
Weak Interactions in the Structures of Newly Synthesized (–)-Cytisine Amino Acid Derivatives



Abstract
1. Introduction
2. Materials and Methods
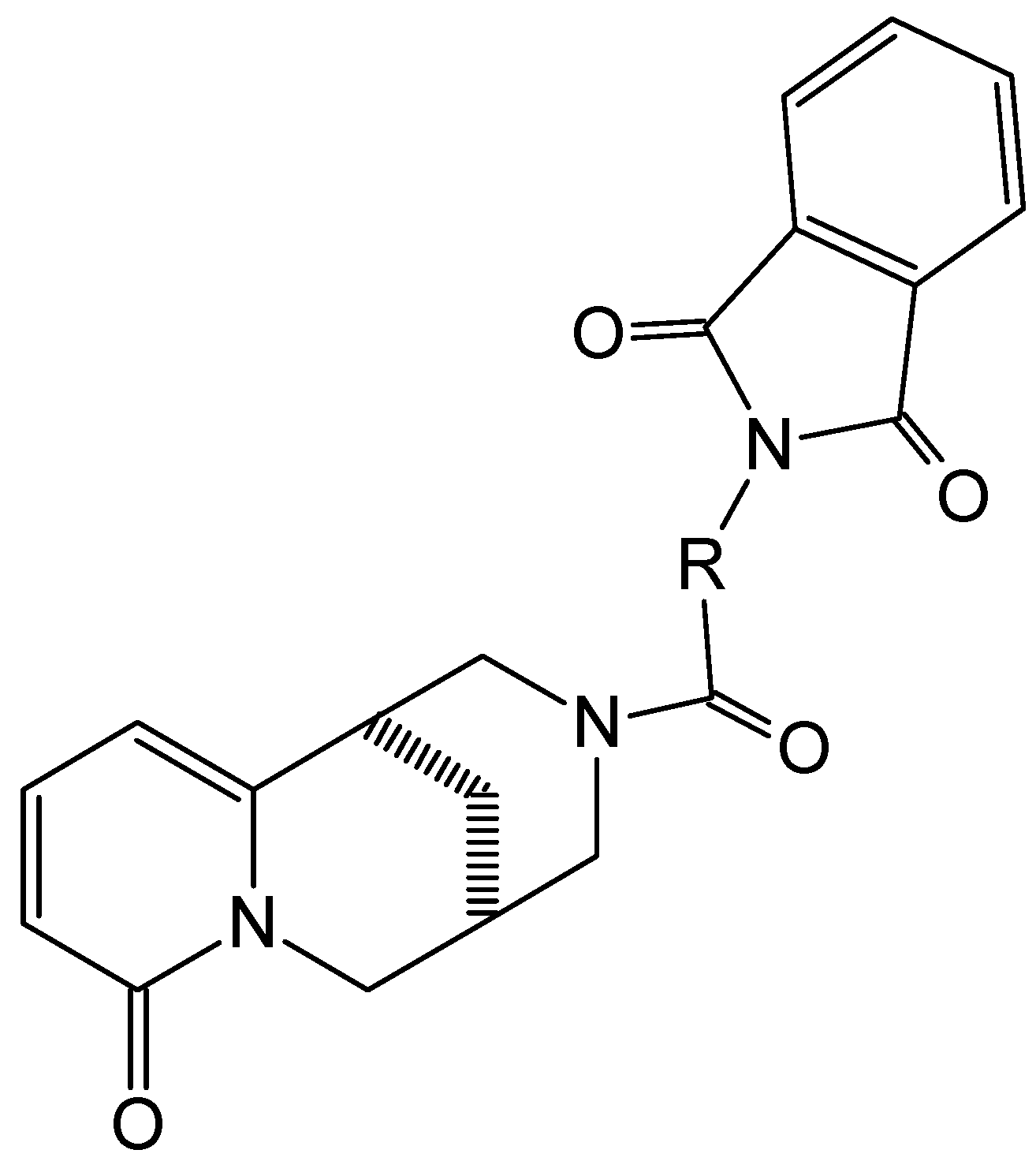
2.1. Synthesis
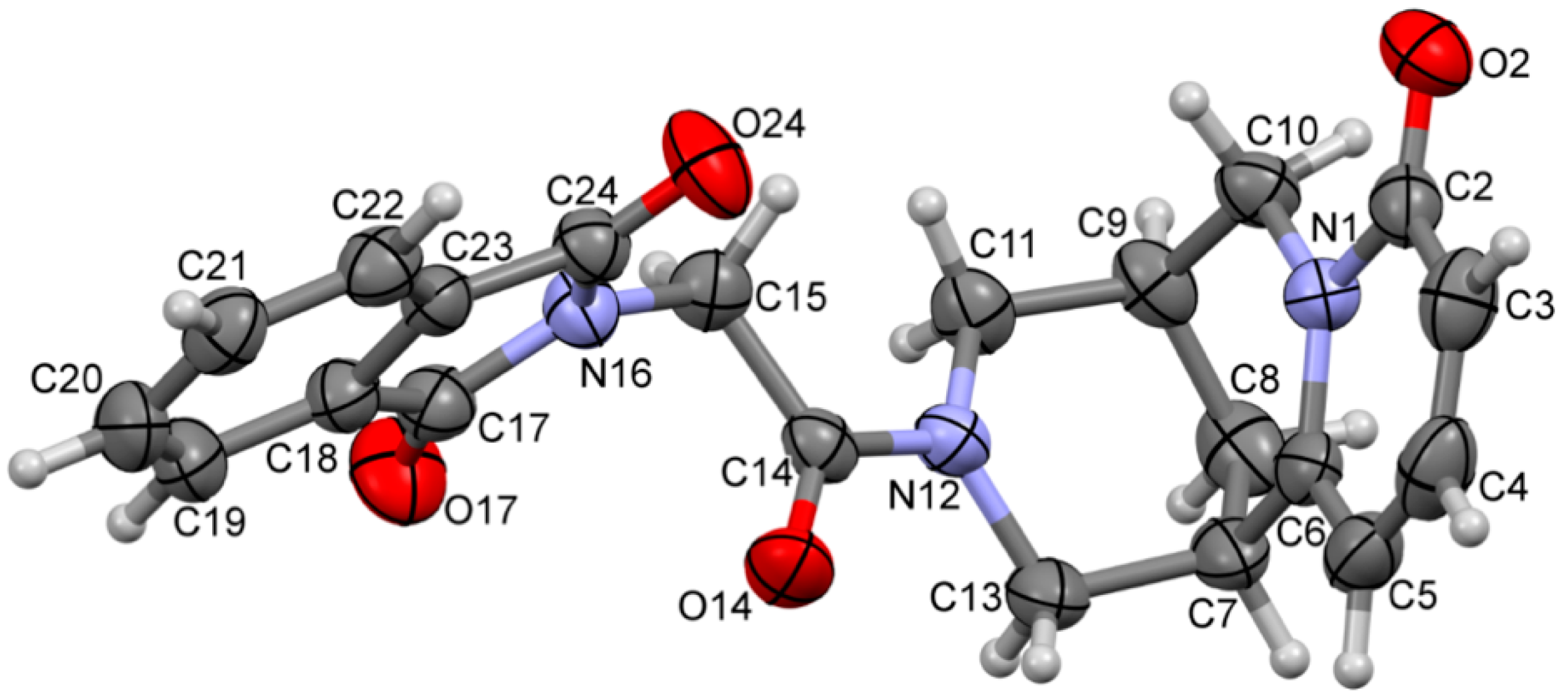
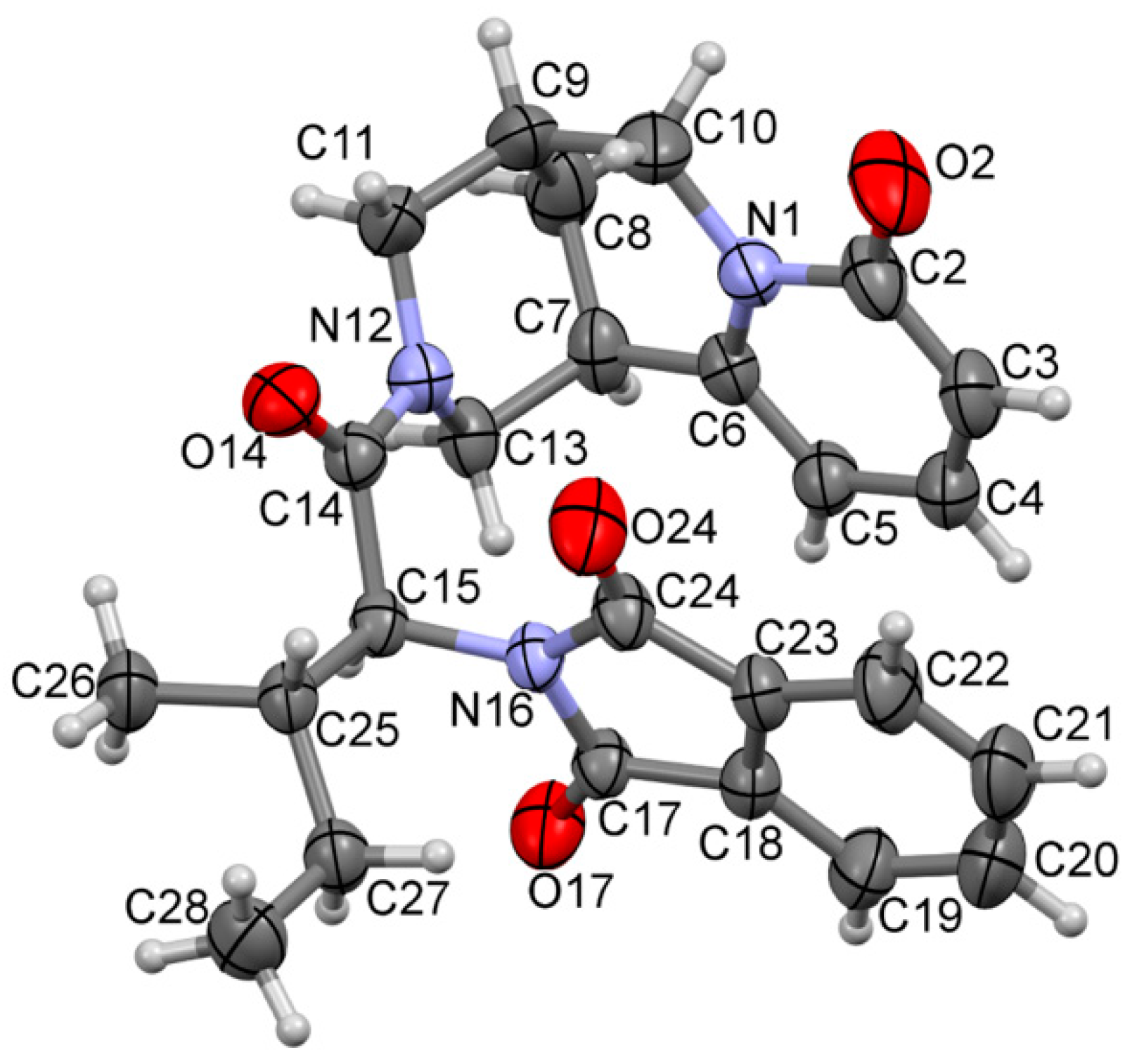
2.2. Crystal Data
- (a)
- Using wavefunctions at B3LYP/6-31G(d,p) level (hereinafter: B3LYP), the energy of interaction was calculated within the CrystalExplorer software [25] in terms of four key components: electrostatic, polarization, dispersion and exchange–repulsion Etot = keleEele + kpolEpol + kdisEdis +k repErep
- (b)
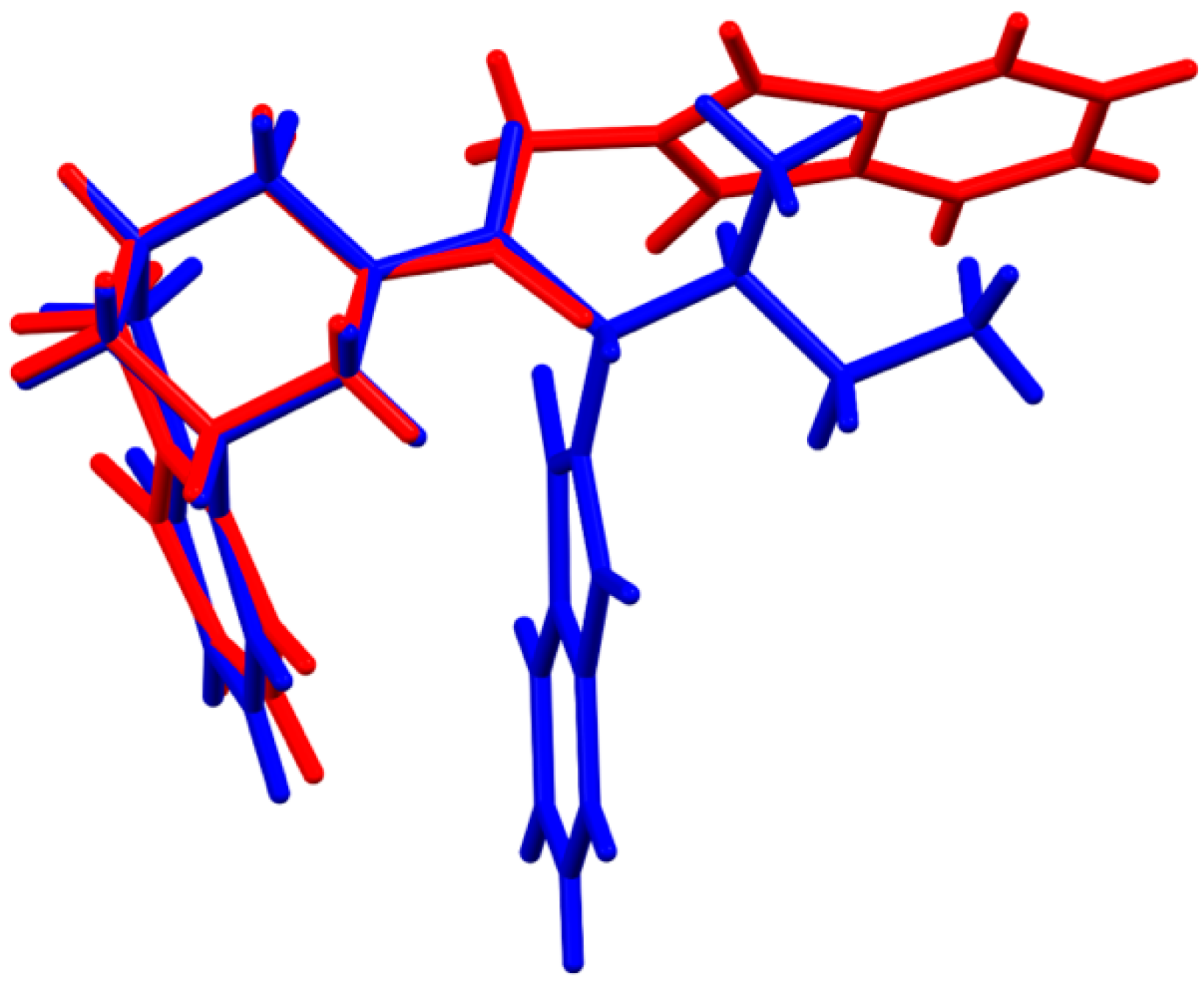
3. Results and Discussion
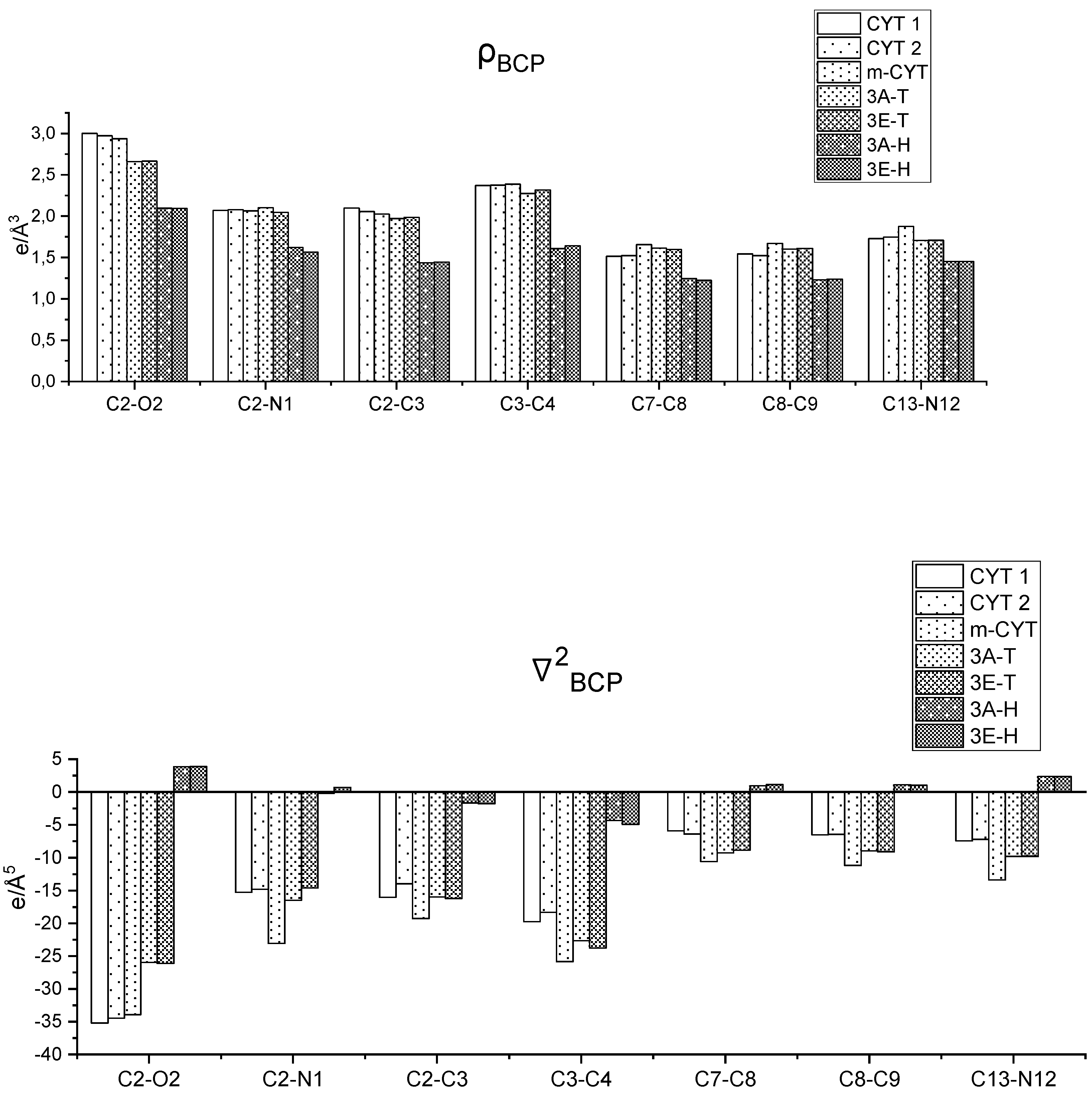
3.1. The Importance of the Quality of the Electron Density Model
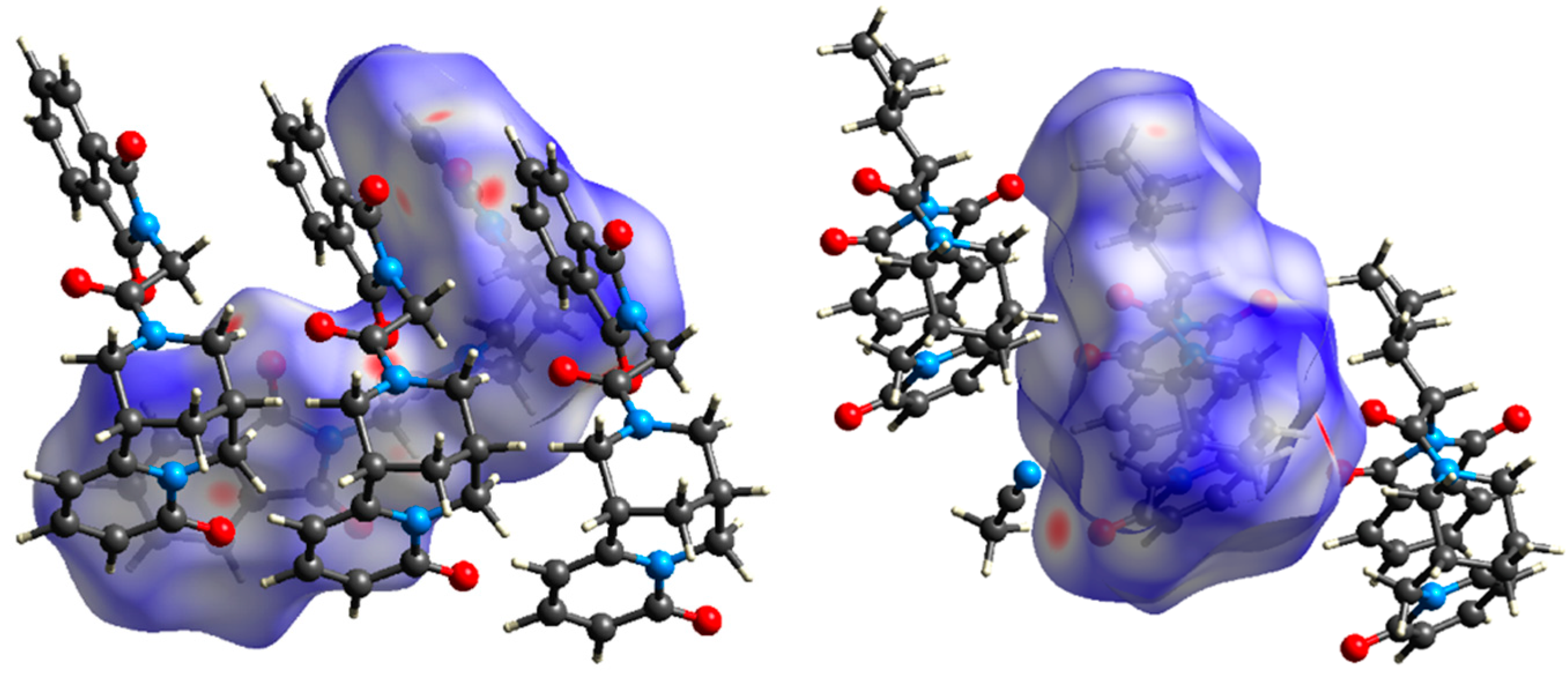
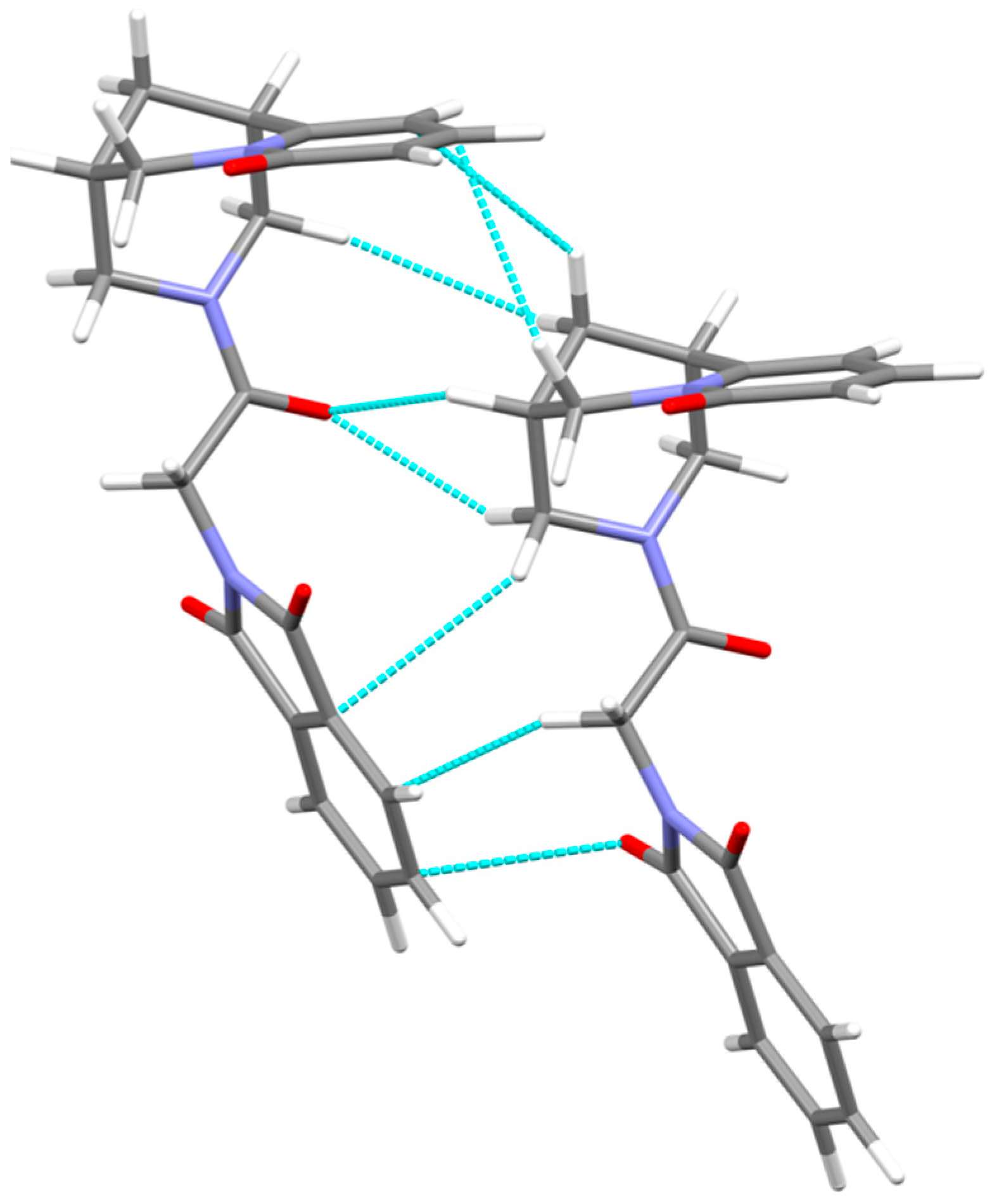
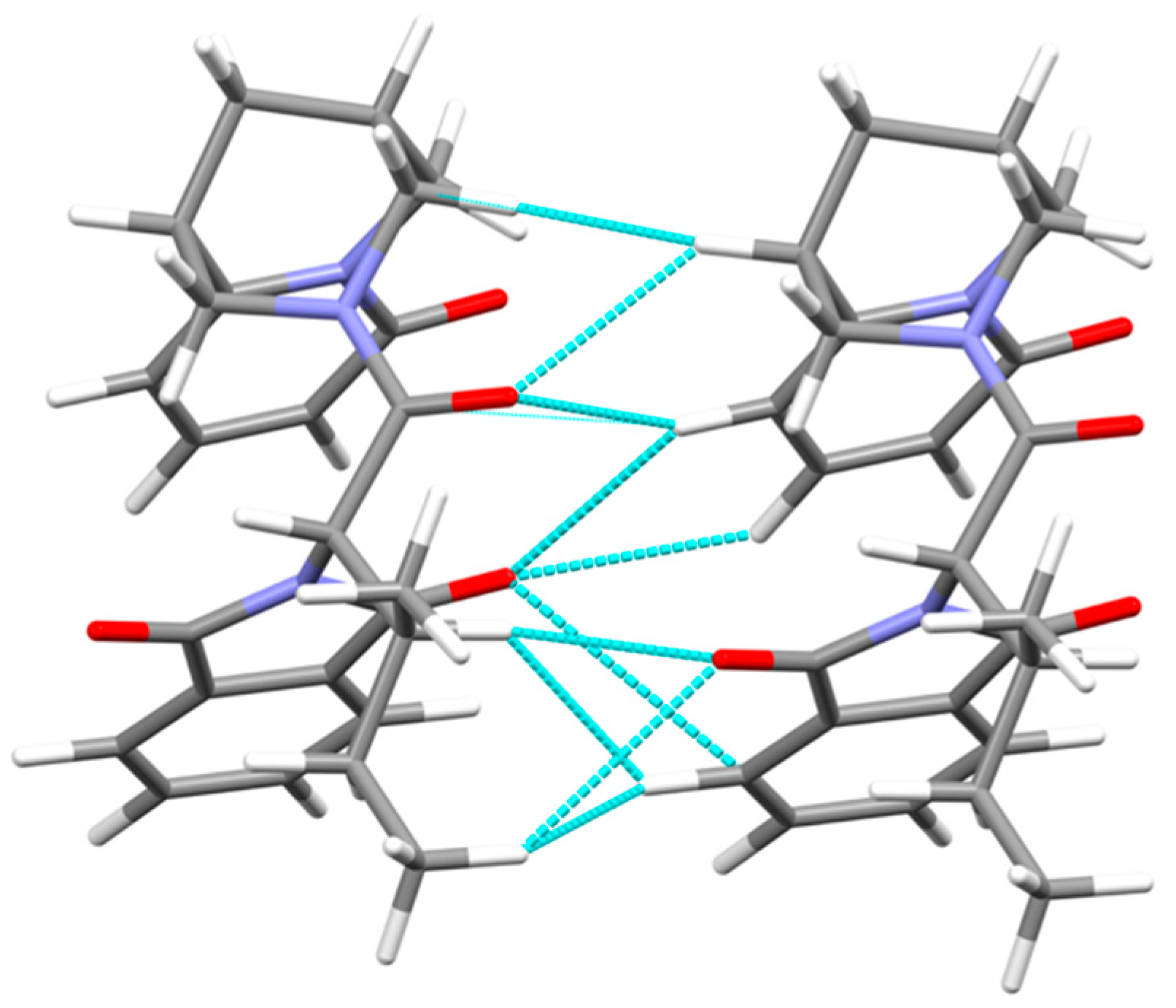
3.2. Crystal Packing, Intermolecular Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dunitz, J.D. Phase transitions in molecular crystals from a chemical viewpoint. Pure Appl. Chem. 1991, 63, 177–185. [Google Scholar] [CrossRef]
- Lehn, J.-M. From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry. Chem. Soc. Rev. 2007, 36, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. The C-H···O hydrogen bond: Structural implications and supramolecular design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The chalcogen bond in crystalline solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2020, 46, 280–288. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding interacion: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The nature and applications of π···π interactions: A perspective. Cryst. Growth Des. 2019, 19, 5230528. [Google Scholar] [CrossRef]
- Mahadevi, A.S.; Sastry, N. Cation-π interaction: Its role and relevance in chemistry, biology, and material science. Chem. Rev. 2013, 113, 2100–2138. [Google Scholar] [CrossRef]
- Wang, D.-X.; Wang, M.-X. Anion-π interactions: Generality, binding strength, and structure. J. Am. Chem. Soc. 2013, 135, 892–897. [Google Scholar] [CrossRef]
- Tutka, P.; Zatoński, W. Cytisine for the treatment of nicotine addiction: From a molecule to therapeutic efficiacy. Pharmacol. Rep. 2005, 58, 777–798. [Google Scholar]
- Rouden, J.; Lasne, M.-C.; Blanchet, J.; Baudoux, J. (−)-Cytisine and derivatives: Synthesis, reactivity, and applications. Chem. Rev. 2014, 114, 712–778. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Chellappan, S.K.; Xiao, Y.; Bajjuri, K.M.; Yuan, H.; Kellar, K.J.; Petukhov, P.A. Chemical medicine: Novel 10-substituted cytisine derivatives with increased selectivity for α4β2 nicotinic acetylcholine receptors. ChemMedChem 2007, 2, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Artyushin, O.I.; Moiseeva, A.A.; Zarubaev, V.V.; Slita, A.V.; Galochkina, A.V.; Muryleva, A.A.; Borisevich, S.S.; Yarovaya, O.I.; Salakhutdinov, N.F.; Brel, V.K. Synthesis of camphecene and cytisine conjugates using click chemistry methodology and study of their antiviral activity. Chem. Biodivers. 2019, 16, e1900340. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.V.; Nagasampagi, B.A.; Sivakumar, M. Chemistry of Natural Products; Springer: Berlin/Heidelberg, Germany, 2005; pp. 237–315. [Google Scholar]
- Juaristi, E.; Beck, A.K.; Hansen, J.; Matt, T.; Mukhopadhyay, T.; Simson, M.; Seebach, D. Enantioselective aldol and Michael additions of achiral enolates in the presence of chiral lithium amides and amines. Synthesis 1993, 1993, 1271–1290. [Google Scholar] [CrossRef]
- Kánai, K.; Podányi, B.; Bokotey, S.; Hajdú, F.; Hermecz, I. Stereoselective sulfoxide formation from a thioproline derivative. Tetrahedron Asymmetry 2002, 13, 491–496. [Google Scholar] [CrossRef]
- Przybył, A.K.; Prukała, W.; Kikut-Ligaj, D. EI-MS study of selected N-amide and N-alkyl derivatives of cytisine. Rapid Commun. Mass Spectrom. 2007, 21, 1409–1413. [Google Scholar] [CrossRef]
- Przybył, A.K.; Nowakowska, Z. EIMS study of Halogenated N-Acetyl- and N-Propionylcytisines. Rapid Commun. Mass Spectrom. 2011, 25, 1193–1197. [Google Scholar] [CrossRef]
- Przybył, A.K.; Kubicki, M. A comparative study of dynamic NMR spectroscopy in analysis of selected N-alkyl-, N-acyl-, and halogenated cytisine derivatives. J. Mol. Struct. 2011, 985, 157–166. [Google Scholar] [CrossRef]
- Przybył, A.K.; Kubicki, M.; Hoffmann, M. The amide protonation of (-)-N-benzoyl-cytisine in its perchlorate salts. Spectrochim. Acta A Mol. Biomol. Spectr. 2014, 129, 1–6. [Google Scholar] [CrossRef]
- CrysAlisPro; Rigaku Oxford Diffraction LTd.: Oxford, UK, 2013.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Part A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Part C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17. University of Western Australia. 2017. Available online: http://crystalexplorer.scb.uwa.edu.au/ (accessed on 29 January 2021).
- Gavezzotti, A. Are crystal structures predictable? Acc. Chem. Res. 1994, 27, 309–314. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Fillippini, G. Geometry of the intermolecular X-H···Y (X, Y = N, O) hydrogen bond and the calibration of empirical hydrogen-bond potentials. J. Phys. Chem. 1994, 98, 4831–4837. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Guillot, B.; Viry, L.; Guillot, R.; Lecomte, C.; Jelsch, C. Refinement of proteins at subatomic resolution with MOPRO. J. Appl. Cryst. 2001, 34, 214–223. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The cambridge structural database. Acta Cryst. Part B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Suponitskii, K.Y.; Tsypysheva, I.P.; Kovalskaya, A.V. Molecular and crystal structure of (1R,5S)-8-oxo-1,5,6,8-tetrahydro-2H-1,5-methanopyrido[1,2-a][1,5]diazocine-3(4H)-carboxamide and (1R,5S)-8-OXO-1,5,6,8-tetrahydro-2H-1,5-methanopyrido[1,2-a][1,5]diazocine-3(4H)-thiocarbo-xamide. J. Struct. Chem. 2015, 56, 188–190. [Google Scholar] [CrossRef]
- Hansen, N.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Cryst. Part A 1978, 71, 3–8. [Google Scholar] [CrossRef]
- Domagala, S.; Fournier, B.; Liebschner, D.; Guillot, B.; Jelsch, C. An improved experimental databank of transferable multipolar atom models—ELMAM2. Construction details and applications. Acta Cryst. Part A 2012, 68, 337–351. [Google Scholar] [CrossRef]
- Paul, A.; Kubicki, M.; Kubas, A.; Jelsch, C.; Fink, K.; Lecomte, C. Charge density analysis of 2-methyl-4-nitro-1-phenyl-1H-imidazole-5-carbonitrile: An experimental and theoretical study of C≡N ···C≡N interactions. J. Phys. Chem. A 2011, 115, 12941–12952. [Google Scholar] [CrossRef] [PubMed]
- Owczarzak, A.; Grześkiewicz, A.M.; Kubicki, M. Experimental studies of charge density distribution in the crystals of cytisine and N-methylcytisine. Inside the fake tobacco. Struct. Chem. 2017, 28, 1359–1367. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Hirshfeld surfaces: A new tool for visualizing and exploring molecular crystals. Chem. Eur. J. 1998, 4, 2136–2141. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. Molecular recognition in organic crystals. Directed intermolecular bonds or nonlocalized bonding? Angew. Chem. Int. Ed. 2005, 44, 1766–1787. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
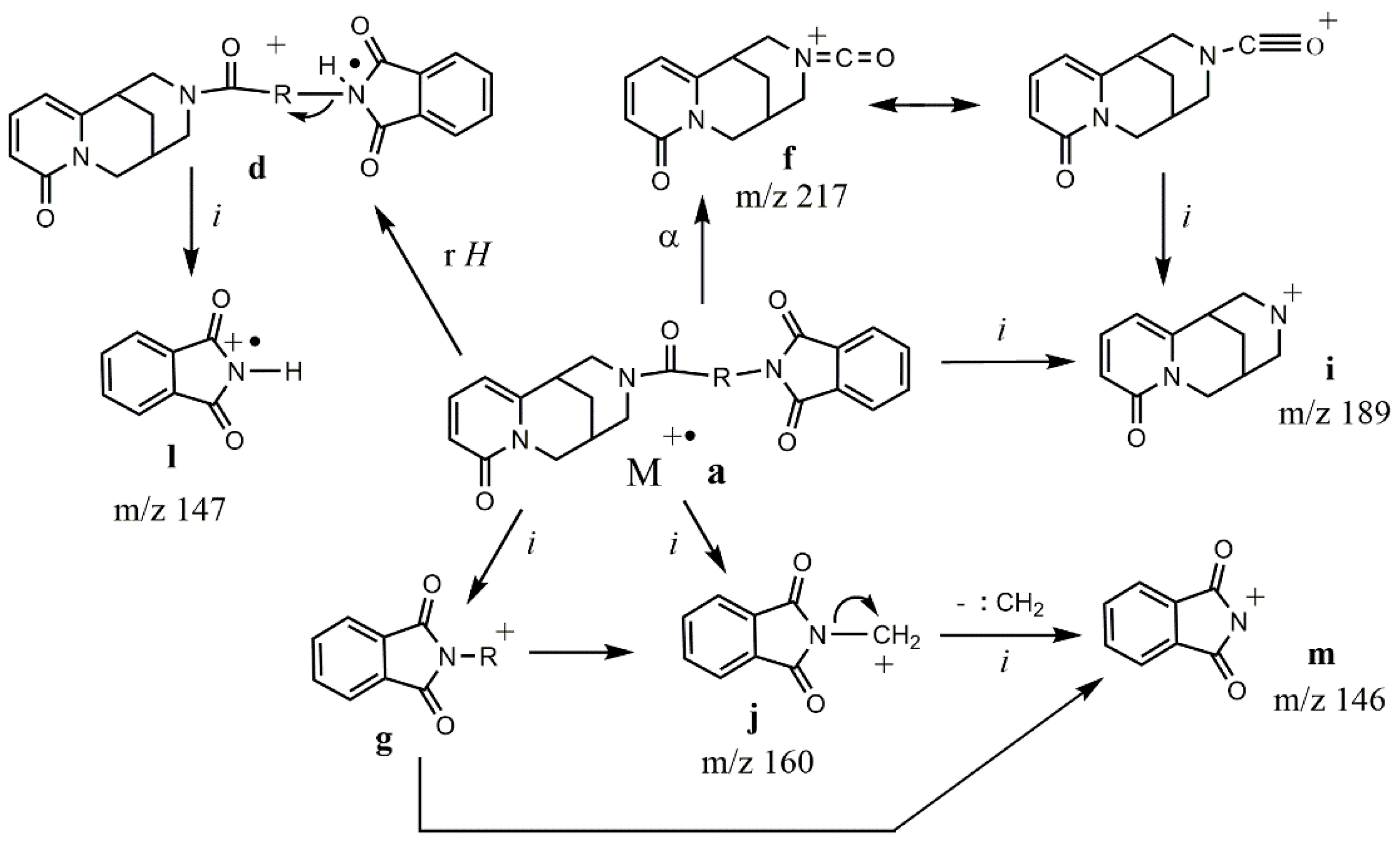
| Ion | Elemental Composition | m/z | 3A | 3B | 3C (D) | 3D (L) | 3E (L) | 3G (D) | 3F (L) | 3H (D) | 3H (L) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M+• a | C21H19N3O4 C22H21N3O4 C24H25N3O4 C25H27N3O4 C28H25N3O4 | 377 391 419 433 467 | 29 - - - | - 21 - - | - 30 - - | - - 7 - | - - - 1 | - - - 16 | - - - 3 | - - - - 27 | - - - - 3 |
| d | C21H19N3O4 | 377 | - | - | 4 | 12 | 70 | 34 | 46 | - | - |
| f | C12H13N2O2 | 217 | 14 | 1 | 8 | 6 | 12 | 10 | 6 | 1 | 3 |
| g | C9H6NO2 C13H14NO2 C10H8NO2 C12H12NO2 | 160 216 174 202 | 100 - - - | - - 5 - | - - - 100 | - - - 100 | - 19 - - | - 24 - - | - 41 - - | 30 - 13 2 | 13 - 1 - |
| i | C11H13N2O | 189 | 35 | 30 | 18 | 27 | 37 | 23 | 21 | 4 | 13 |
| j | C9H6NO2 | 160 | 100 | 73 | 63 | 67 | 100 | 100 | 100 | 30 | 13 |
| l | C8H5NO2 | 147 | 2 | 100 | 56 | 64 | 75 | 72 | 36 | 3 | 74 |
| m | C8H4NO2 | 146 | 87 | 81 | 64 | 58 | 55 | 54 | 36 | 35 | 38 |
| 1 | 3A | 3B | 3D cis/trans | 3E cis/trans | 3F | 3H | |
|---|---|---|---|---|---|---|---|
| At C | δ (ppm) | ||||||
| 2 | 162.26 | 165.1/164.6 | 163.6/163.5 | 163.2/162.4 | 163.3/162.4 | 162.9/162.9 | 162.9/162.9 |
| 3 | 115.04 | 118.3/117.7 | 117.4/116.9 | 117.3/116.9 | 117.3/116.8 | 117.9/117.3 | 118.1/117.2 |
| 4 | 138.64 | 138.8/138.4 | 139.5/139.1 | 139.0/136.8 | 139.0/136.9 | 138.6/137.4 | 137.0/136.8 |
| 5 | 103.80 | 105.6/104.7 | 106.6/105.9 | 105.8/104.0 | 105.8/104.1 | 105.1/104.3 | 105.9/105.1 |
| 6 | 152.34 | 147.7/147.7 | 148.4/148.2 | 134.2/131.4 | 148.1/148.1 | 147.9/147.9 | 134.1/133.9 |
| 7 | 34.68 | 34.8/34.1 | 34.7/34.0 | 27.6/26.8 | 33.3/32.9 | 30.0/29.6 | 35.2/34.3 |
| 8 | 25.78 | 26.1/25.9 | 25.71/25.6 | 20.8/20.0 | 26.1/25.9 | 24.9/24.8 | 26.1/25.9 |
| 9 | 27.13 | 27.3/27.3 | 27.2/27.0 | 26.7/26.2 | 27.6/26.7 | 27.2/27.0 | 27.4/26.8 |
| 10 | 49.39 | 48.7/48.7 | 49.9/48.5 | 49.4/48.9 | 51.5/51.1 | 49.2/48.7 | 51.7/51.3 |
| 11 | 52.53 | 49.4/48.3 | 48.4/47.5 | 51.3/51.0 | 48.9/48.5 | 50.6/50.2 | 48.748.7 |
| 13 | 53.45 | 52.3/50.9 | 52.7/51.43 | 55.8/55.3 | 54.7/54.3 | 51.6/51.6 | 51.7/51.3 |
| 3A | 3E | 3A | 3E | ||
|---|---|---|---|---|---|
| N1–C2 | 1.400(3) | 1.418(4) | N1–C6 | 1.380(3) | 1.369(4) |
| N1–C10 | 1.473(3) | 1.478(5) | C2–O2 | 1.241(3) | 1.241(5) |
| C11–N12 | 1.464(3) | 1.463(4) | N12–C13 | 1.462(3) | 1.461(4) |
| N12–C14 | 1.352(3) | 1.364(4) | C14–O14 | 1.222(3) | 1.218(4) |
| C15–N16 | 1.437(3) | 1.470(4) | N16–C17 | 1.395(3) | 1.397(4) |
| N16–C24 | 1.389(3) | 1.406(4) | C17–O17 | 1.209(3) | 1.199(4) |
| C24–O24 | 1.203(3) | ||||
| C2–N1–C6 | 122.4(2) | 122.2(3) | C2–N1–C10 | 113.9(2) | 114.1(3) |
| C6–N1–C10 | 123.7(2) | 123.6(3) | C11–N12–C13 | 115.9(2) | 114.6(3) |
| C11–N12–C14 | 123.6(2) | 116.8(3) | C13–N12–C14 | 117.6(2) | 124.8(3) |
| C15–N16–C17 | 124.0(2) | 122.3(2) | C15–N16–C24 | 124.4(2) | 124.6(2) |
| C17–N16–C24 | 111.5(2) | 111.8(3) | |||
| C7–C6–N1–C10 | 3.2(3) | −0.4(5) | C6–C7–C8–C9 | −57.7(3) | −61.8(4) |
| C7–C8–C9–C10 | 68.3(3) | 63.6(4) | C8–C9–C10–N1 | −43.3(3) | −33.5(5) |
| C9–C10–N1–C6 | 7.5(3) | 1.4(5) | C7–C8–C9–C11 | −59.0(3) | −61.0(4) |
| C8–C9–C11–N12 | 49.6(3) | 57.2(4) | C9–C11–N12–C13 | −45.0(3) | −53.0(5) |
| C11–N12–C13–C7 | 49.6(3) | 52.2(4) | N12–C13–C7–C8 | −60.2(3) | −56.3(4) |
| C13–C7–C8–C9 | 64.8(3) | 60.8(4) | C9–C11–N12–C14 | 154.7(2) | 147.9(4) |
| C7–C13–N12–C14 | −148.9(2) | −150.6(3) | C11–N12–C14–C15 | −13.1(3) | 177.9(3) |
| C13–N12–C14–C15 | −173.1(2) | 21.1(4) | N12–C14–C15–N16 | −176.7(2) | 60.4(3) |
| C11–N12–C14–O14 | 167.2(2) | −2.6(5) | C13–N12–C14–O14 | 7.2(4) | −159.4(3) |
| C14–C15–N16–C17 | −77.6(3) | 99.2(3) | C14–C15–N16–C24 | 97.4(3) | 57.9(4) |
| D | H | A | D–H | H···A | D···A | D–H···A | ||
|---|---|---|---|---|---|---|---|---|
| C9 | H9 | O14i | 1.09 | 2.39 | 3.181 | 129 | ||
| C15 | H15B | C22i | 1.09 | 2.73 | 3.896 | 117 | ||
| C11 | H11B | O14i | 1.09 | 2.61 | 3.310 | 122 | ||
| C8 | H8B | C5i | 1.09 | 3.31 | 3.854 | 144 | ||
| C10 | H10A | C4i | 1.09 | 2.99 | 3.963 | 150 | ||
| C11 | H11A | C23i | 1.09 | 3.14 | 3.896 | 127 | ||
| contacts | ||||||||
| O17 | C20i | 3.826 | ||||||
| H8A | H13Bi | 2.90 | ||||||
| Atom 1 | Atom 2 | Gcp | Vcp | D12 | Dcp1 | Dcp2 | Den | Lap |
| H9 | O14i | 23.65 | −17.62 | 2.3798 | 0.9848 | 1.3951 | 0.072 | 1.09 |
| H15B | C22i | 15.46 | −11.13 | 2.7260 | 1.0967 | 1.6605 | 0.052 | 0.727 |
| H11B | O14i | 14.63 | −9.44 | 2.6056 | 1.1224 | 1.5003 | 0.039 | 0.727 |
| H8B | C5i | 7.76 | −5.04 | 2.9847 | 1.1959 | 1.8014 | 0.027 | 0.385 |
| H10A | C4i | 5.61 | −3.91 | 2.9823 | 1.2298 | 1.7607 | 0.026 | 0.269 |
| H11A | C23i | 6.2 | −3.91 | 3.1346 | 1.2537 | 1.9289 | 0.022 | 0.312 |
| O17 | C20i | 5.48 | −3.46 | 3.5235 | 1.7248 | 1.8222 | 0.020 | 0.275 |
| H8A | H13Bi | 2.88 | −1.72 | 2.8997 | 1.4358 | 1.4647 | 0.012 | 0.149 |
| D | H | A | D–H | H···A | D···A | D–H···A | ||
|---|---|---|---|---|---|---|---|---|
| C5 | H5 | O14i | 1.09 | 2.23 | 3.257 | 157 | ||
| C7 | H7 | O14i | 1.09 | 2.65 | 3.798 | 136 | ||
| C25i | H25i | O17 | 1.09 | 2.72 | 3.537 | 132 | ||
| C4 | H4 | O24i | 1.09 | 2.81 | 3.551 | 125 | ||
| C5 | H5 | O24i | 1.09 | 2.86 | 3.566 | 123 | ||
| C28i | H28Bi | O17 | 1.09 | 3.04 | 3.913 | 138 | ||
| contacts | ||||||||
| C19 | O24i | 3.526 | ||||||
| H19 | H25 | 2.558 | ||||||
| H19 | H28B | 2.605 | ||||||
| H7 | H11Ai | 2.459 | ||||||
| Atom 1 | Atom 2 | Gcp | Vcp | D12 | Dcp1 | Dcp2 | Den | Lap |
| H5 | O14 | 27.12 | −22.4 | 2.2334 | 0.8943 | 1.3399 | 0.093 | 1.169 |
| H7 | O14 | 11.61 | −7.79 | 2.6429 | 1.1201 | 1.5251 | 0.038 | 0.566 |
| O17 | H25 | 10.21 | −6.68 | 2.7114 | 1.5597 | 1.153 | 0.033 | 0.505 |
| H7 | H11A | 5.72 | −4.09 | 2.4475 | 1.2347 | 1.2151 | 0.028 | 0.27 |
| H4 | O24 | 7.83 | −4.76 | 2.8178 | 1.2065 | 1.6115 | 0.023 | 0.4 |
| H5 | O24 | 7.32 | −4.43 | 2.8605 | 1.2263 | 1.636 | 0.021 | 0.375 |
| C19 | O24 | 5.83 | −3.56 | 3.5262 | 1.8699 | 1.6669 | 0.019 | 0.297 |
| H19 | H25 | 5.44 | −3.36 | 2.5544 | 1.2681 | 1.2919 | 0.019 | 0.276 |
| H19 | H28B | 4.47 | −2.71 | 2.6173 | 1.3034 | 1.3178 | 0.016 | 0.229 |
| O17 | H28B | 4.34 | −2.65 | 3.0464 | 1.7272 | 1.3338 | 0.016 | 0.222 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przybył, A.K.; Grzeskiewicz, A.M.; Kubicki, M. Weak Interactions in the Structures of Newly Synthesized (–)-Cytisine Amino Acid Derivatives. Crystals 2021, 11, 146. https://doi.org/10.3390/cryst11020146
Przybył AK, Grzeskiewicz AM, Kubicki M. Weak Interactions in the Structures of Newly Synthesized (–)-Cytisine Amino Acid Derivatives. Crystals. 2021; 11(2):146. https://doi.org/10.3390/cryst11020146
Chicago/Turabian StylePrzybył, Anna K., Anita M. Grzeskiewicz, and Maciej Kubicki. 2021. "Weak Interactions in the Structures of Newly Synthesized (–)-Cytisine Amino Acid Derivatives" Crystals 11, no. 2: 146. https://doi.org/10.3390/cryst11020146
APA StylePrzybył, A. K., Grzeskiewicz, A. M., & Kubicki, M. (2021). Weak Interactions in the Structures of Newly Synthesized (–)-Cytisine Amino Acid Derivatives. Crystals, 11(2), 146. https://doi.org/10.3390/cryst11020146