
Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation

 , , , and
, , , and 
Abstract
1. Introduction
2. Theoretical and Computational Details
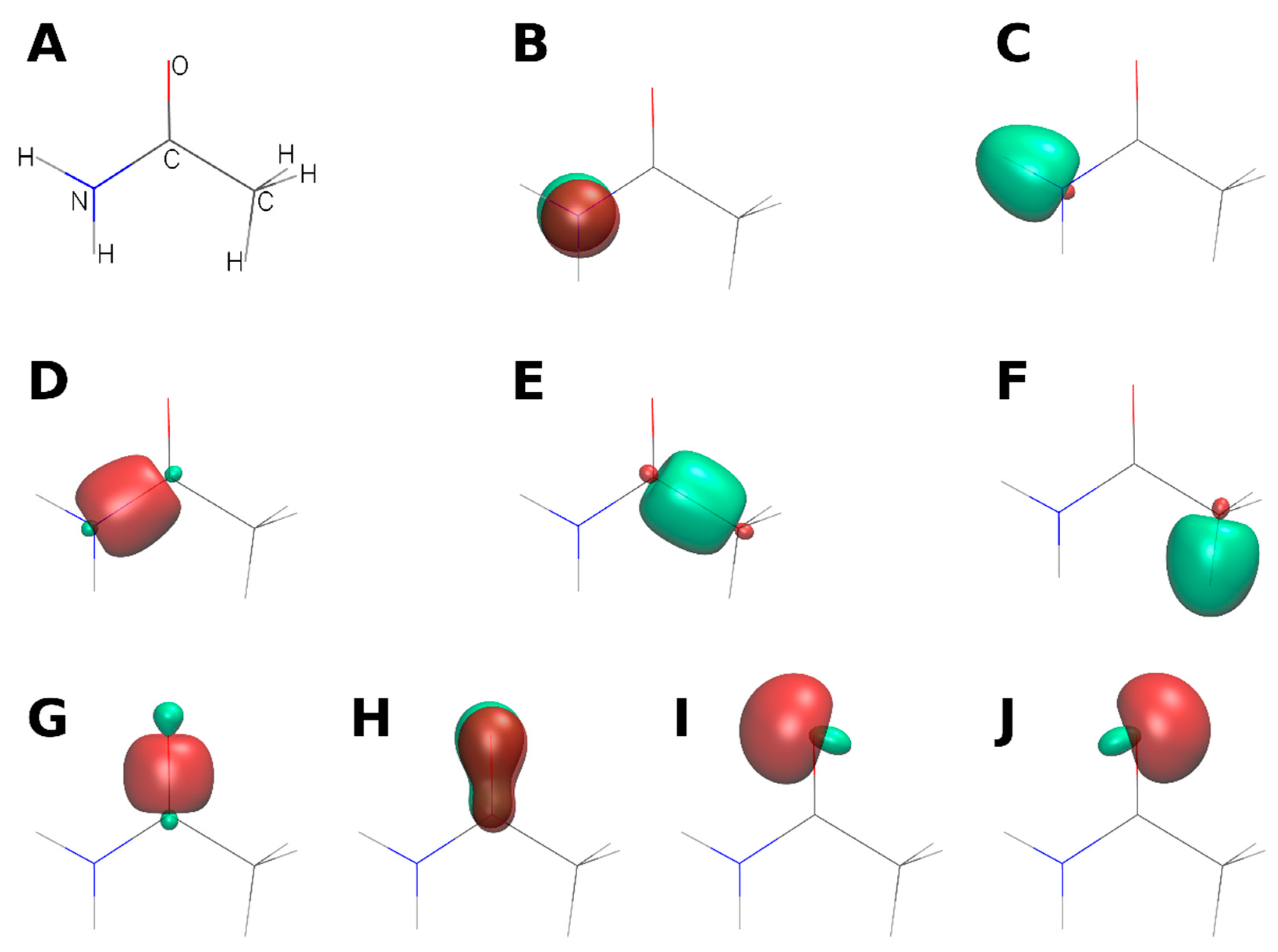
2.1. Extremely Localized Molecular Orbitals and ELMO Libraries
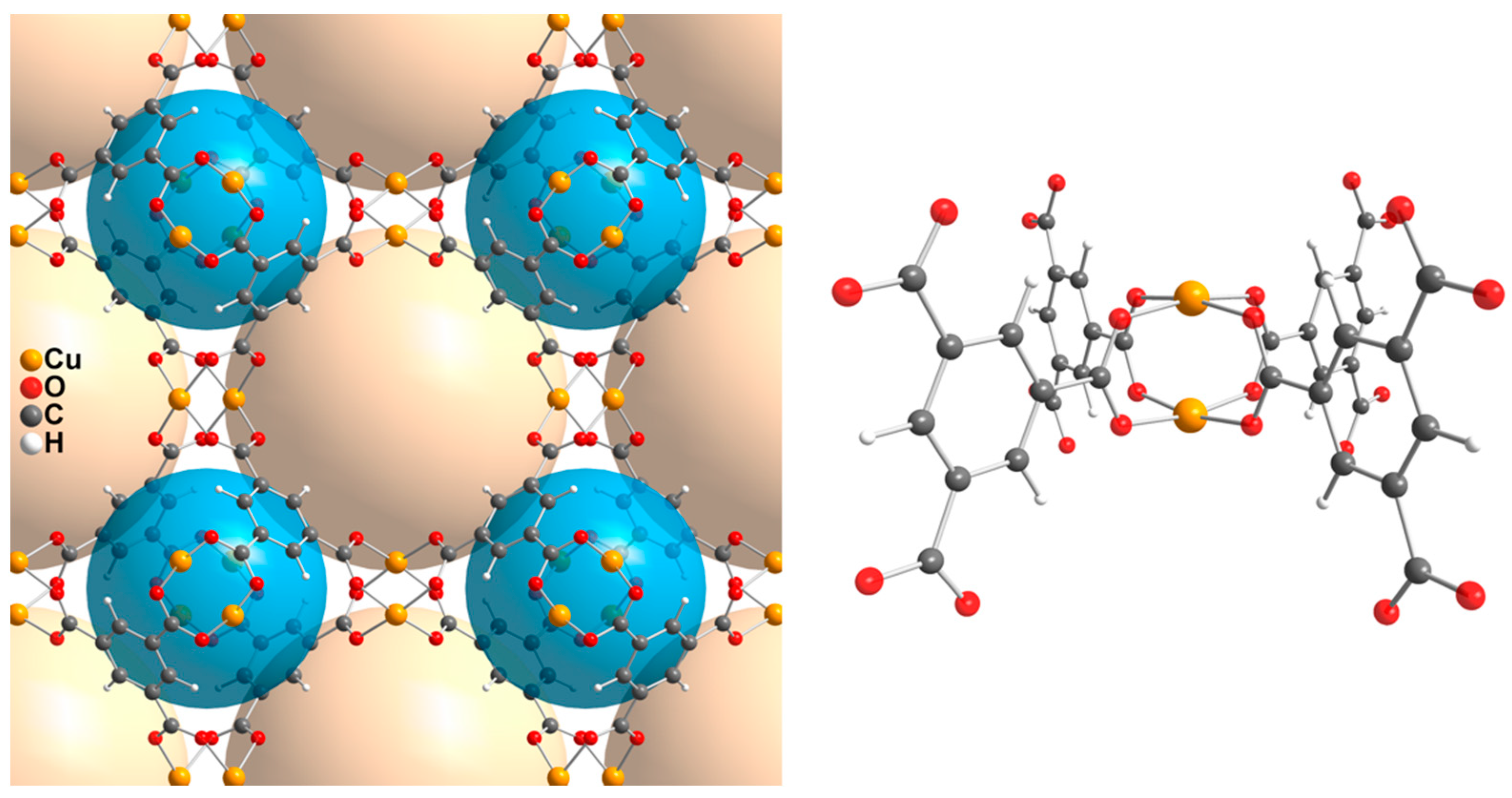
2.2. ELMO-Protocol for MOFs
- Construct a library of ELMOs describing the elementary fragments of the most common linkers employed for MOFs design.
- For each MOF crystal structure, perform ad hoc QM/ELMO calculations on model systems consisting of a SBU (at QM level) and the connected linkers (using ELMOs computed at step 1).
- The ELMOs for the linkers (see point 1) and the localized molecular orbitals for the SBU (obtained from point 2) will be finally transferred to the symmetry-related positions of the crystal structure using the ELMOdb program [61], thus quickly obtaining an approximate wavefunction/electron density for the periodic system.
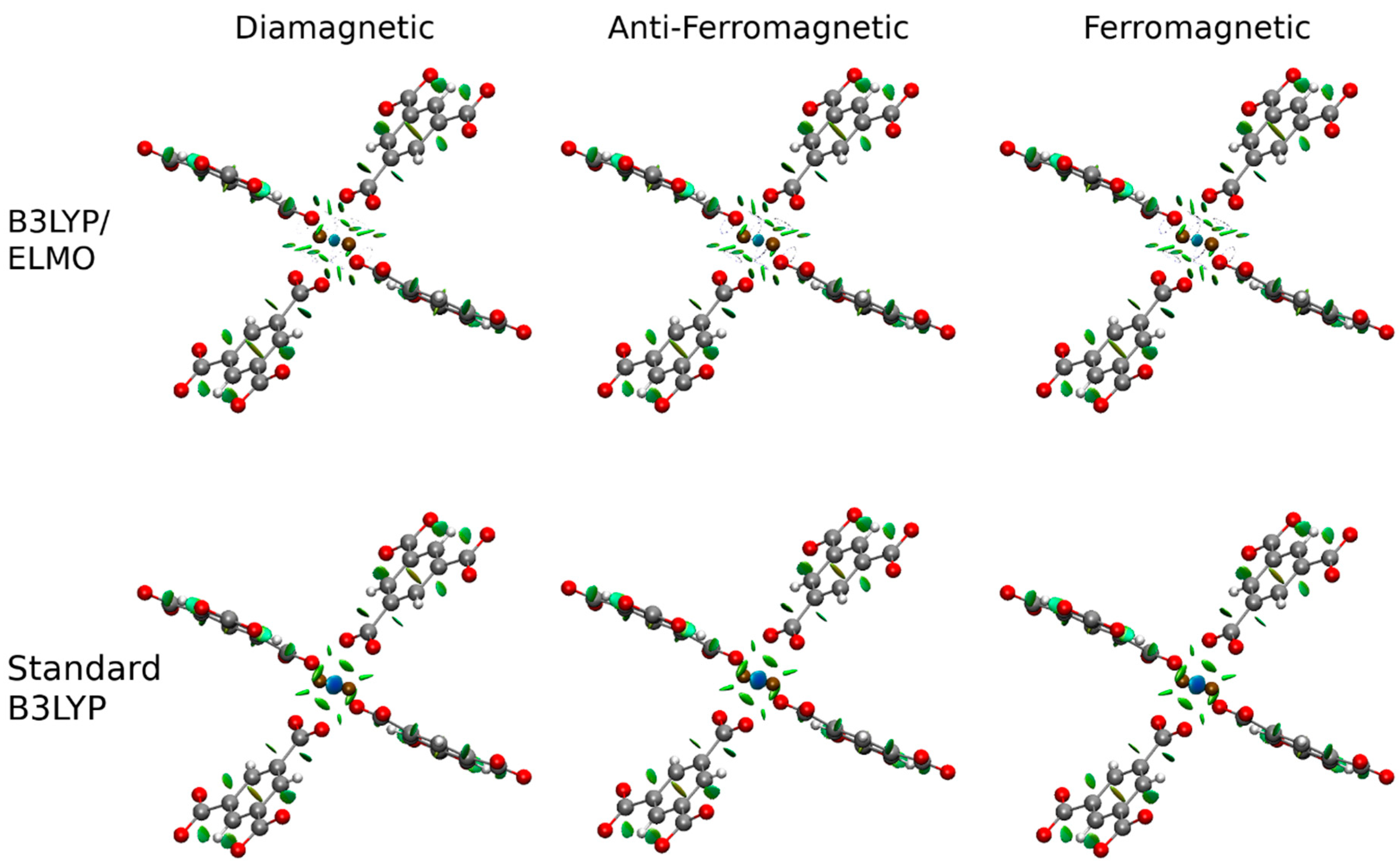
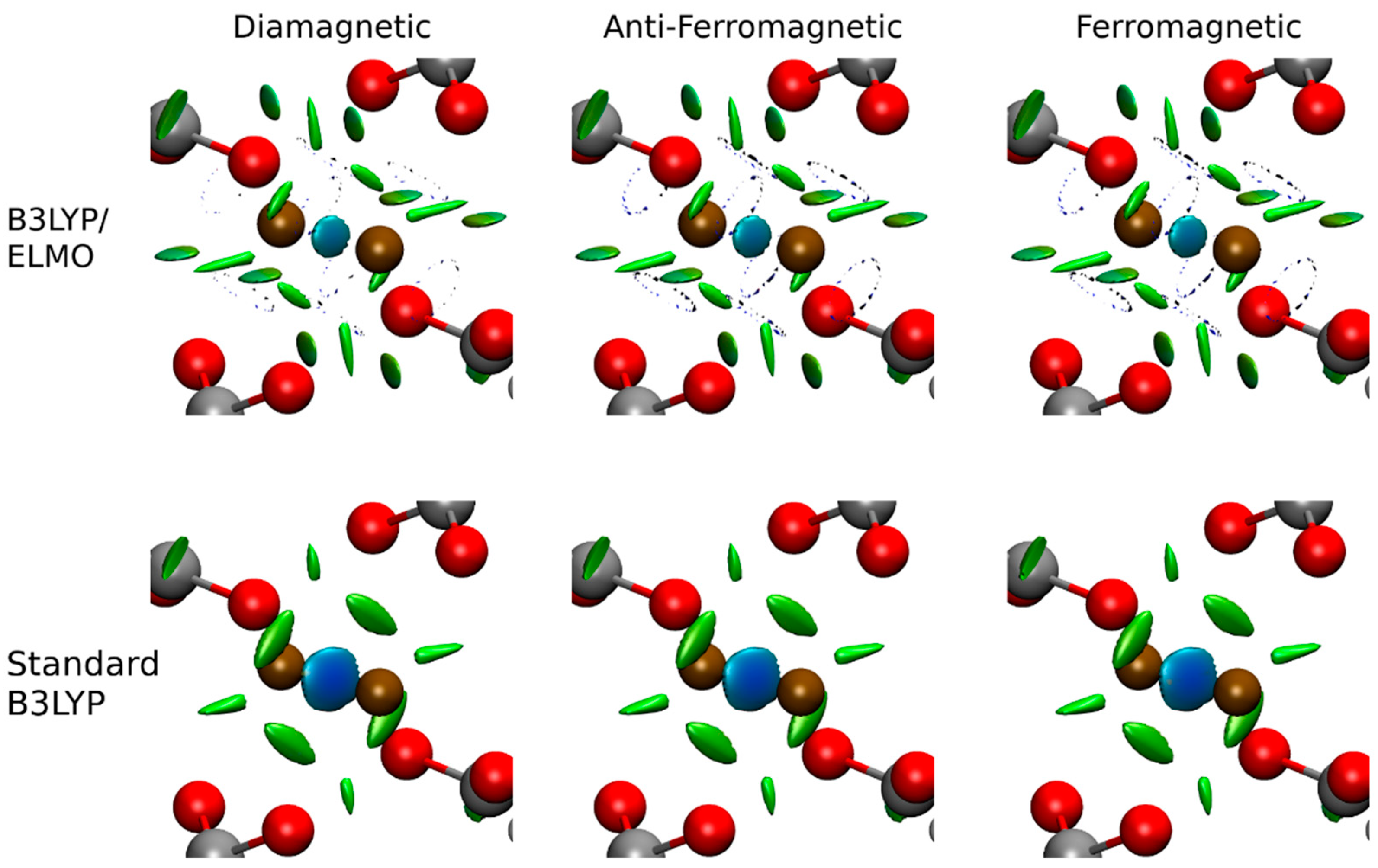
3. Results and Discussion
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gordon, M.S.; Slipchenko, L.V. Introduction: Calculations on Large Systems. Chem. Rev. 2015, 115, 5605–5606. [Google Scholar] [CrossRef]
- Jones, L.O.; Mosquera, M.A.; Schatz, G.C.; Ratner, M.A. Embedding Methods for Quantum Chemistry: Applications from Materials to Life Sciences. J. Am. Chem. Soc. 2020, 142, 3281–3295. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Car-bonium ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A Combined Quantum Mechanical and Molecular Mechanical Potential for Molecular Dynamics Simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Gao, J. Methods and Applications of Combined Quantum Mechanical and Molecular Mechanical Potentials. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; VCH Publishers, Inc.: Weinheim, Germany, 1996; Volume 7, pp. 119–186. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- Warshel, A. Multiscale Modeling of Biological Functions: From Enzymes to Molecular Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 10020–10031. [Google Scholar] [CrossRef]
- Levitt, M. Birth and Future of Multiscale Modeling for Macromolecular Systems (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 10006–10018. [Google Scholar] [CrossRef]
- Karplus, M. Development of Multiscale Models for Complex Chemical Systems: From H+H2 to Biomolecules (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 9992–10005. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels−Alder Reactions and Pt(P(t-Bu)3)2+ H2 Oxidative Addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, T.A.; Warshel, A. Frozen density functional approach for ab initio calculations of solvated molecules. J. Phys. Chem. 1993, 97, 8050–8053. [Google Scholar] [CrossRef]
- Wesolowski, T.A. Embedding a multideterminantal wave function in an orbital-free environment. Phys. Rev. A 2008, 77, 012504. [Google Scholar] [CrossRef]
- Wesolowski, T.A.; Shedge, S.; Zhou, X. Frozen-Density Embedding Strategy for Multilevel Simulations of Electronic Structure. Chem. Rev. 2015, 115, 5891–5928. [Google Scholar] [CrossRef]
- Manby, F.R.; Stella, M.; Goodpaster, J.D.; Miller, T.F., III. A Simple, Exact Density-Functional-Theory Embedding Scheme. J. Chem. Theory Comput. 2012, 8, 2564–2568. [Google Scholar] [CrossRef]
- Barnes, T.A.; Goodpaster, J.D.; Manby, F.R.; Miller, T.F., III. Accurate basis set truncation for wavefunction embedding. J. Chem. Phys. 2013, 139, 24103. [Google Scholar] [CrossRef]
- Goodpaster, J.D.; Barnes, T.A.; Manby, F.R.; Miller, T.F., III. Accurate and systematically improvable density functional theory embedding for correlated wave functions. J. Chem. Phys. 2014, 140, 18A507. [Google Scholar] [CrossRef]
- Bennie, S.J.; Stella, M.; Miller, T.F., III; Manby, F.R. Accelerating wavefunction in density-functional-theory embedding by truncating the active basis set. J. Chem. Phys. 2015, 143, 024105. [Google Scholar] [CrossRef]
- Pennifold, R.C.R.; Bennie, S.J.; Miller, T.F., III; Manby, F.R. Correcting density-driven errors in projection-based embedding. J. Chem. Phys. 2017, 146, 084113. [Google Scholar] [CrossRef]
- Welborn, M.; Manby, F.R.; Miller, T.F., IIII. Even-handed subsystem selection in projection-based embedding. J. Chem. Phys. 2018, 149, 144101. [Google Scholar] [CrossRef] [PubMed]
- Chulhai, D.V.; Goodpaster, J.D. Improved Accuracy and Efficiency in Quantum Embedding through Absolute Localization. J. Chem. Theory Comput. 2017, 13, 1503–1508. [Google Scholar] [CrossRef]
- Chulhai, D.V.; Goodpaster, J.D. Projection-Based Correlated Wave Function in Density Functional Theory Embedding for Periodic Systems. J. Chem. Theory Comput. 2018, 14, 1928–1942. [Google Scholar] [CrossRef]
- Bennie, S.J.; van der Kamp, M.W.; Pennifold, R.C.R.; Stella, M.; Manby, F.R.; Mulholland, A.J. A Projector-Embedding Approach for Multiscale Coupled-Cluster Calculations Applied to Citrate Synthase. J. Chem. Theory Comput. 2016, 12, 2689–2697. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.R.; Welborn, M.; Manby, F.R.; Miller, T.F., III. Projection-Based Wavefunction-in-DFT Embedding. Acc. Chem. Res. 2019, 52, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Direct calculation of electron density in density-functional theory. Phys. Rev. Lett. 1991, 66, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Direct calculation of electron density in density-functional theory: Implementation for benzene and a tetrapeptide. Phys. Rev. A 1991, 44, 7823–7826. [Google Scholar] [CrossRef]
- Dixon, S.L.; Merz, K.M., Jr. Semiempirical molecular orbital calculations with linear system size scaling. J. Chem. Phys. 1996, 104, 6643–6649. [Google Scholar] [CrossRef]
- Dixon, S.L.; Merz, K.M., Jr. Fast, accurate semiempirical molecular orbital calculations for macromolecules. J. Chem. Phys. 1997, 107, 879–893. [Google Scholar] [CrossRef]
- He, X.; Merz, K.M., Jr. Divide and Conquer Hartree−Fock Calculations on Proteins. J. Chem. Theory Comput. 2010, 6, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Gadre, S.R.; Shirsat, R.N.; Limaye, A.C. Molecular Tailoring Approach for Simulation of Electrostatic Properties. J. Phys. Chem. 1994, 98, 9165–9169. [Google Scholar] [CrossRef]
- Sahu, N.; Gadre, S.R. Molecular Tailoring Approach: A Route for ab Initio Treatment of Large Clusters. Acc. Chem. Res. 2014, 47, 2739–2747. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Nakano, T.; Kaminuma, T.; Sato, T.; Akiyama, Y.; Uebayasi, M.; Kitaura, K. Fragment molecular orbital method: Application to polypeptides. Chem. Phys. Lett. 2000, 318, 614–618. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Theoretical development of the fragment molecular orbital (FMO) method. In Modern Methods for Theoretical Physical Chemistry and Biopolymers; Starikov, E.B., Lewis, J.P., Tanaka, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Chapter 1; pp. 3–38. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Theoretical Background of the Fragment Molecular Orbital (FMO) Method and Its Implementation in GAMESS. In The Fragment Molecular Orbital Method: Practical Applications to Large Molecular Systems; Fedorov, D.G., Kitaura, K., Eds.; CRC Press-Taylor & Francis Group: Boca Raton, FL, USA, 2009; Chapter 2; pp. 5–36. [Google Scholar] [CrossRef]
- Huang, L.; Massa, L.; Karle, J. Kernel energy method illustrated with peptides. Int. J. Quantum Chem. 2005, 103, 808–817. [Google Scholar] [CrossRef]
- Huang, L.; Massa, L.; Karle, J. Kernel energy method applied to vesicular stomatitis virus nucleoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Bohorquez, H.; Matta, C.F.; Massa, L. The Kernel Energy Method: Application to Graphene and Extended Aromatics. Int. J. Quantum Chem. 2011, 111, 4150–4157. [Google Scholar] [CrossRef]
- Zhang, D.W.; Zhang, J.Z.H. Molecular fractionation with conjugate caps for full quantum mechanical calculation of protein-molecule interaction energy. J. Chem. Phys. 2003, 119, 3599–3605. [Google Scholar] [CrossRef]
- Gao, A.M.; Zhang, D.W.; Zhang, J.Z.H.; Zhang, Y. An efficient linear scaling method for ab initio calculation of electron density of proteins. Chem. Phys. Lett. 2004, 394, 293–297. [Google Scholar] [CrossRef]
- He, X.; Zhang, J.Z.H. The generalized molecular fractionation with conjugate caps/molecular mechanics method for direct calculation of protein energy. J. Chem. Phys. 2006, 124, 184703. [Google Scholar] [CrossRef]
- Li, S.; Li, W.; Fang, T. An Efficient Fragment-Based Approach for Predicting the Ground-State Energies and Structures of Large Molecules. J. Am. Chem. Soc. 2005, 127, 7215–7226. [Google Scholar] [CrossRef]
- Walker, P.D.; Mezey, P.G. Ab Initio Quality Electron Densities for Proteins: A MEDLA Approach. J. Am. Chem. Soc. 1994, 116, 12022–12032. [Google Scholar] [CrossRef]
- Exner, T.E.; Mezey, P.G. Ab Initio-Quality Electrostatic Potentials for Proteins: An Application of the ADMA Approach. J. Phys. Chem. A 2002, 106, 11791–11800. [Google Scholar] [CrossRef]
- Exner, T.E.; Mezey, P.G. Ab initio quality properties for macromolecules using the ADMA approach. J. Comput. Chem. 2003, 24, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Breneman, C.M.; Thompson, T.R.; Rhem, M.; Dung, M. Electron density modeling of large systems using the transferable atom equivalent method. Comput. Chem. 1995, 19, 161–179. [Google Scholar] [CrossRef]
- Chang, C.; Bader, R.F.W. Theoretical construction of a polypeptide. J. Phys. Chem. 1992, 96, 1654–1662. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Martín, F.J. Interdeterminancy of basin and surface properties of an open system. Can. J. Chem. 1998, 76, 284–291. [Google Scholar] [CrossRef]
- Matta, C.F. Theoretical Reconstruction of the Electron Density of Large Molecules from Fragments Determined as Proper Open Quantum Systems: The Properties of the Oripavine PEO, Enkephalins, and Morphine. J. Phys. Chem. A 2001, 105, 11088–11101. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Stoll, H.; Wagenblast, G.; Preuβ, H. On the use of local basis sets for localized molecular orbitals. Theor. Chim. Acta 1980, 57, 169–178. [Google Scholar] [CrossRef]
- Fornili, A.; Sironi, M.; Raimondi, M. Determination of Extremely Localized Molecular Orbitals and Their Application to Quantum Mechanics/Molecular Mechanics Methods and to the Study of Intramolecular Hydrogen Bonding. J. Mol. Struct. 2003, 632, 157–172. [Google Scholar] [CrossRef]
- Sironi, M.; Genoni, A.; Civera, M.; Pieraccini, S.; Ghitti, M. Extremely localized molecular orbitals: Theory and applications. Theor. Chem. Acc. 2007, 117, 685–698. [Google Scholar] [CrossRef]
- Genoni, A.; Sironi, M. A novel approach to relax extremely localized molecular orbitals: The extremely localized molecular orbital-valence bond method. Theor. Chem. Acc. 2004, 112, 254–262. [Google Scholar] [CrossRef]
- Genoni, A.; Fornili, A.; Sironi, M. Optimal Virtual Orbitals to Relax Wave Functions Built Up with Transferred Extremely Localized Molecular Orbitals. J. Comput. Chem. 2005, 26, 827–835. [Google Scholar] [CrossRef]
- Genoni, A.; Ghitti, M.; Pieraccini, S.; Sironi, M. A novel extremely localized molecular orbitals based technique for the one-electron density matrix computation. Chem. Phys. Lett. 2005, 415, 256–260. [Google Scholar] [CrossRef]
- Genoni, A.; Merz, K.M., Jr.; Sironi, M. A Hylleras functional based perturbative technique to relax extremely localized molecular orbitals. J. Chem. Phys. 2008, 129, 054101. [Google Scholar] [CrossRef]
- Sironi, M.; Ghitti, M.; Genoni, A.; Saladino, G.; Pieraccini, S. DENPOL: A new program to determine electron densities of poly-peptides using extremely localized molecular orbitals. J. Mol. Struct. 2009, 898, 8–16. [Google Scholar] [CrossRef]
- Meyer, B.; Guillot, B.; Ruiz-Lopez, M.F.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 1. Model Molecules Approximation and Molecular Orbitals Transferability. J. Chem. Theory Comput. 2016, 12, 1052–1067. [Google Scholar] [CrossRef]
- Meyer, B.; Guillot, B.; Ruiz-Lopez, M.F.; Jelsch, C.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 2. Comparison with the Pseudoatoms Transferability. J. Chem. Theory Comput. 2016, 12, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 3. Construction and Preliminary Assessment of the New Databanks. J. Phys. Chem. A 2018, 122, 8965–8981. [Google Scholar] [CrossRef]
- Genoni, A.; Bučinský, L.; Claiser, N.; Contreras-García, J.; Dittrich, B.; Dominiak, P.M.; Espinosa, E.; Gatti, C.; Giannozzi, P.; Gillet, J.-M.; et al. Quantum Crystallography: Current Developments and Future Perspectives. Chem. Eur. J. 2018, 24, 10881–10905. [Google Scholar] [CrossRef]
- Grabowsky, S.; Genoni, A.; Bürgi, H.-B. Quantum crystallography. Chem. Sci. 2017, 8, 4159–4176. [Google Scholar] [CrossRef] [PubMed]
- Genoni, A.; Macchi, P. Quantum Crystallography in the Last Decade: Developments and Outlooks. Crystals 2020, 10, 473. [Google Scholar] [CrossRef]
- Grabowsky, S.; Genoni, A.; Thomas, S.P.; Jayatilaka, D. The Advent of Quantum Crystallography: Form and Structure Factors from Quantum Mechanics for Advanced Structure Refinement and Wavefunction Fitting. In 21st Century Challenges in Chemical Crystallography II. Structure and Bonding; Springer International Publishing: Berlin/Heidelberg, Germany, 2020; pp. 65–144. [Google Scholar] [CrossRef]
- Macchi, P. The connubium between crystallography and quantum mechanics. Crystallogr. Rev. 2020, 26, 209–268. [Google Scholar] [CrossRef]
- Massa, L.; Matta, C.F. Quantum crystallography: A perspective. J. Comput. Chem. 2018, 39, 1021–1028. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Dittrich, B. X-ray structure refinement using aspherical atomic density functions obtained from quantum me-chanical calculations. Acta Crystallogr. Sect. A 2008, 64, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld atom refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and pre-cisely by x-ray crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef]
- Fugel, M.; Jayatilaka, D.; Hupf, E.; Overgaard, J.; Hathwar, V.R.; Macchi, P.; Turner, M.J.; Howard, J.A.K.; Dolomanov, O.V.; Puschmann, H.; et al. Probing the accuracy and precision of Hirshfeld atom refinement with HARt interfaced with Olex2. IUCrJ 2018, 5, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Wieduwilt, E.K.; Macetti, G.; Malaspina, L.A.; Jayatilaka, D.; Grabowsky, S.; Genoni, A. Post-Hartree-Fock methods for Hirshfeld atom refinement: Are they necessary? Investigation of a strongly hydrogen-bonded molecular crystal. J. Mol. Struct. 2020, 1209, 127934. [Google Scholar] [CrossRef]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Borhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate Crystal Structures and Chemical Properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Malaspina, L.A.; Wieduwilt, E.K.; Bergmann, J.; Kleemiss, F.; Meyer, B.; Ruiz-López, M.F.; Pal, R.; Hupf, E.; Beckmann, J.; Piltz, R.O.; et al. Fast and Accurate Quantum Crystallography: From Small to Large, from Light to Heavy. J. Phys. Chem. Lett. 2019, 10, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately Extracting the Signature of In-termolecular Interactions Present in the NCI Plot of the Reduced Density Gradient versus Electron Density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Ponce-Vargas, M.; Lefebvre, C.; Boisson, J.-C.; Hénon, E. Atomic Decomposition Scheme of Noncovalent Interactions Applied to Host–Guest Assemblies. J. Chem. Inf. Model. 2020, 60, 268–278. [Google Scholar] [CrossRef]
- Klein, J.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. New Way for Probing Bond Strength. J. Phys. Chem. A 2020, 124, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Arias-Olivares, D.; Wieduwilt, E.K.; Contreras-García, J.; Genoni, A. NCI-ELMO: A New Method to Quickly and Accurately Detect Noncovalent Interactions in Biosystems. J. Chem. Theory Comput. 2019, 15, 6456–6470. [Google Scholar] [CrossRef]
- Wieduwilt, E.K.; Boisson, J.-C.; Terraneo, G.; Hénon, E.; Genoni, A. A Step toward the Quantification of Noncovalent Interactions in Large Biological Systems: The Independent Gradient Model-Extremely Localized Molecular Orbital Approach. J. Chem. Inf. Model. 2021. [Google Scholar] [CrossRef] [PubMed]
- Macetti, G.; Genoni, A. Quantum Mechanics/Extremely Localized Molecular Orbital Method: A Fully Quantum Mechanical Embedding Approach for Macromolecules. J. Phys. Chem. A 2019, 123, 9420–9428. [Google Scholar] [CrossRef] [PubMed]
- Macetti, G.; Wieduwilt, E.K.; Assfeld, X.; Genoni, A. Localized Molecular Orbital-Based Embedding Scheme for Correlated Methods. J. Chem. Theory Comput. 2020, 16, 3578–3596. [Google Scholar] [CrossRef]
- Macetti, G.; Genoni, A. Quantum Mechanics/Extremely Localized Molecular Orbital Embedding Strategy for Excited States: Coupling to Time-Dependent Density Functional Theory and Equation-of-Motion Coupled Cluster. J. Chem. Theory Comput. 2020, 16, 7490–7506. [Google Scholar] [CrossRef]
- Macetti, G.; Wieduwilt, E.K.; Genoni, A. QM/ELMO: A Multi-Purpose Fully Quantum Mechanical Embedding Scheme Based on Extremely Localized Molecular Orbitals. J. Phys. Chem. A. submitted.
- Wieduwilt, E.K.; Macetti, G.; Genoni, A. Climbing Jacob’s Ladder of Structural Refinement: Introduction of a Localized Molecular Orbital-Based Embedding for Accurate X-ray Determinations of Hydrogen Atom Positions. J. Phys. Chem. Lett. 2021, 12, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [PubMed]
- Ockwig, N.W.; Delgado-Friedrichs, O.; O’Keeffe, M.; Yaghi, O.M. Reticular Chemistry: Occurrence and Taxonomy of Nets and Grammar for the Design of Frameworks. Acc. Chem. Res. 2005, 38, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, N.W.; Chae, H.K.; Eddaoudi, M.; Kim, J. Reticular Synthesis and the Design of New Materials. Nature 2003, 423, 705–714. [Google Scholar] [CrossRef]
- Zagorodniy, K.; Seifert, G.; Hermann, H. Metal-organic frameworks as promising candidates for future ultralow-k dielectrics. Appl. Phys. Lett. 2010, 97, 251905. [Google Scholar] [CrossRef]
- Boys, S.F. Construction of Some Molecular Orbitals to Be Approximately Invariant for Changes from One Molecule to Another. Rev. Mod. Phys. 1960, 32, 296–299. [Google Scholar] [CrossRef]
- Foster, J.M.; Boys, S.F. Canonical Configurational Interaction Procedure. Rev. Mod. Phys. 1960, 32, 300–302. [Google Scholar] [CrossRef]
- Edmiston, C.; Ruedenberg, K. Localized Atomic and Molecular Orbitals. Rev. Mod. Phys. 1963, 35, 457–464. [Google Scholar] [CrossRef]
- Edmiston, C.; Ruedenberg, K. Localized Atomic and Molecular Orbitals. II. J. Chem. Phys. 1965, 43, S97–S116. [Google Scholar] [CrossRef]
- Von Niessen, W. Density Localization of Atomic and Molecular Orbitals. I. J. Chem. Phys. 1972, 56, 4290. [Google Scholar] [CrossRef]
- Pipek, J.; Mezey, P.G. A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions. J. Chem. Phys. 1989, 90, 4916–4926. [Google Scholar] [CrossRef]
- McWeeny, R. The density matrix in many-electron quantum mechanics I. Generalized product functions. Factorization and physical interpretation of the density matrices. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1959, 253, 242–259. [Google Scholar] [CrossRef]
- Adams, W.H. On the Solution of the Hartree-Fock Equation in Terms of Localized Orbitals. J. Chem. Phys. 1961, 34, 89. [Google Scholar] [CrossRef]
- Huzinaga, S.; Cantu, A. A Theory of Separability of Many-Electron Systems. J. Chem. Phys. 1971, 55, 5543–5549. [Google Scholar] [CrossRef]
- Gilbert, T.L. Multiconfiguration Self-Consistent-Field Theory for Localized Orbitals. II. Overlap constraints, Lagrangian multipliers, and the screened interaction field. J. Chem. Phys. 1974, 60, 3835–3844. [Google Scholar] [CrossRef]
- Matsuoka, O. Expansion methods for Adams-Gilbert equations. I. Modified Adams-Gilbert equation and common and fluctuating basis sets. J. Chem. Phys. 1977, 66, 1245–1254. [Google Scholar] [CrossRef]
- Smits, G.F.; Altona, C. Calculation and properties of non-orthogonal, strictly local molecular orbitals. Theor. Chem. Acc. 1985, 67, 461–475. [Google Scholar] [CrossRef]
- Francisco, E.; Martín Pendás, A.; Adams, W.H. Generalized Huzinaga building-block equations for nonorthogonal electronic groups: Relation to the Adams–Gilbert theory. J. Chem. Phys. 1992, 97, 6504–6508. [Google Scholar] [CrossRef]
- Ordejón, P.; Drabold, D.A.; Grumbach, M.P.; Martin, R.M. Unconstrained minimization approach for electronic computations that scales linearly with system size. Phys. Rev. B 1993, 48, 14646–14649. [Google Scholar] [CrossRef]
- Couty, M.; Bayse, C.A.; Hall, M.B. Extremely localized molecular orbitals (ELMO): A non-orthogonal Hartree-Fock method. Theor. Chem. Acc. 1997, 97, 96–109. [Google Scholar] [CrossRef]
- Philipp, D.M.; Friesner, R.A. Mixed Ab Initio QM/MM Modeling Using Frozen Orbitals and Tests with Alanine Dipeptide and Tetrapeptide. J. Comput. Chem. 1999, 20, 1468–1494. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Guntern, Y.T.; Macchi, P. Electron Density and Dielectric Properties of Highly Porous MOFs: Binding and Mobility of Guest Molecules in Cu3(BTC)2 and Zn3(BTC)2. J. Am. Chem. Soc. 2019, 141, 9382–9390. [Google Scholar] [CrossRef]
- Guest, M.F.; Bush, I.J.; van Dam, H.J.J.; Sherwood, P.; Thomas, J.M.H.; van Lenthe, J.H.; Havenith, R.W.A.; Kendrick, J. The GAMESS-UK electronic structure package: Algorithms, developments and applications. Mol. Phys. 2005, 103, 719–747. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and ex-perimental electron densities. Coord. Chem. Rev. 2003, 238–239, 383–412. [Google Scholar] [CrossRef]
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states-I—Orbital degeneracy. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1937, 161, 220–235. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
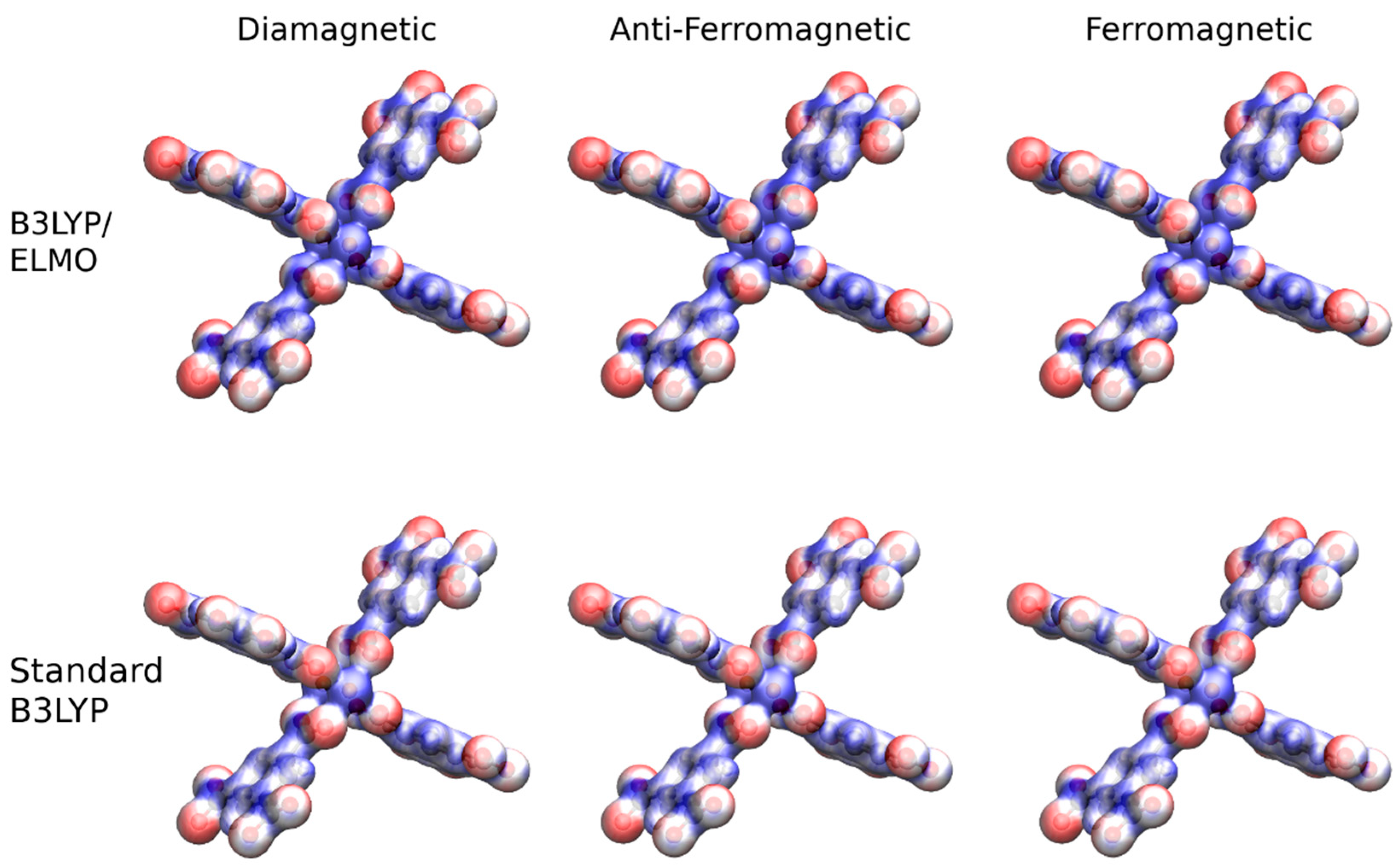{kind=link}
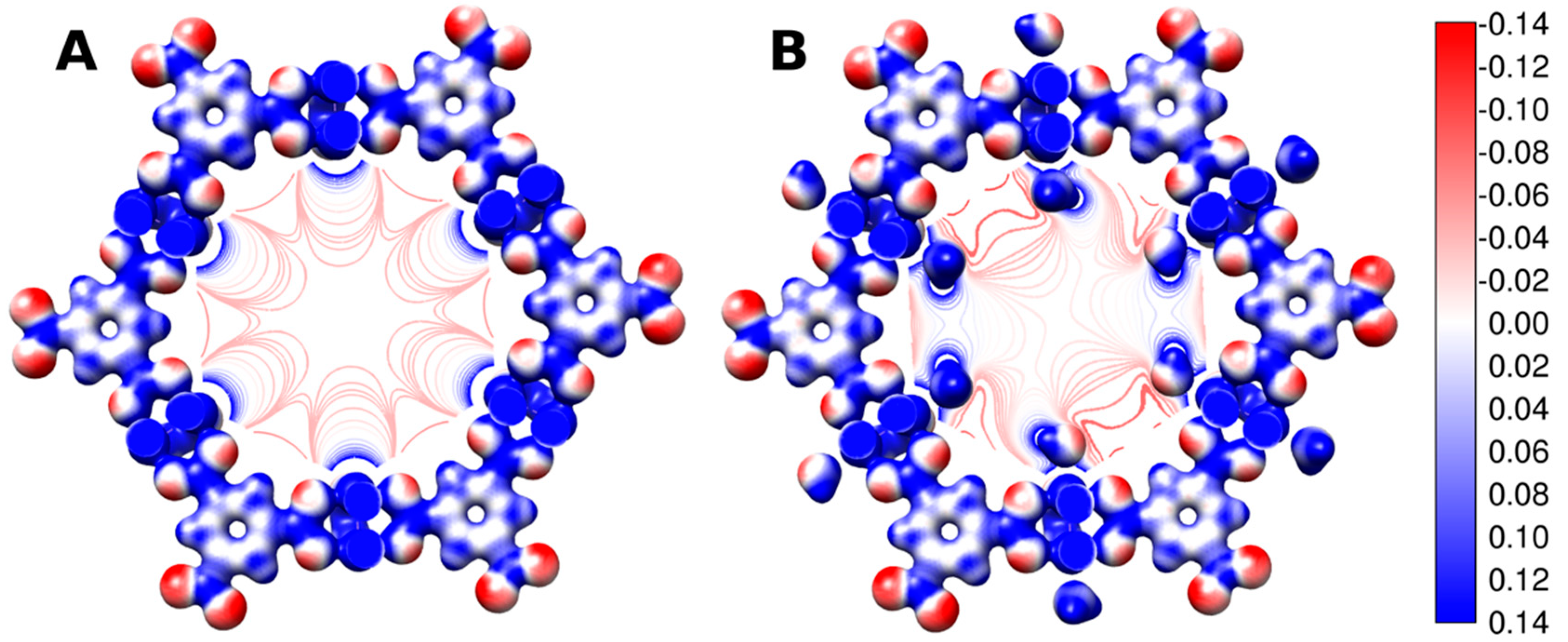{kind=link}
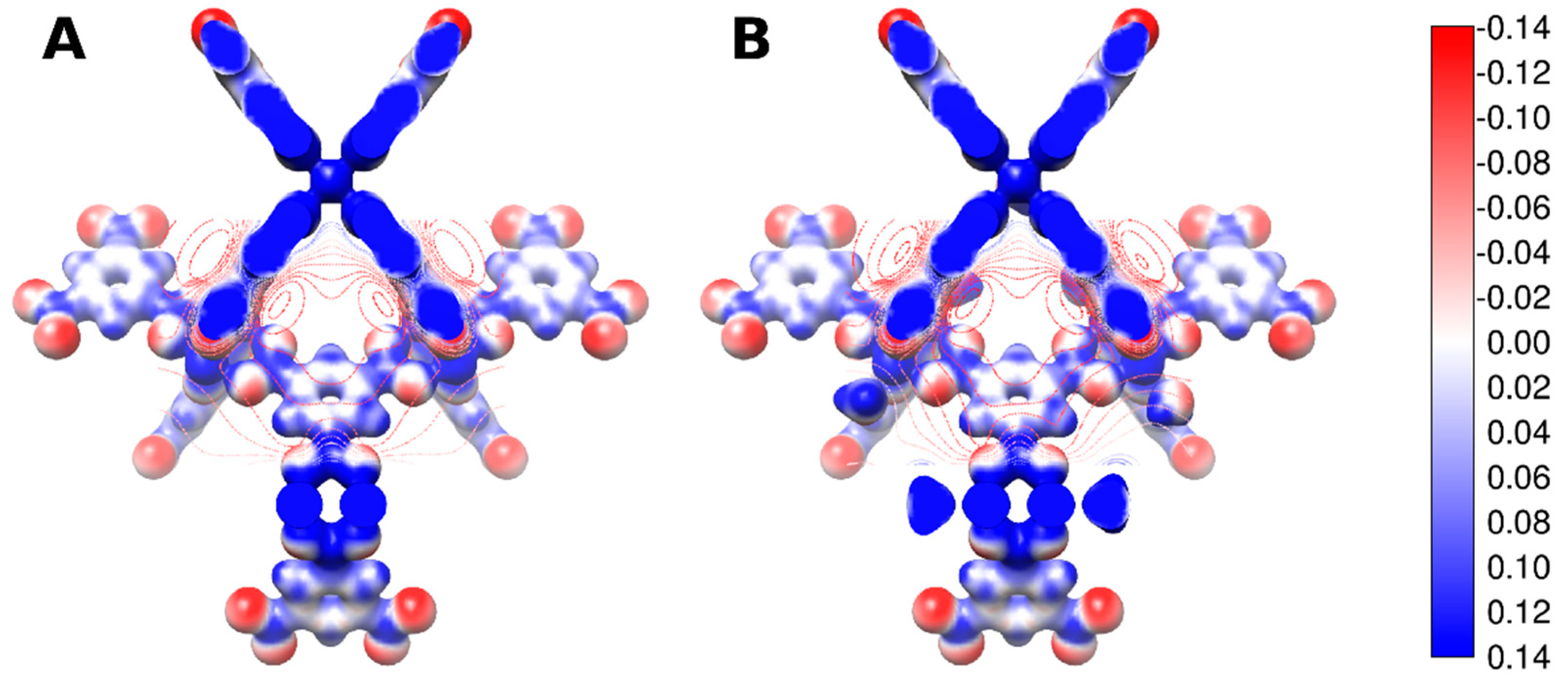{kind=link}
| Interaction | Spin State | Calculation | |||||
|---|---|---|---|---|---|---|---|
| Cu-Cu | Diamagnetic | B3LYP/ELMO | 0.029 | 0.075 | 1.344 | −0.0098 | 1.031 |
| Standard B3LYP | 0.036 | 0.069 | 1.423 | −0.0127 | 0.426 | ||
| Anti-Ferromagnetic | B3LYP/ELMO | 0.029 | 0.074 | 1.348 | −0.0099 | 0.100 | |
| Standard B3LYP | 0.035 | 0.070 | 1.410 | −0.0122 | 0.146 | ||
| Ferromagnetic | B3LYP/ELMO | 0.029 | 0.074 | 1.348 | −0.0099 | 0.100 | |
| Standard B3LYP | 0.035 | 0.070 | 1.409 | −0.0121 | 0.144 | ||
| Cu-O | Diamagnetic | B3LYP/ELMO | 0.066 | 0.614 | 0.956 | 0.0065 | 0.227 |
| Standard B3LYP | 0.095 | 0.462 | 1.140 | −0.0189 | 0.429 | ||
| Anti-Ferromagnetic | B3LYP/ELMO | 0.065 | 0.626 | 0.943 | 0.0084 | 0.233 | |
| Standard B3LYP | 0.093 | 0.477 | 1.127 | −0.0174 | 0.466 | ||
| Ferromagnetic | B3LYP/ELMO | 0.065 | 0.626 | 0.943 | 0.0084 | 0.233 | |
| Standard B3LYP | 0.093 | 0.478 | 1.127 | −0.0173 | 0.466 |
| Spin State | Calculation | ||||||
|---|---|---|---|---|---|---|---|
| Diamagnetic | B3LYP/ELMO | 1.79 | 1.79 | −1.42 | 61.50 | 61.50 | 109.45 |
| Standard B3LYP | 1.10 | 1.10 | −1.13 | 75.03 | 75.03 | 104.62 | |
| Anti-Ferromagnetic | B3LYP/ELMO | 1.79 | 1.79 | −1.42 | 61.52 | 61.52 | 109.46 |
| Standard B3LYP | 1.20 | 1.20 | −1.16 | 73.41 | 73.41 | 104.95 | |
| Ferromagnetic | B3LYP/ELMO | 1.79 | 1.79 | −1.42 | 61.52 | 61.52 | 109.46 |
| Standard B3LYP | 1.21 | 1.21 | −1.16 | 73.35 | 73.35 | 104.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieduwilt, E.K.; Macetti, G.; Scatena, R.; Macchi, P.; Genoni, A. Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation. Crystals 2021, 11, 207. https://doi.org/10.3390/cryst11020207
Wieduwilt EK, Macetti G, Scatena R, Macchi P, Genoni A. Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation. Crystals. 2021; 11(2):207. https://doi.org/10.3390/cryst11020207
Chicago/Turabian StyleWieduwilt, Erna K., Giovanni Macetti, Rebecca Scatena, Piero Macchi, and Alessandro Genoni. 2021. "Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation" Crystals 11, no. 2: 207. https://doi.org/10.3390/cryst11020207
APA StyleWieduwilt, E. K., Macetti, G., Scatena, R., Macchi, P., & Genoni, A. (2021). Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation. Crystals, 11(2), 207. https://doi.org/10.3390/cryst11020207