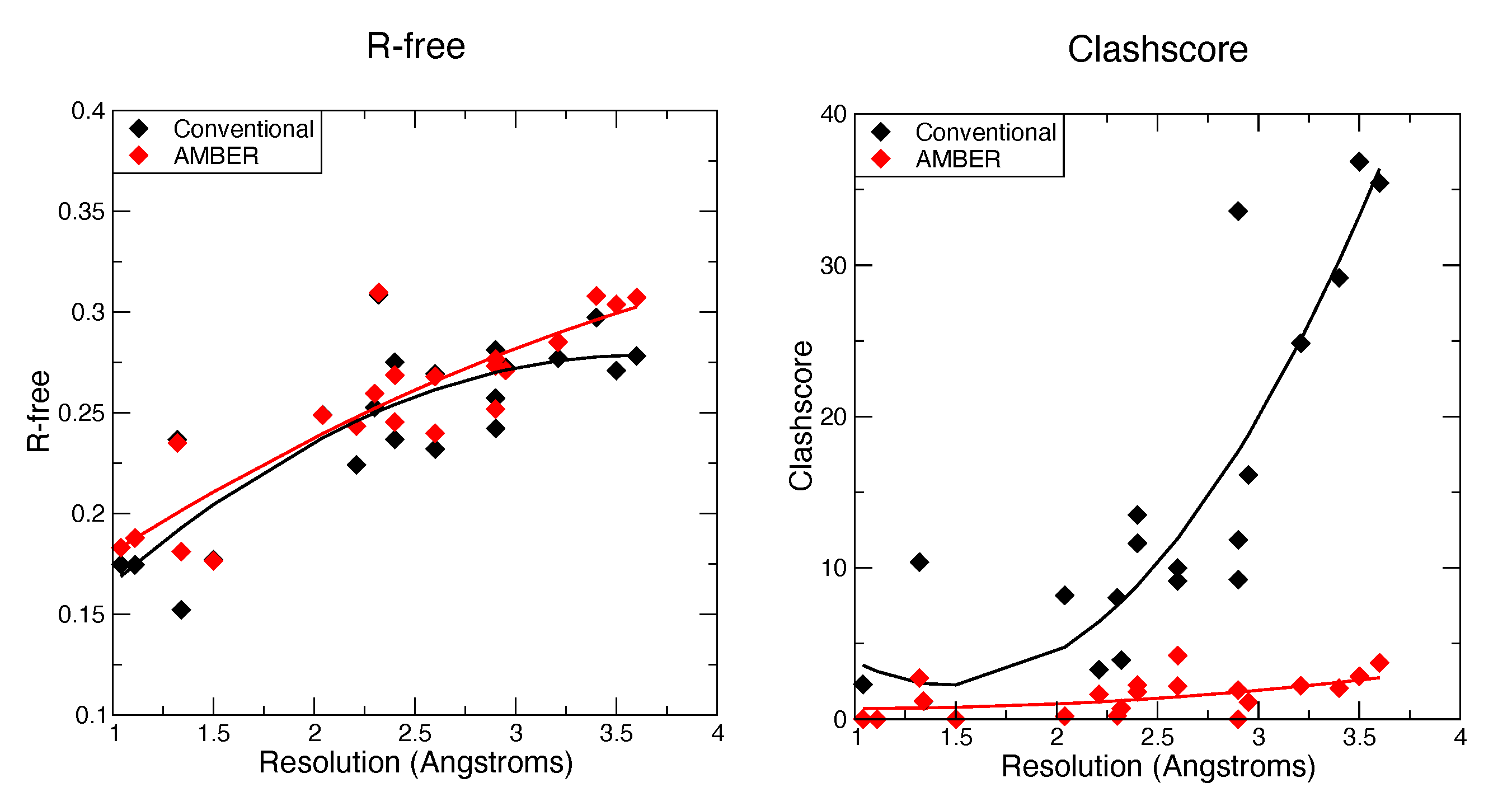
3.1. Refinements Using Standard Phenix Approaches
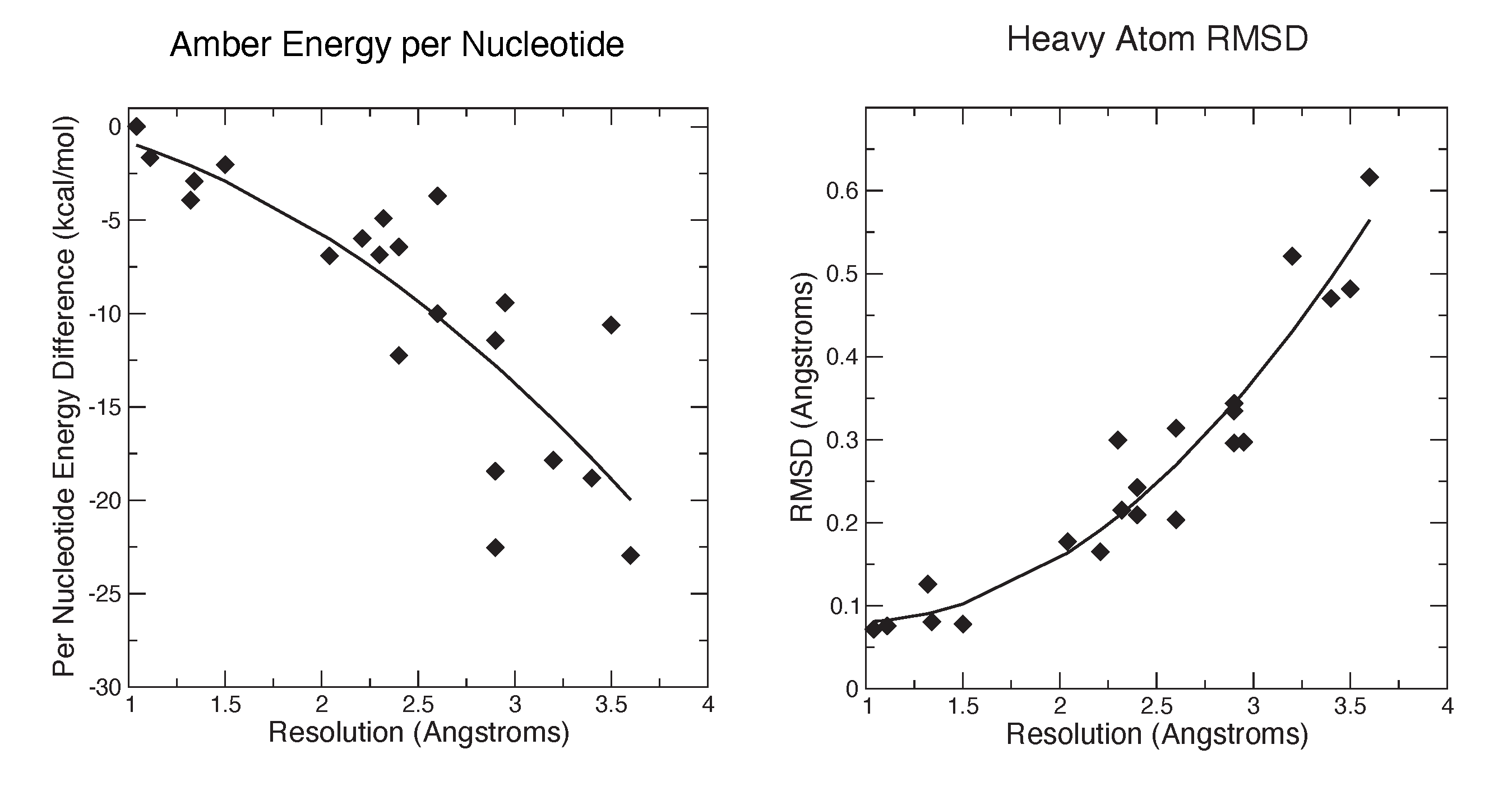
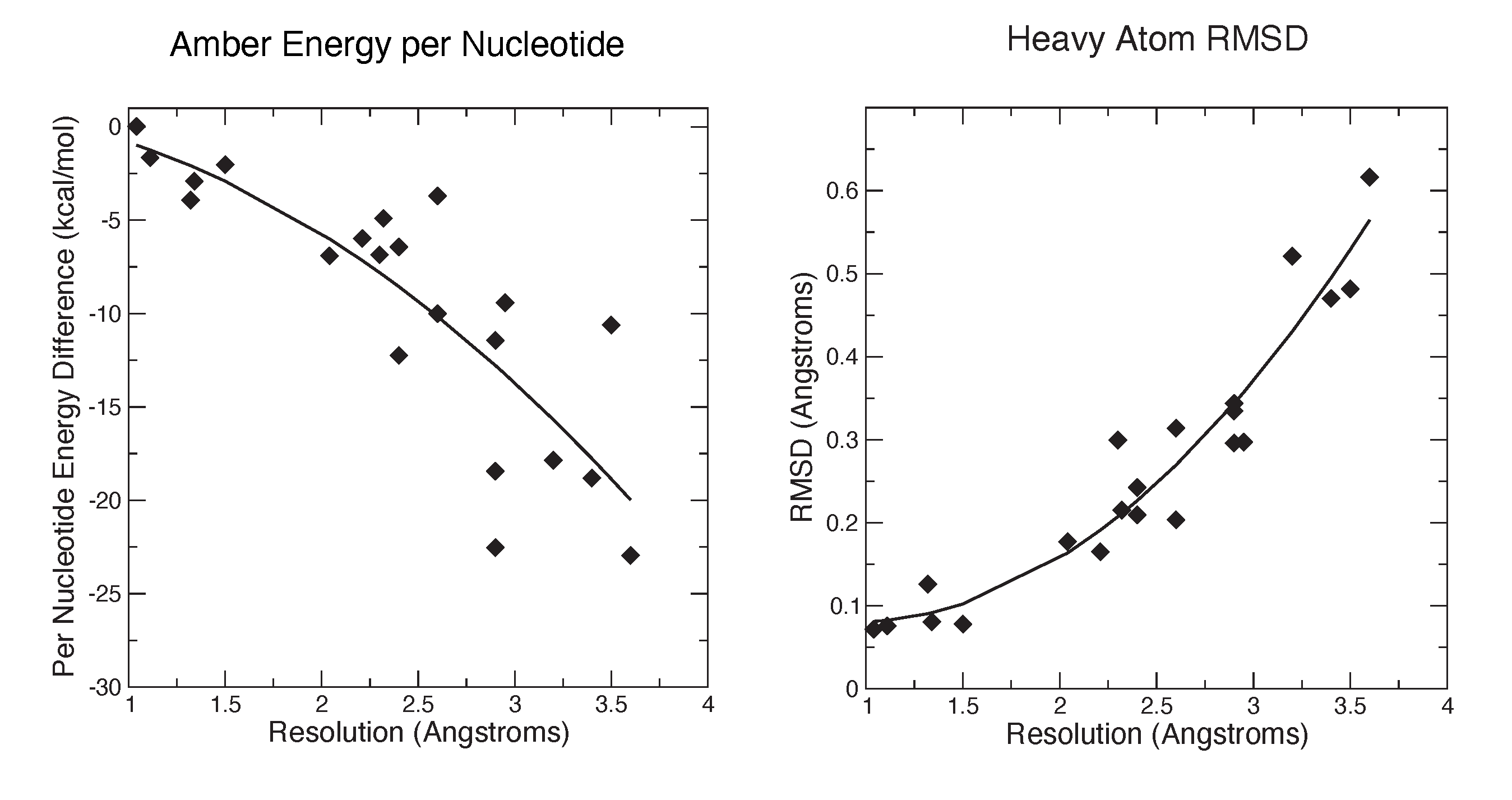
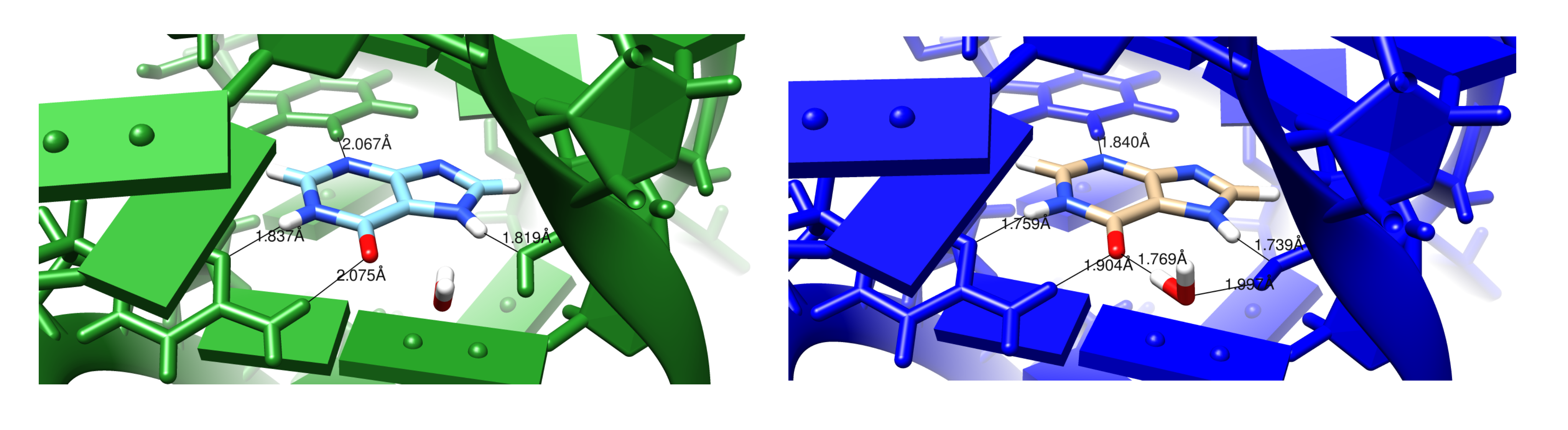
3.2. Refinements Using an Integral Equation Solvent Model
4. Discussion
- 1.
- The best relative weight of X-ray and geometric restraints is always a thorny issue in refinement. Heuristic approaches are often used, which are based on experience with particular types of geometric restraints. An alternate idea, used here, is to assume that the most likely prior distribution (in the absence of X-ray data) assumes a Boltzmann form with some effective temperature. This can be combined with a maximum likelihood estimate for the X-ray terms to establish the relative weighting. Further study of these alternatives is warranted, although in practice various relative weights often give quite similar results.
- 2.
- We have not optimized the parameters involved in integral equation models for bulk solvent. There is flexibility in choosing the solvent composition, the grid spacing, and the closure relations needed for these models [22,23,24]. Beyond this, entirely different approaches to incorporating bulk solvent electrostatics exist, including Poisson-Boltzmann and generalized Born models [31]. These alternatives are not presently implemented for compact periodic systems like macromolecular crystals, but this possibility might be worth exploring.
- 3.
- The coordinate updates used here were restricted to minimization, in our case starting from the deposited atomic model. This may well end up with atomic models that are trapped in local minima. More robust procedures, such as simulated annealing, should be explored to increase the radius of convergence from what might be an imperfect starting structure. Such extensions might make it useful to apply force-field restraints at earlier stages of refinement.
- 4.
- The calculations used here assume that the entire solvent can be described by the correlation functions arising from the 3D-RISM model. In practice, some “localized” waters or ions can often be identified from electron density maps. In such cases, a mixed model that includes some explicit waters and ions could be combined with an integral equation model for what is left. Figuring out the optimal combination will depend a lot on resolution and the nature of the solvent channels in any particular crystal.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chou, F.C.; Sripakdeevong, P.; Dibrov, S.; Hermann, T.; Das, R. Correcting pervasive errors in RNA crystallography through enumerative structure prediciton. Nat. Methods 2013, 10, 74–76. [Google Scholar] [CrossRef]
- Read, R.J.; Adams, P.D.; Arendall, W.B., III; Brunger, A.T.; Emsley, P.; Joosten, R.P.; Kleywegt, G.J.; Krissinel, E.B.; Lütteke, T.; Otwinowski, Z.; et al. A New Generation of Crystallographic Validation Tools for the Protein Data Bank. Structure 2011, 19, 1395–1412. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004, 32, W615–W619. [Google Scholar] [CrossRef]
- Keating, K.S.; Pyle, A.M. Semiautomated model building for RNA crystallography using a directed rotameric approach. Proc. Natl. Acad. Sci. USA 2010, 107, 8177–8182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty, N.; Tronrud, D.; Adams, P.; Karplus, P. A new default restraint library for the protein backbone in Phenix: A conformation-dependent geometry goes mainstream. Acta Cryst. 2016, D72, 176–179. [Google Scholar]
- Jain, S.; Richardson, D.; Richardson, J. Computational methods for RNA structure validation and improvement. Meth. Enzymol. 2015, 558, 181–212. [Google Scholar]
- Jack, A.; Levitt, M. Refinement of large structures by simultaneous minimization of energy and R factor. Acta Cryst. A 1978, 34, 931–935. [Google Scholar] [CrossRef]
- Brünger, A.; Karplus, M. Molecular dynamics simulations with experimental restraints. Acc. Chem. Res. 1991, 24, 54–61. [Google Scholar] [CrossRef]
- Schnieders, M.; Fenn, T.; Pande, V.; Brunger, A. Polarizable atomic multipole X-ray refinement: Application to peptide crystals. Acta Cryst. D 2009, 65, 952–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenn, T.; Schnieders, M.; Mastyakimov, M.; Wu, C.; Langan, P.; Pande, V.; Brunger, A. Reintroducing Electrostatics into Macromolecular Crystallographic Refinement: Application to Neutron Crystallography and DNA Hydration. Structure 2011, 19, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, N.W.; Janowski, P.A.; Swails, J.M.; Nguyen, H.; Richardson, J.S.; Case, D.A.; Adams, P.D. Improved chemistry restraints for crystallographic refinement by integrating the Amber force field into Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldararu, O.; Manzoni, F.; Oksanen, E.; Logan, D.; Ryde, U. Refinement of protein structures using a combination of quantum-mechanical calculations with neutron and X-ray crystallographic data. Acta Cryst. D 2019, 75, 368–380. [Google Scholar] [CrossRef]
- Borbulevych, O.; Martin, R.; Westerhoff, L. High-throughput quantum-mechanics/molecular-mechanics (ONIOM) macromolecular crystallographic refinement with PHENIX/DivCon: The impact of mixed Hamiltonian methods on ligand and protein structure. Acta Cryst. D 2018, 74, 1063–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borbulevych, O.; Martin, R.; Westerhoff, L. The critical role of QM/MM X-ray refinement and accurate tautomer/protomer determination in structure-based drug design. J. Comput. Aided Mol. Des. 2021, 35, 433–451. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.; Baker, M.; Bunkoczi, G.; Chen, V.; Croll, T.; Hintze, B.; Hung, L.; Jain, S.; McCoy, A.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Cryst. D 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, B.L.; Kim, H.; Chase, E. Crystal structure of a phage Twort group I ribozyme–product complex. Nat. Struct. Mol. Biol. 2005, 12, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Pallan, P.S.; Marshall, W.S.; Harp, J.; Jewett, F.C.; Wawrzak, Z.; Brown, B.A.; Rich, A.; Egli, M. Crystal structure of a luteoviral RNA pseudoknot and model for a minimal ribosomal frameshifting motif. Biochemistry 2005, 44, 11315–11322. [Google Scholar] [CrossRef] [Green Version]
- Robertson, M.P.; Scott, W.G. The structural basis of ribozyme-catalyzed RNA assembly. Science 2007, 315, 1549–1553. [Google Scholar] [CrossRef] [Green Version]
- Lipchock, S.V.; Strobel, S.A. A relaxed active site after exon ligation by the group I intron. Proc. Natl. Acad. Sci. USA 2008, 105, 5699–5704. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Adams, P.D. A robust bulk-solvent correction and anisotropic scaling procedure. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Pannu, N.; Read, R. Improved structure refinement through maximum likelihood. Acta Cryst. 1996, A52, 659–668. [Google Scholar] [CrossRef]
- Luchko, T.; Joung, I.; Case, D. Integral equation theory of biomolecules and electrolytes. In Innovations in Biomolecular Modeling and Simulation, Volume 1; Schlick, T., Ed.; Royal Society of Chemistry: London, UK, 2012; pp. 51–86. [Google Scholar]
- Ratkova, E.; Palmer, D.; Fedorov, M. Solvation Thermodynamics of Organic Molecules by the Molecular Integral Equation Theory: Approaching Chemical Accuracy. Chem. Rev. 2015, 115, 6312–6356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalenko, A. Molecular theory of solvation: Methodology summary and illustrations. Cond. Matter Phys. 2015, 18, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Luchko, T.; Gusarov, S.; Roe, D.R.; Simmerling, C.; Case, D.A.; Tuszynski, J.; Kovalenko, A. Three-dimensional molecular theory of solvation coupled with molecular dynamics in Amber. J. Chem. Theory Comput. 2010, 6, 607–624. [Google Scholar] [CrossRef] [Green Version]
- Gray, J. Using Molecular Mechanics Force Fields and RISM Densities to Improve Macromolecular Models. Ph.D. Thesis, Rutgers University, New Brunswick, NJ, USA, 2020. [Google Scholar]
- Sponer, J.; Bussi, G.; Krepl, M.; Banás, P.; Bottaro, S.; Cunha, R.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurecka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [Green Version]
- Batey, R.T.; Gilbert, S.D.; Montange, R.K. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 2004, 432, 411. [Google Scholar] [CrossRef] [PubMed]
- Montange, R.; Batey, R. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature 2006, 441, 1172–1175. [Google Scholar] [CrossRef]
- Chen, V.; Arendall, W.; Headd, J.; Keedy, D.; Immormino, R.; Kapral, G.; Murray, L.; Richardson, J.; Richardson, D. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.; Case, D. Generalized Born implicit solvent models for biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Subramaniam, S.; Case, D.; Wu, K.; Brooks, B. Map constrained self-guided Langevin dynamics. J. Struct. Biol. 2013, 183, 429–440. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Description | Resolution |
|---|---|---|
| 1q9a | sarcin/ricin domain from E.coli 23S rRNA | 1.04 |
| 1y0q | active group I ribozyme-product complex [16] | 3.60 |
| 2a43 | luteoviral pseudoknot [17] | 1.34 |
| 2gdi | thiamine pyrophosphate-specific riboswitch | 2.04 |
| 2gis | SAM riboswitch mRNA regulatory element | 2.90 |
| 2oiu | L1 ribozyme ligase circular adduct [18] | 2.60 |
| 2pn3 | Hep C IRES subdomain 2a | 2.90 |
| 2pn4 | Hep C IRES subdomain 2a | 2.32 |
| 2qus | hammerhead G12A mutant pre-cleavage | 2.40 |
| 2ygh | SAM-I riboswitch, with S-adenosylmethionine | 2.60 |
| 3bo3 | group I intron (U1) [19] | 3.40 |
| 3e5e | SMK box (SAM-III) riboswitch with SAH | 2.90 |
| 3f2q | FMN riboswitch with FMN | 2.95 |
| 3gx5 | T. tencongensis SAM-I riboswitch | 2.40 |
| 3iwn | bacterial c-di-GMP riboswitch (U1) | 3.20 |
| 3mxh | c-di-GMP riboswitch from V. cholerae (U1) | 2.30 |
| 3r4f | prohead RNA | 3.50 |
| 3tzr | riboswitch complex from Hep C IRES | 2.21 |
| 480d | sarcin/ricin domain from E. coli 23S rRNA | 1.50 |
| 483d | sarcin/ricin domain from E. coli 23S rRNA | 1.11 |
| 483d | sarcin/ricin domain from E. coli 23S rRNA | 1.11 |
| 4fe5 | xpt-pbuX guanine riboswitch aptamer domain | 1.32 |
| PDB ID | Spacing | Grid Size | Solvent |
|---|---|---|---|
| 1y0q | 1.0 | 96 × 160 × 224 | 0.1 M NaCl |
| 2oiu | 0.5 | 96 × 224 × 144 | 0.1 M MgCl2, 1.29 M NaCl |
| 3bo3 | 1.0 | 112 × 112 × 256 | 0.1 M NaCl |
| 3e5e | 1.0 | 100 × 112 × 96 | 0.02 M MgCl2, 0.14 M KCl |
| 3f2q | 0.8 | 90 × 96 × 192 | 0.1 M NaCl |
| 3iwn | 1.0 | 32 × 96 × 188 | 0.1 M NaCl |
| 3r4f | 1.0 | 108 × 112 × 224 | 0.1 M NaCl |
| 4fe5 | 0.8 | 168 × 48 × 64 | 0.02 M MgCl2, 0.14 M KCl |
| 1y0q | Phenix_cdl | Phenix-Amber | 3D-RISM | |
|---|---|---|---|---|
| molprobity score | 3.35 | 3.18 | 2.31 | 1.91 |
| clashscore | 53.7 | 35.4 | 3.7 | 0.9 |
| pucker outliers (%) | 8.6 | 8.6 | 10.7 | 8.2 |
| suite outliers (%) | 22.5 | 27.0 | 27.5 | 18.9 |
| average suiteness | 0.492 | 0.414 | 0.307 | 0.557 |
| R-work | 0.277 | 0.221 | 0.264 | 0.251 |
| R-free | 0.310 | 0.278 | 0.307 | 0.293 |
| RMS from deposited | 0.00 | 0.36 | 0.71 | 0.37 |
| 2oiu | RISM | 3r4f | RISM | 3e5e | RISM | 3f2q | RISM | 3iwn | RISM | 3bo3 | RISM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Resolution, Å | 2.6 | 3.5 | 2.9 | 2.95 | 3.2 | 3.4 | ||||||
| molprobity score | 2.59 | 1.73 | 3.21 | 1.65 | 2.22 | 2.07 | 2.50 | 1.84 | 3.63 | 1.59 | 2.33 | 1.59 |
| clashscore | 8.0 | 0.2 | 37.8 | 0.0 | 2.8 | 1.7 | 6.4 | 0.6 | 42.6 | 0.5 | 6.1 | 2.0 |
| pucker outliers | 4.2 | 3.5 | 7.6 | 7.6 | 3.8 | 5.8 | 2.8 | 0.9 | 12.4 | 9.7 | 6.6 | 9.1 |
| suite outliers | 20.4 | 11.3 | 24.2 | 16.7 | 13.5 | 9.6 | 19.4 | 8.3 | 28.5 | 22.0 | 37.2 | 28.6 |
| average suiteness | 0.447 | 0.621 | 0.449 | 0.534 | 0.537 | 0.534 | 0.456 | 0.507 | 0.480 | 0.513 | 0.370 | 0.433 |
| R-work | 0.203 | 0.208 | 0.239 | 0.232 | 0.222 | 0.190 | 0.200 | 0.218 | 0.222 | 0.209 | 0.282 | 0.245 |
| R-free | 0.238 | 0.244 | 0.271 | 0.245 | 0.259 | 0.225 | 0.243 | 0.247 | 0.292 | 0.253 | 0.325 | 0.275 |
| RMS vs deposited | 0.24 | 0.42 | 0.52 | 0.36 | 0.46 | 0.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gray, J.G.; Case, D.A. Refinement of RNA Structures Using Amber Force Fields. Crystals 2021, 11, 771. https://doi.org/10.3390/cryst11070771
Gray JG, Case DA. Refinement of RNA Structures Using Amber Force Fields. Crystals. 2021; 11(7):771. https://doi.org/10.3390/cryst11070771
Chicago/Turabian StyleGray, Jonathon G., and David A. Case. 2021. "Refinement of RNA Structures Using Amber Force Fields" Crystals 11, no. 7: 771. https://doi.org/10.3390/cryst11070771
APA StyleGray, J. G., & Case, D. A. (2021). Refinement of RNA Structures Using Amber Force Fields. Crystals, 11(7), 771. https://doi.org/10.3390/cryst11070771