
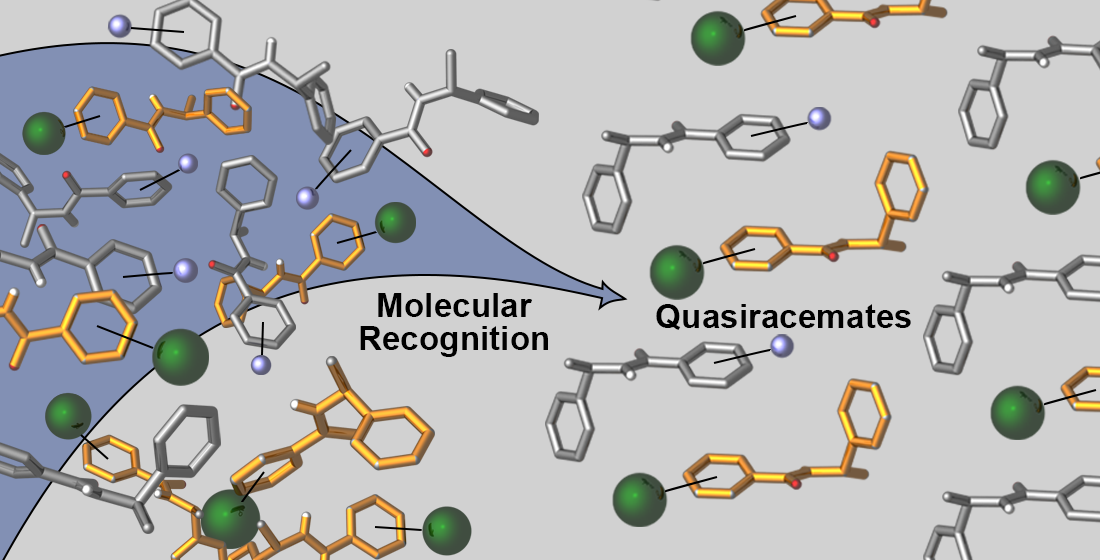
Molecular Recognition and Shape Studies of 3- and 4-Substituted Diarylamide Quasiracemates

 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthetic and Crystal Growth Procedures
2.2. Hot Stage Polarized Thermomicroscopy
2.3. Single Crystal X-ray Crystallography
2.4. Differential Scanning Calorimetry
2.5. Lattice Energy Calculations
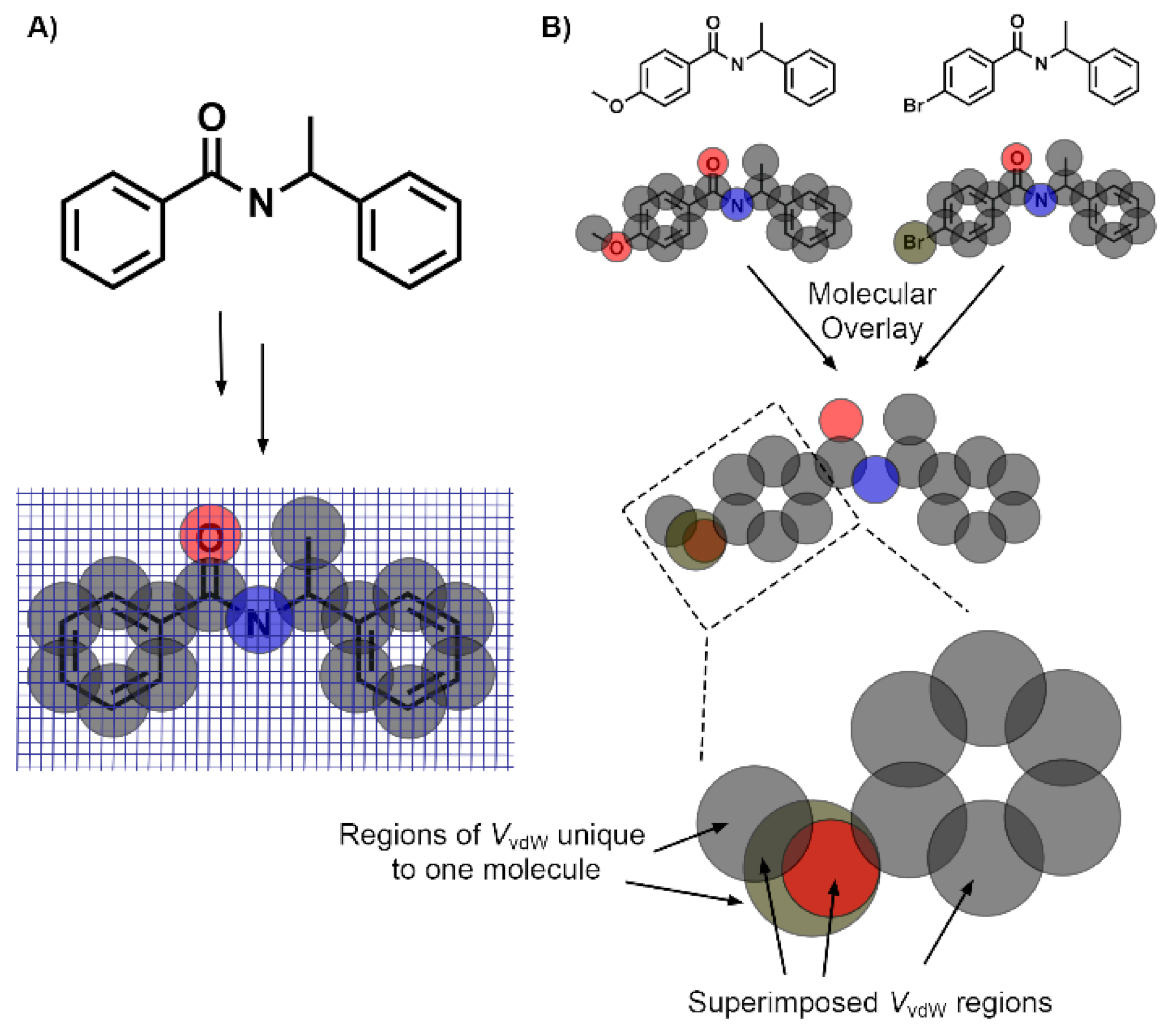
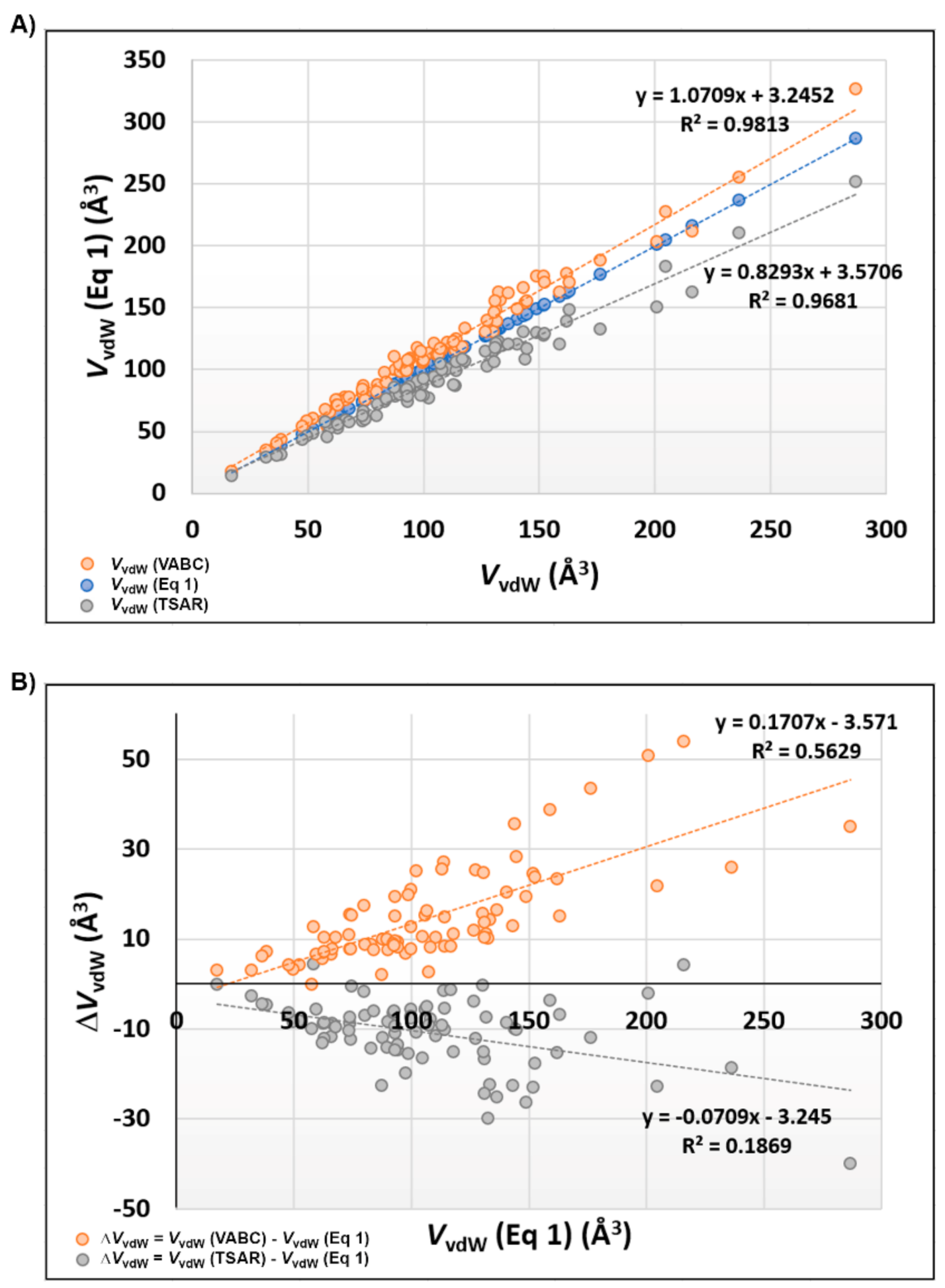
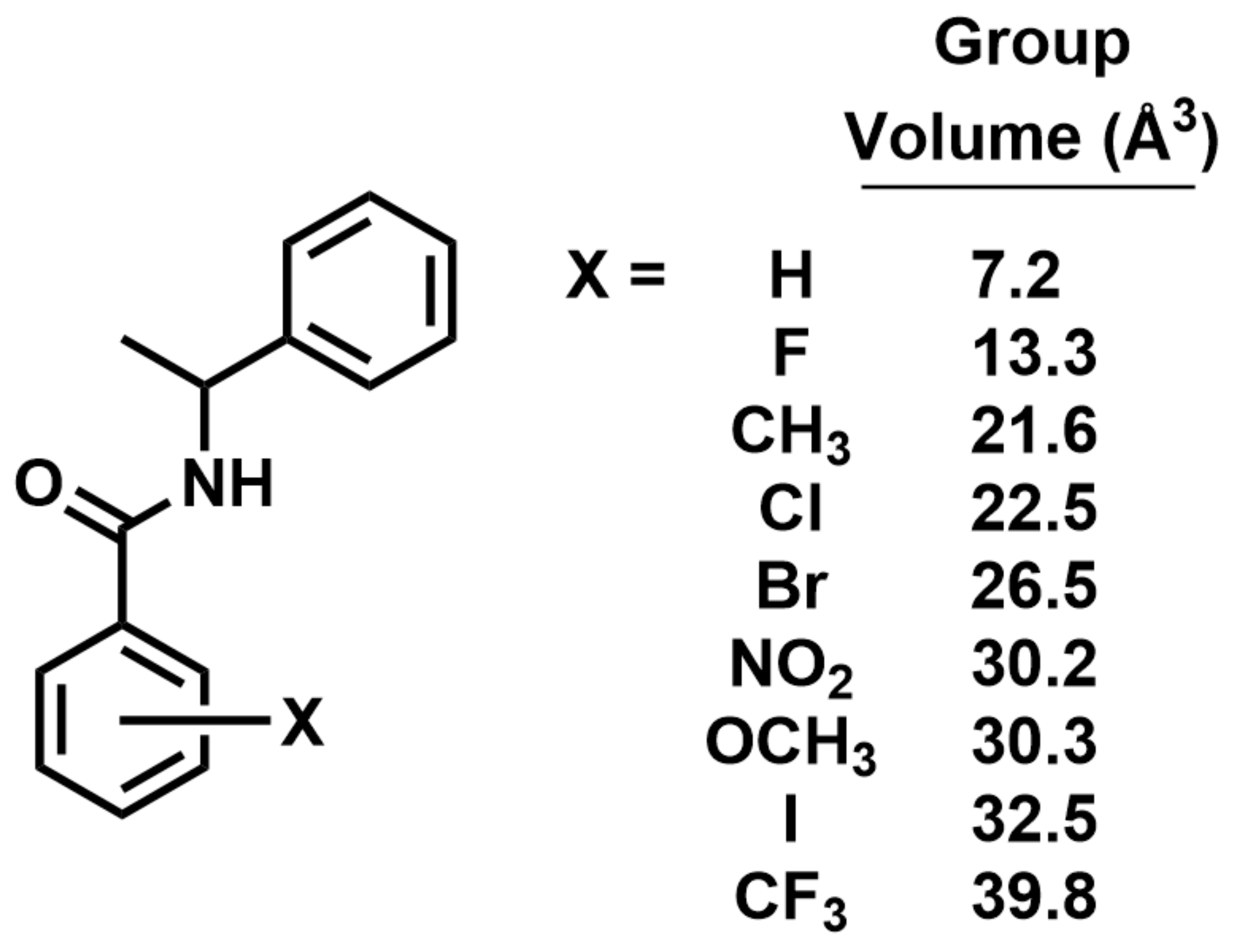
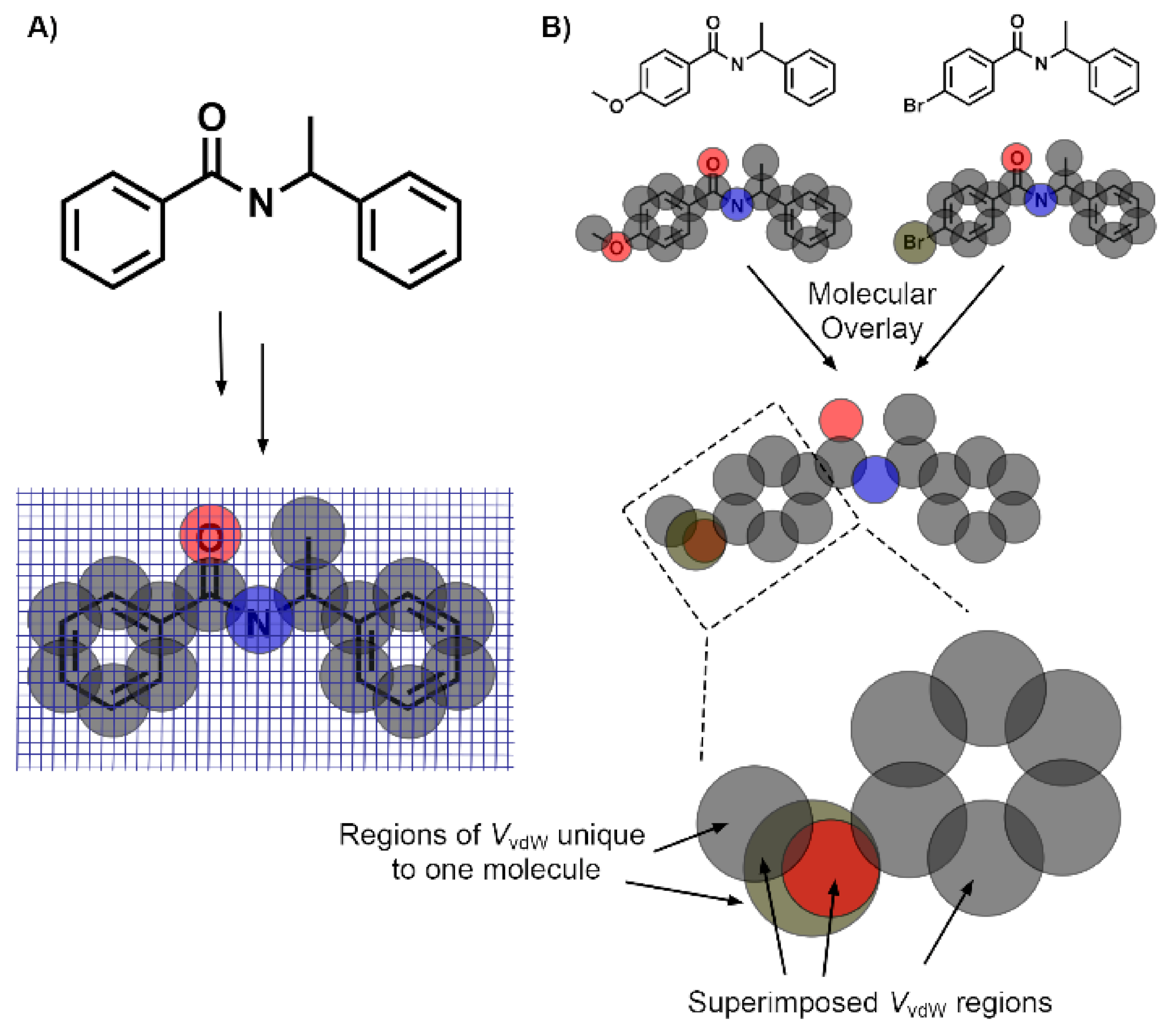
2.6. Molecular Volume and Shape Difference Determinations
- Δx = grid mesh size (Å3)
- n = number of grid points that coincide with the molecule
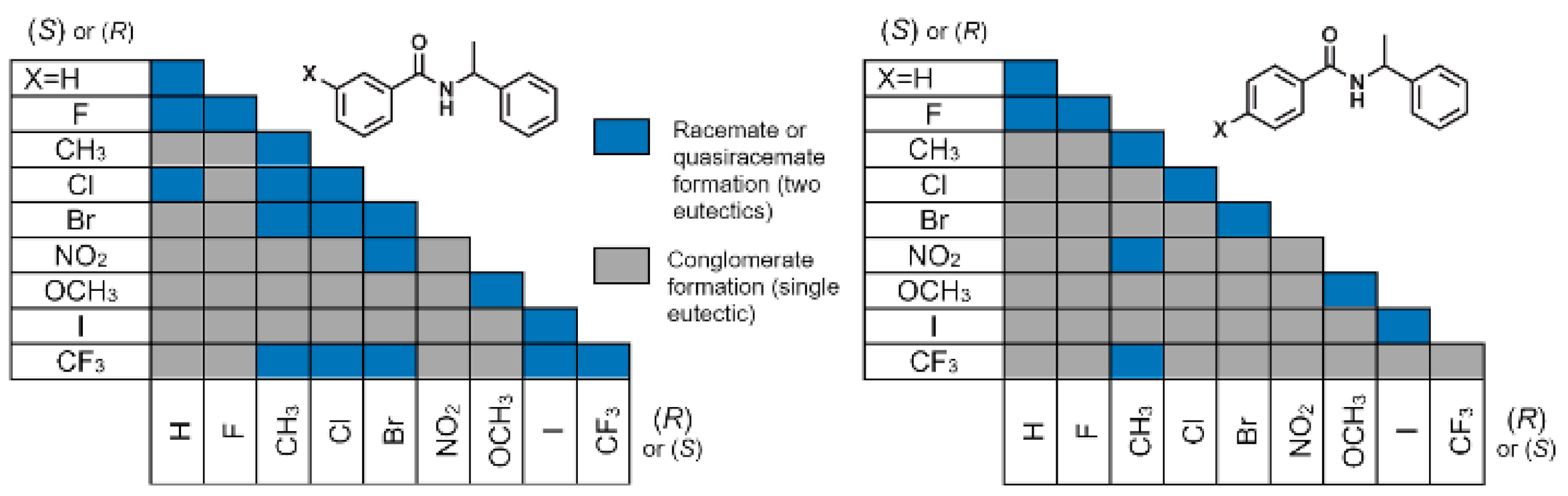
3. Results and Discussion
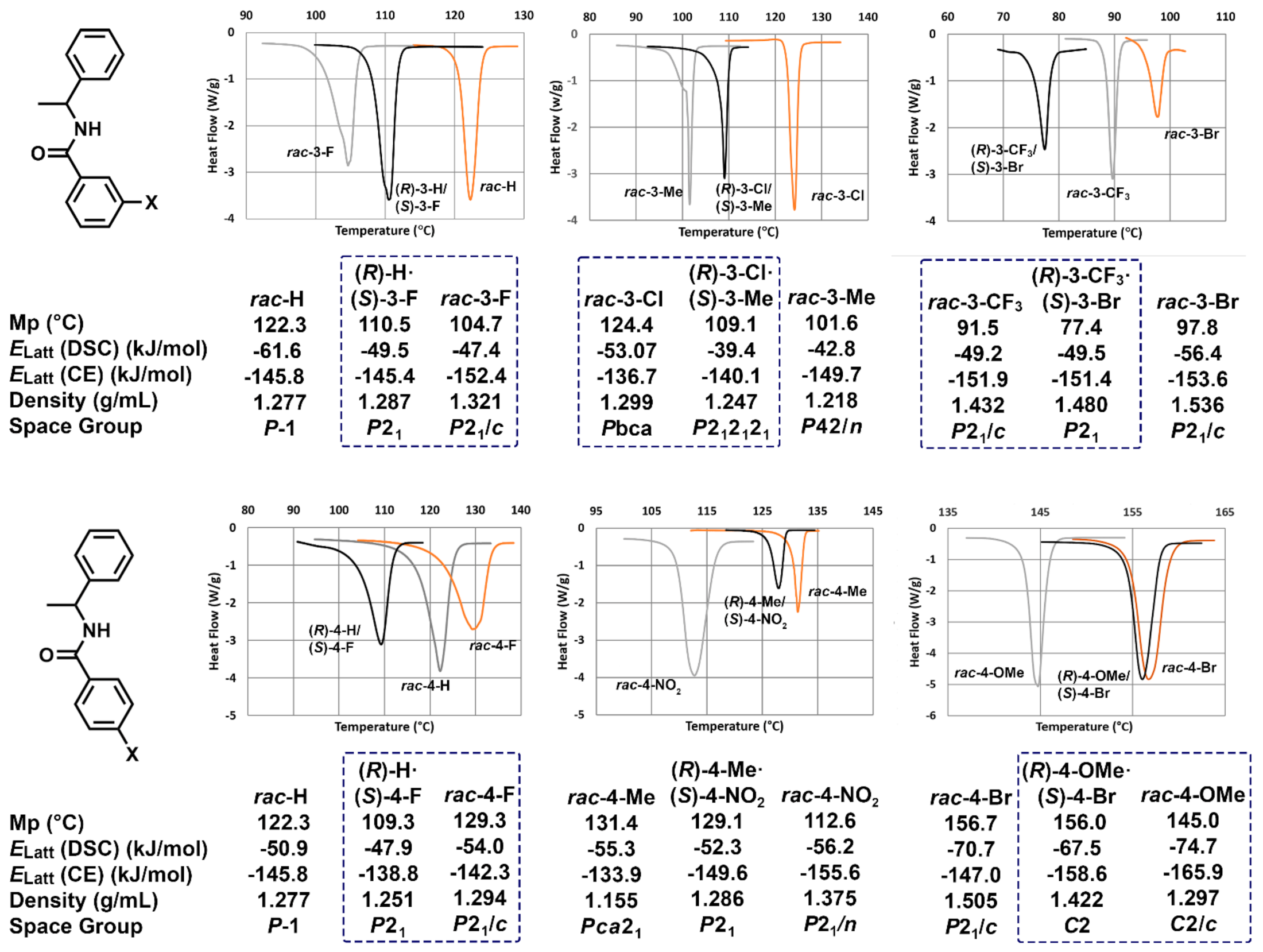
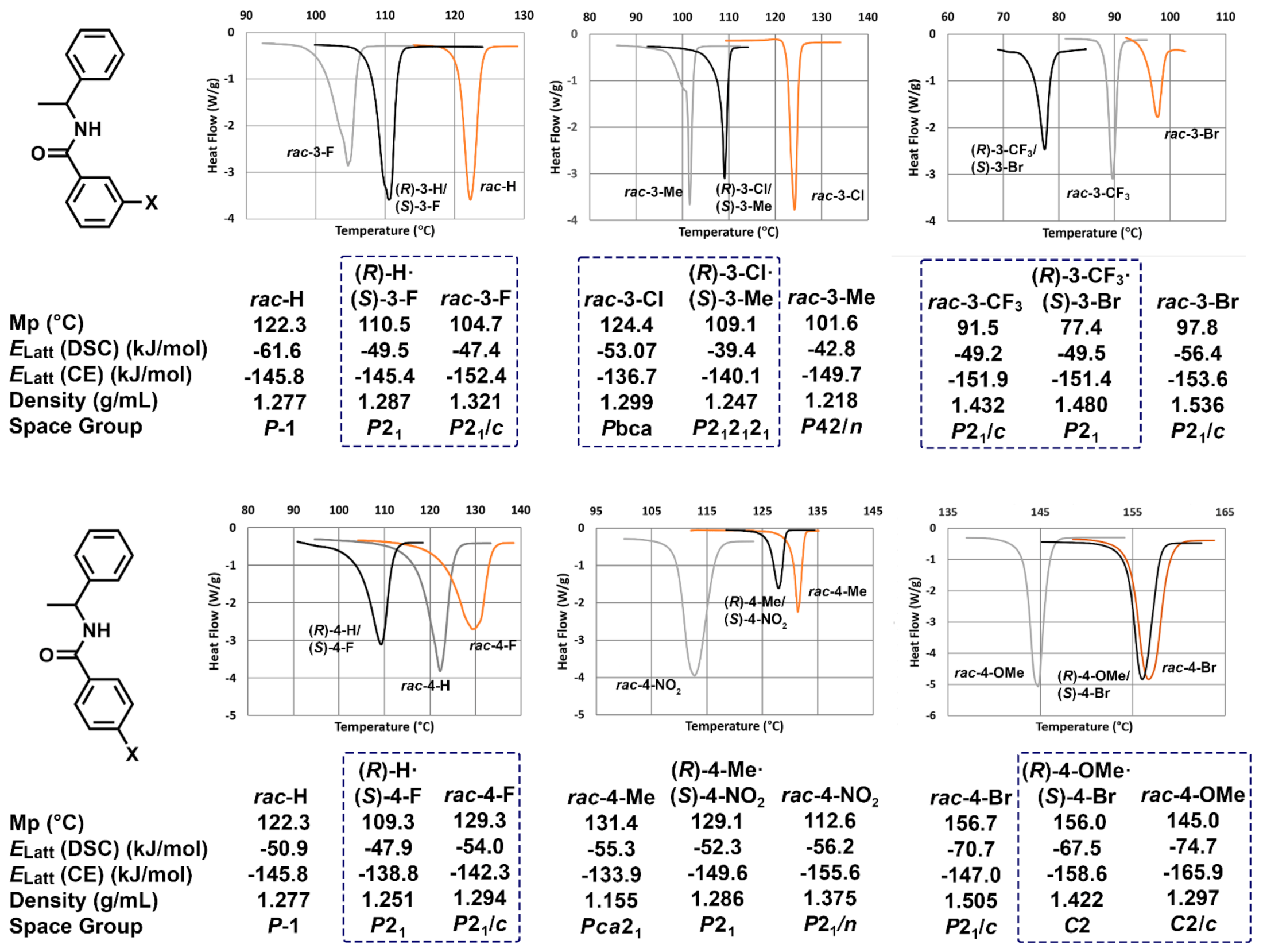
3.1. Hot Stage Thermomicroscopy
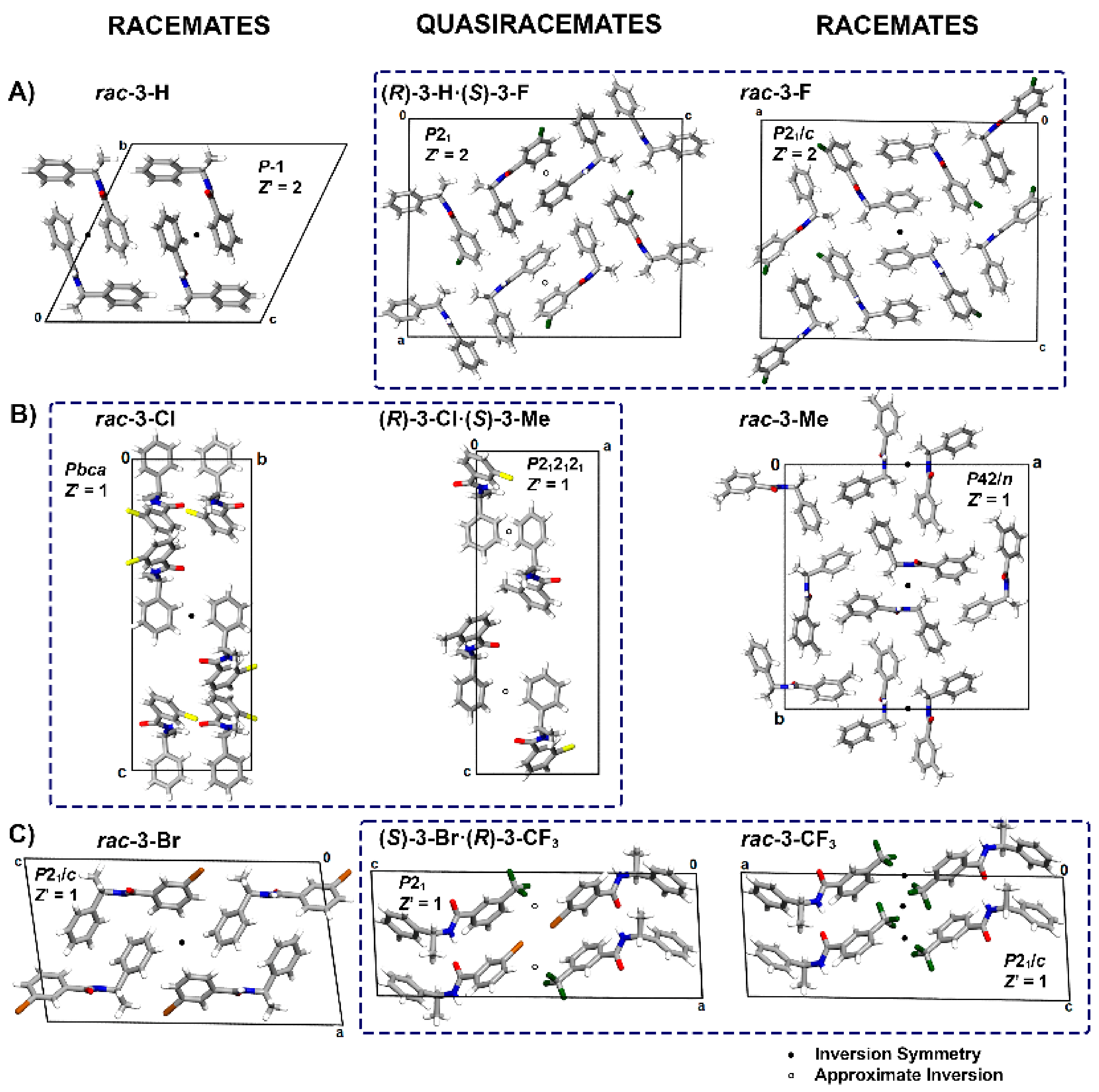
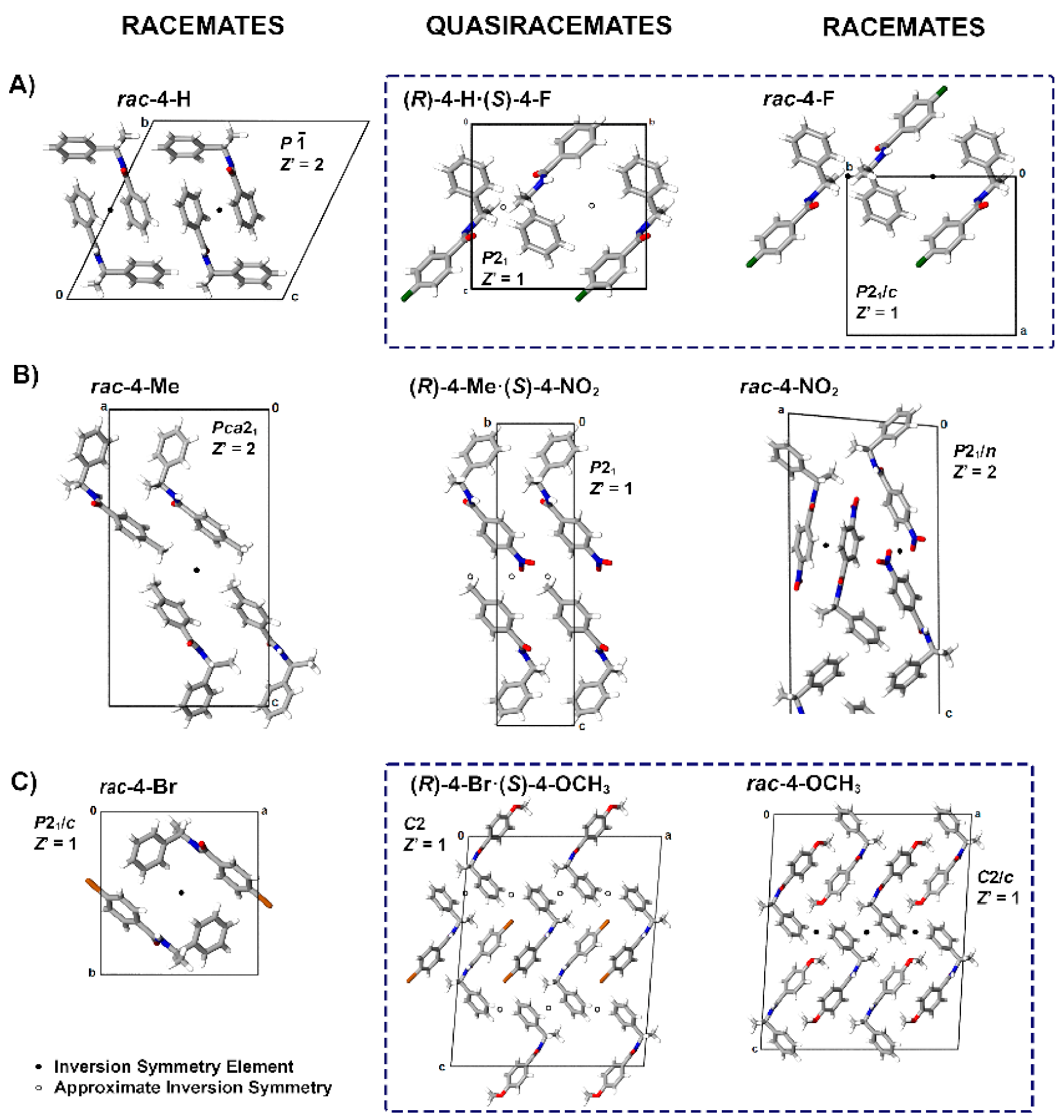
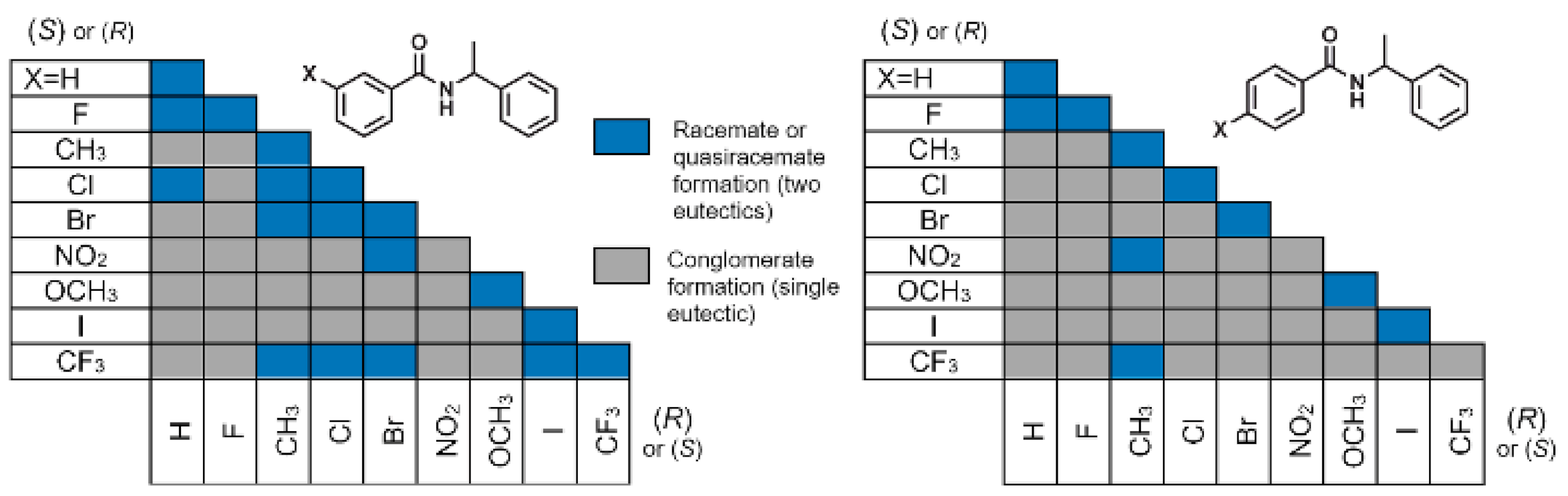
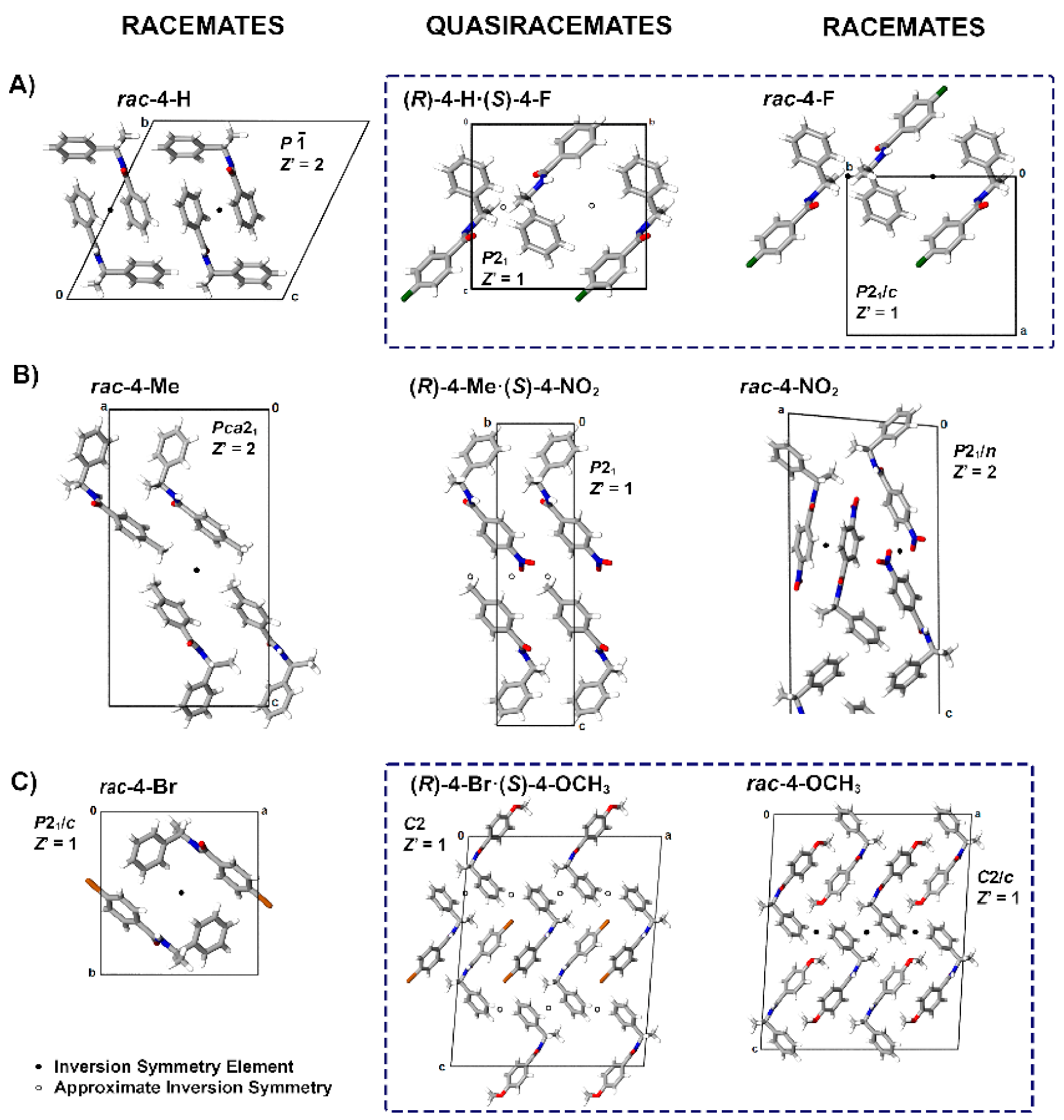
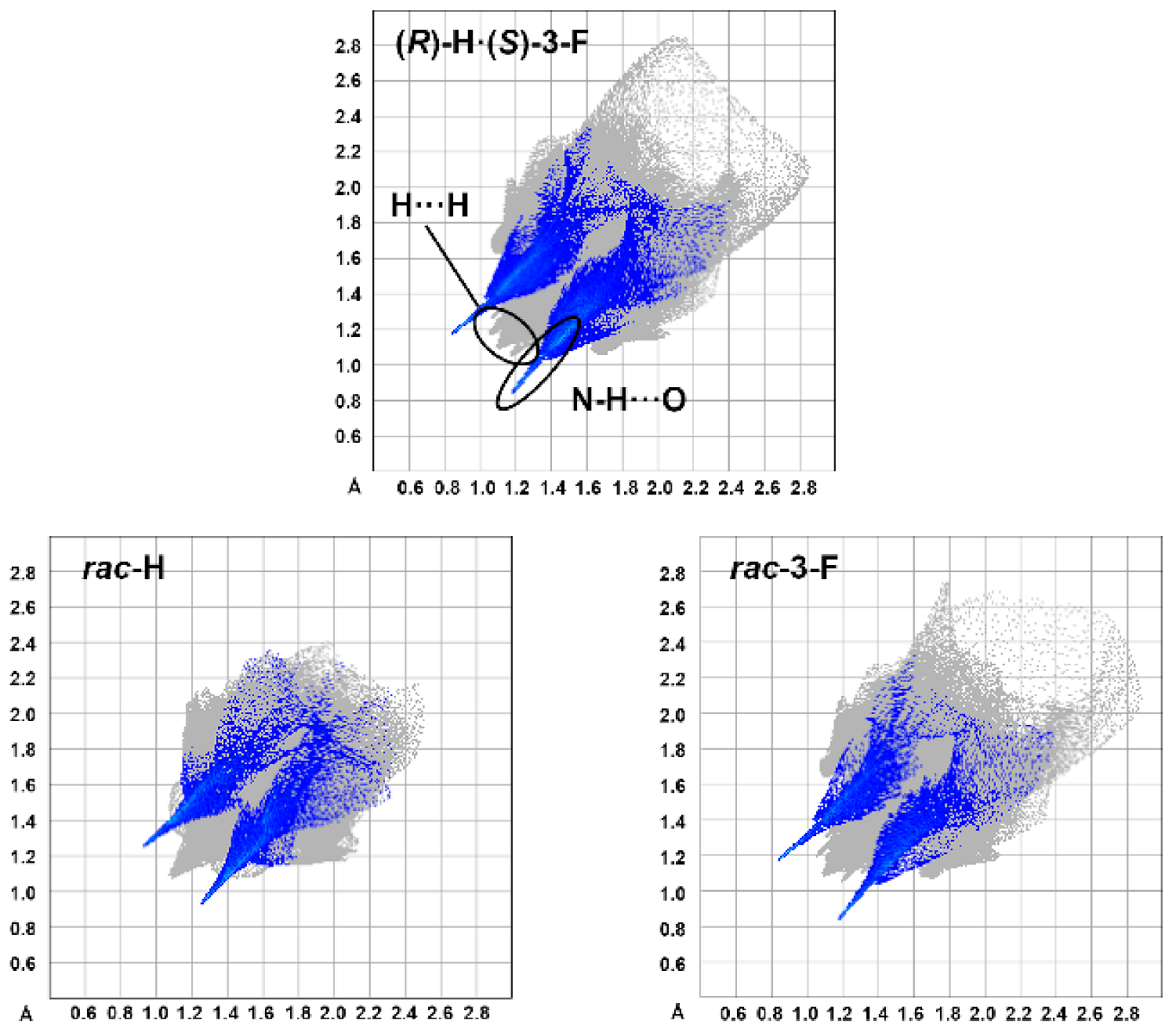
3.2. Crystal Structure Assessment
3.3. Crystal Lattice Energy Determinations
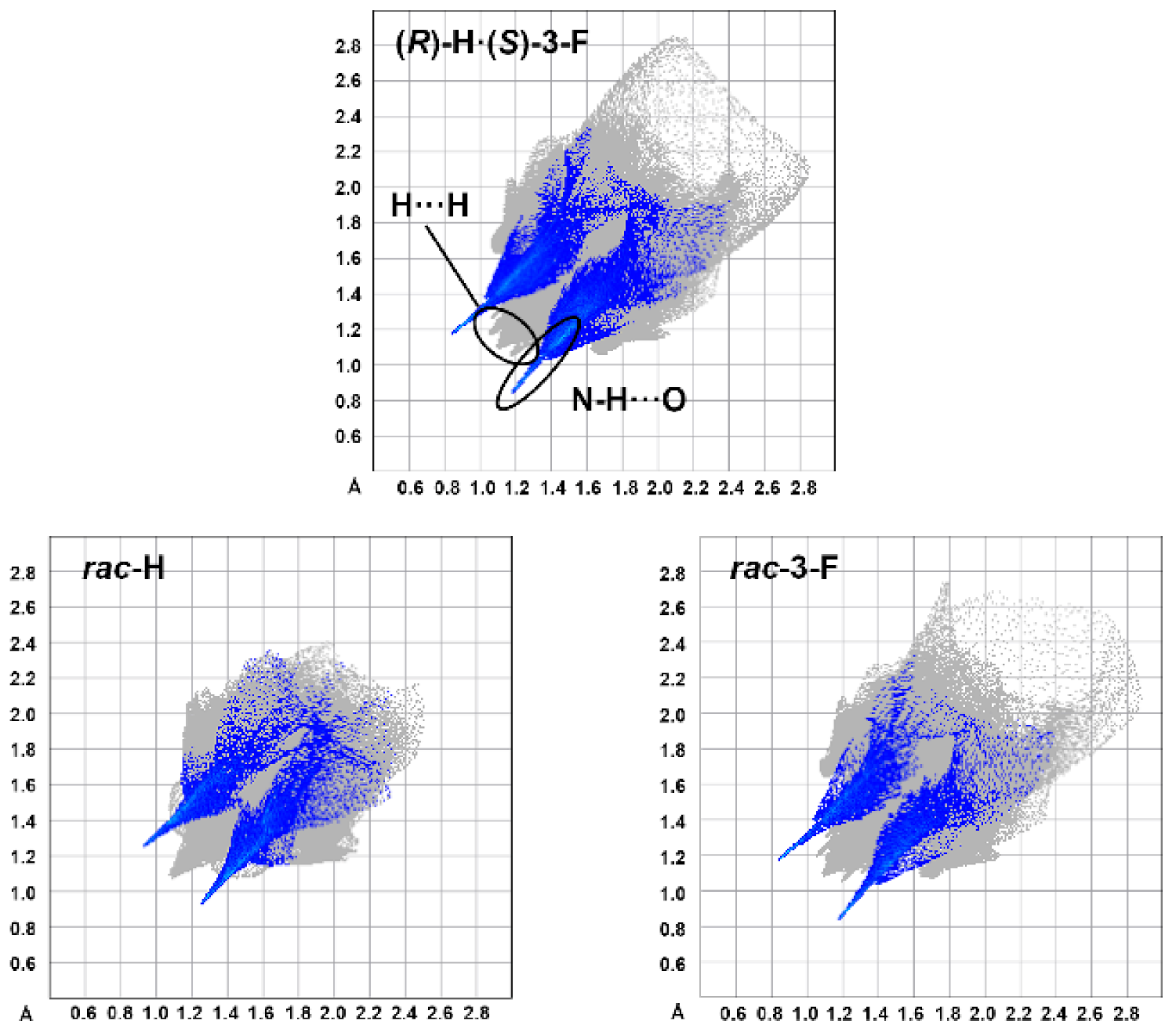
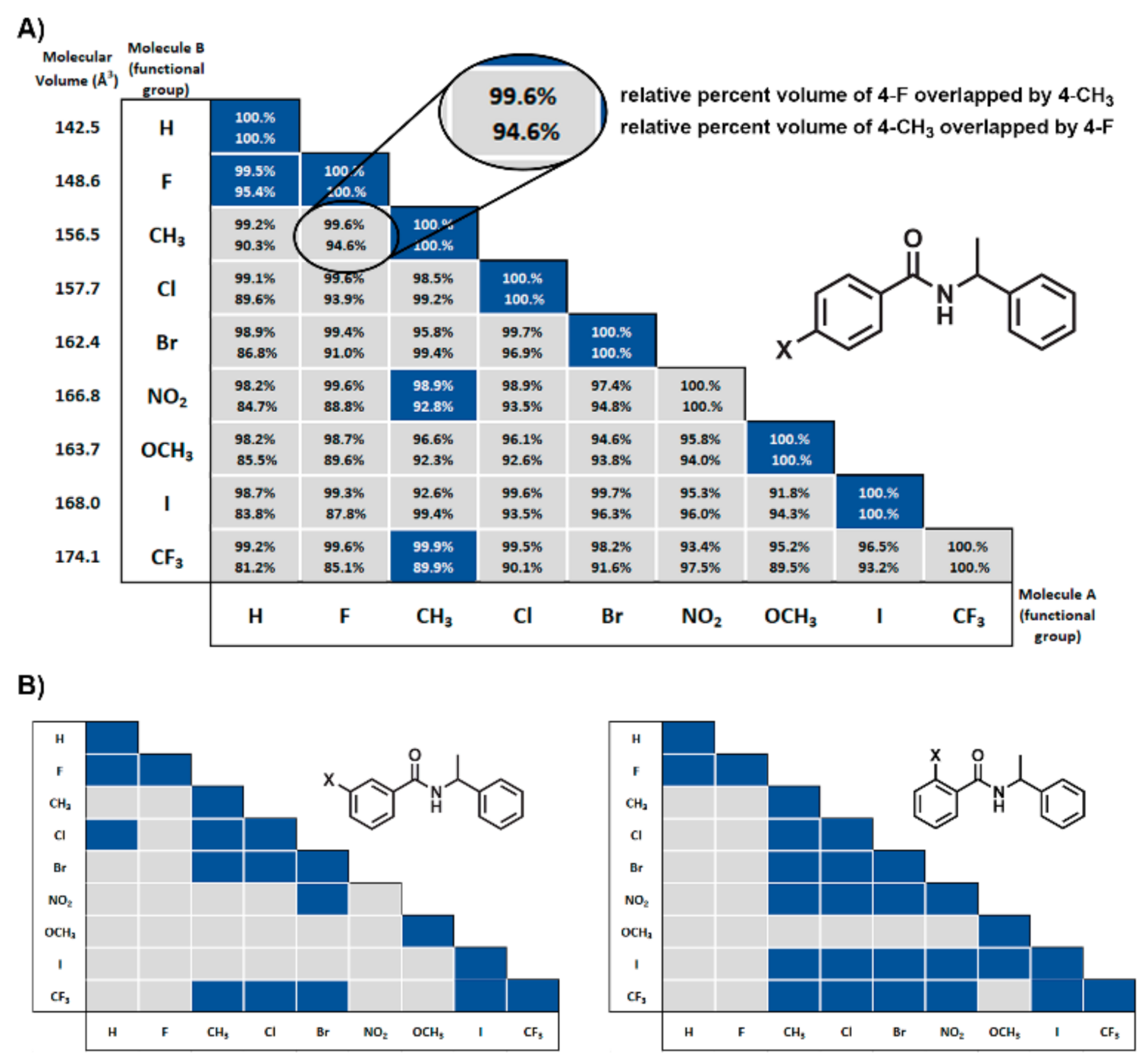
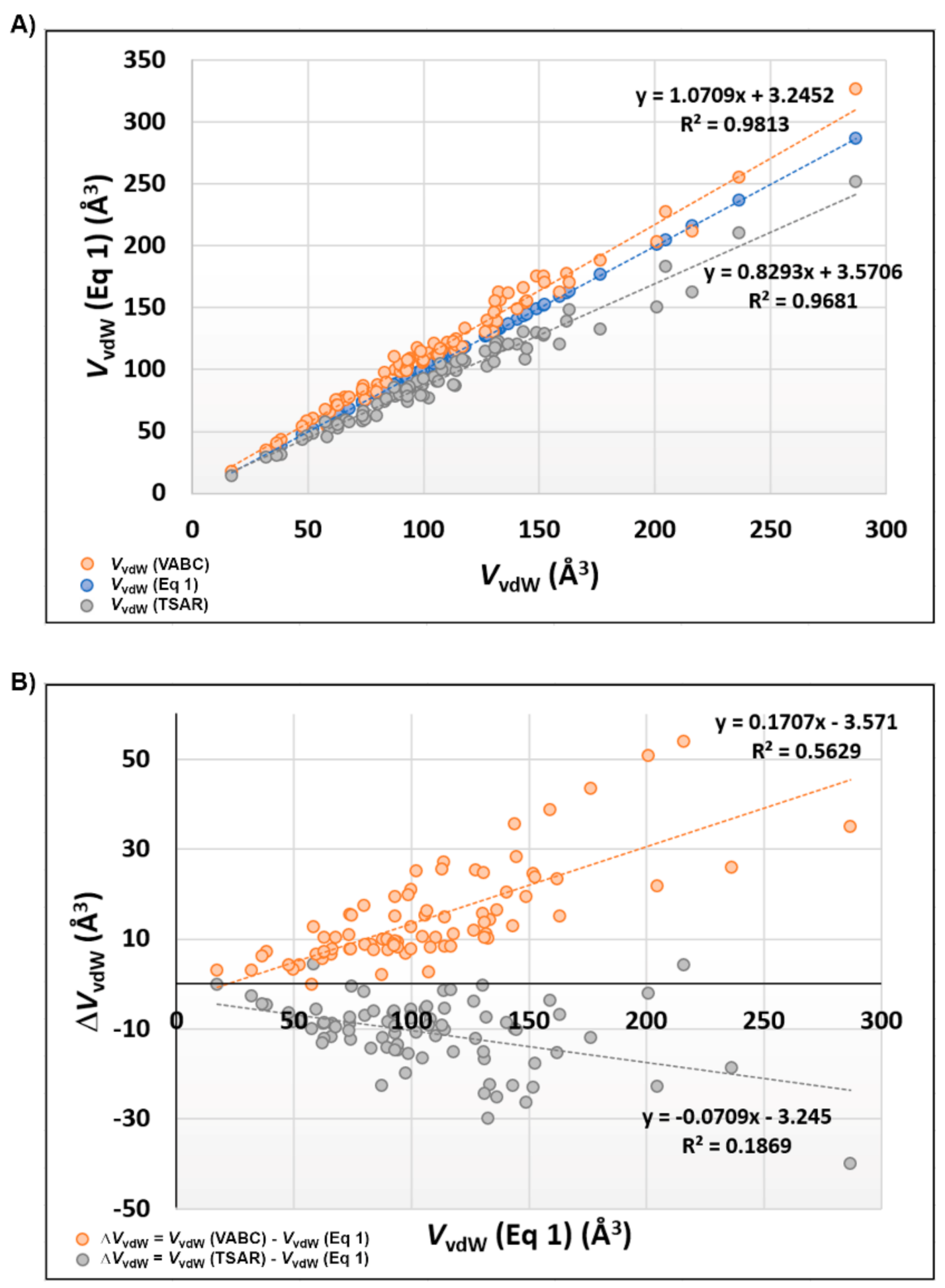
3.4. Molecular Volume and Shape Difference Determinations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fredga, A. Quasiracemic compounds and their use for studying the configuration of optically active compounds. Bull. De La Soc. Chim. Fr. 1973, 1, 173–182. [Google Scholar]
- Zhang, Q.; Curran, D.P. Quasienantiomers and quasiracemates: New tools for identification, analysis, separation, and synthesis of enantiomers. Chem. Eur. J. 2005, 11, 4866–4880. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Understanding Hydrogen Bonds: Theoretical and Experimental Views; Royal Society of Chemistry: Cambridge, UK, 2021. [Google Scholar]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Wei, S.-H.; Zhang, C. Review of the Intermolecular Interactions in Energetic Molecular Cocrystals. Cryst. Growth Des. 2020, 20, 7065–7079. [Google Scholar] [CrossRef]
- Remsing, R.C.; Klein, M.L. Halogen bond structure and dynamics from molecular simulations. J. Phys. Chem. B 2019, 123, 6266–6273. [Google Scholar] [CrossRef] [Green Version]
- Mir, N.A.; Dubey, R.; Desiraju, G.R. Strategy and methodology in the synthesis of multicomponent molecular solids: The quest for higher cocrystals. Acc. Chem. Res. 2019, 52, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef]
- Liao, W.-Q.; Tang, Y.-Y.; Li, P.-F.; You, Y.-M.; Xiong, R.-G. Competitive halogen bond in the molecular ferroelectric with large piezoelectric response. J. Am. Chem. Soc. 2018, 140, 3975–3980. [Google Scholar] [CrossRef]
- Zhao, Y.; Sarnello, E.S.; Robertson, L.A.; Zhang, J.; Shi, Z.; Yu, Z.; Bheemireddy, S.R.; Li, T.Y.Z.; Assary, R.S.; Cheng, L.; et al. Competitive Pi-Stacking and H-Bond piling increase solubility of heterocyclic redoxmers. J. Phys. Chem. B 2020, 124, 10409–10418. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Benchmark calculations of interaction energies in noncovalent complexes and their applications. Chem. Rev. 2016, 116, 5038–5071. [Google Scholar] [CrossRef]
- Biedermann, F.; Schneider, H.-J. Experimental binding energies in supramolecular complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef]
- Taylor, C.R.; Day, G.M. Evaluating the energetic driving force for cocrystal formation. Cryst. Growth Des. 2018, 18, 892–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavezzotti, A. Collective variables for the simulation of crystallization of organic compounds: Some case studies. Isr. J. Chem. 2021, 61, 498–506. [Google Scholar] [CrossRef]
- Spackman, M.A. How reliable are intermolecular interaction energies estimated from topological analysis of experimental electron densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Görbitz, C.H.; Dalhus, B.; Day, G.M. Pseudoracemic amino acid complexes: Blind predictions for flexible two-component crystals. Phys. Chem. Chem. Phys. 2010, 12, 8466–8477. [Google Scholar] [CrossRef]
- Craddock, D.E.; Parks, M.J.; Taylor, L.A.; Wagner, B.L.; Ruf, M.; Wheeler, K.A. Increasing the structural boundary of quasiracemate formation: 4-substituted naphthylamides. CrystEngComm 2021, 23, 210–215. [Google Scholar] [CrossRef]
- Pinter, E.N.; Cantrell, L.S.; Day, G.M.; Wheeler, K.A. Pasteur’s tartaramide/malamide quasiracemates: New entries and departures from near inversion symmetry. CrystEngComm 2018, 20, 4213–4220. [Google Scholar] [CrossRef] [Green Version]
- Wells, R.G.; Sahlstrom, K.D.; Wheeler, K.A. Amino acid hydrogen oxalate quasiracemates—Sulfur containing side chains. CrystEngComm 2021, 23, 8061–8070. [Google Scholar] [CrossRef]
- Kreitler, D.F.; Mortenson, D.E.; Forest, K.T.; Gellman, S.H. Effects of single α-to-β residue replacements on structure and stability in a small protein: Insights from quasiracemic crystallization. J. Am. Chem. Soc. 2016, 138, 6498–6505. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Ye, L.; Xu, W.; Liu, L. Recent advances in racemic protein crystallography. Bioorganic Med. Chem. 2017, 25, 4953–4965. [Google Scholar] [CrossRef]
- Mandal, K.; Dhayalan, B.; Avital-Shmilovici, M.; Tokmakoff, A.; Kent, S.B.H. Crystallization of enantiomerically pure proteins from quasi-racemic mixtures: Structure determination by X-ray diffraction of isotope-labeled ester insulin and human insulin. ChemBioChem 2016, 17, 421–425. [Google Scholar] [CrossRef]
- Kurgan, K.W.; Kleman, A.F.; Bingman, C.A.; Kreitler, D.F.; Weisblum, B.; Forest, K.T.; Gellman, S.H. Retention of native quaternary structure in racemic melittin crystals. J. Am. Chem. Soc. 2019, 141, 7704–7708. [Google Scholar] [CrossRef]
- Smets, M.M.H.; Kalkman, E.; Krieger, A.; Tinnemans, P.; Meekes, H.; Vlieg, E.; Cuppen, H.M. On the mechanism of solid-state phase transitions in molecular crystals—The role of cooperative motion in (quasi)racemic linear amino acids. IUCrJ 2020, 7, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasell, T.; Little, M.A.; Chong, S.Y.; Schmidtmann, M.; Briggs, M.E.; Santolini, V.; Jelfs, K.E.; Cooper, A.I. Chirality as a tool for function in porous organic cages. Nanoscale 2017, 9, 6783–6790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, B.; Shen, R.; Kubota, T.; Mandal, K.; Bezanilla, F.; Roux, B.; Kent, S.B.H. Inversion of the side-chain stereochemistry of indvidual thr or ile residues in a protein molecule: Impact on the folding, stability, and structure of the shk toxin. Angew. Chem. Int. Ed. 2017, 56, 3324–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, U.K.; Golliher, A.E.; Newar, T.D.; Holguin, F.O.; Maio, W.A. Asymmetric total synthesis and revision of absolute stereochemistry for (+)-taumycin a: An approach that exploits orthogonally protected quasienantiomers. J. Org. Chem. 2021, 86, 11086–11099. [Google Scholar] [CrossRef]
- Mane, S. Racemic drug resolution: A comprehensive guide. Anal. Methods 2016, 8, 7567–7586. [Google Scholar] [CrossRef]
- Coulbeck, E.; Eames, J. Parallel kinetic resolution of active esters using a quasi-enantiomeric combination of (R)-4-phenyl-oxazolidin-2-one and (S)-4,5,5-triphenyl-oxazolidin-2-one. Tetrahedron Asymmetry 2008, 19, 2223–2233. [Google Scholar] [CrossRef]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Speck, J.M.; Rüffer, T.; Schaarschmidt, D. 1H NMR spectroscopic elucidation in solution of the kinetics and thermodynamics of spin crossover for an exceptionally robust Fe2+ complex. Dalton Trans. 2016, 45, 13798–13809. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, I.C.; Spaniol, J.M.; Wheeler, K.A. Mapping the structural boundaries of quasiracemate fractional crystallization using 2-substituted diarylamides. Chem. Commun. 2017, 53, 4601–4604. [Google Scholar] [CrossRef]
- Woodley, S.M.; Catlow, R. Crystal structure prediction from first principles. Nat. Mater. 2008, 7, 937–946. [Google Scholar] [CrossRef]
- Price, S. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SADABS and TWINABS—Program for Area Detector Absorption Corrections; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Barbour, L.J. X-Seed 4: Updates to a program for small-molecule supramolecular crystallography. J. Appl. Cryst. 2020, 53, 1141–1146. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Cryst. A 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The cambridge structural database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Stroustrup, B. The C++ Programming Language, 4th ed.; Addison-Wesley: Upper Saddle River, NJ, USA, 2013; ISBN 978-0-321-56384-2. [Google Scholar]
- Braun, D.E.; Hald, P.; Kahlenberg, V.; Griesser, U.J. Expanding the solid form landscape of bipyridines. Cryst. Growth Des. 2021, 21, 7201–7217. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, P.; Nanda, A. Hot stage microscopy and its applications in pharmaceutical characterization. Appl. Microsc. 2020, 50, 12. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.; Wang, L.; Chen, M.; Fang, W.; Gao, Z.; Chai, Y.; Zhao, P.; Bao, Y. Amino acid based pharmaceutical cocrystals and hydrate cocrystals of the chlorothiazide: Structural studies and physicochemical properties. J. Mol. Struct. 2020, 1217, 128432. [Google Scholar] [CrossRef]
- Kofler, L.; Kofler, A. Thermal Micromethods for the Study of Organic Ompounds and Their Mixtures; Wagner: Innsbruck, Austria, 1952; McCorne, W.C., Translator; McCorne Research Institute: Chicago, IL, USA, 1980. [Google Scholar]
- Kelley, S.P.; Fábián, L.; Brock, C.P. Failures of fractional crystallization: Ordered co-crystals of isomers and near isomers. Acta Crystallogr. Sect. B Struct. Sci. 2011, 67, 79–93. [Google Scholar] [CrossRef]
- Rekis, T. Crystallization of chiral molecular compounds: What can be learned from the Cambridge Structural Database? Acta Cryst. B 2020, 76, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Hendi, M.S.; Hooter, P.; Davis, R.E.; Lynch, V.M.; Wheeler, K.A. Structural studies of enantiomers, racemates, and quasiracemates: N-(4-methylbenzoyl) methylbenzylamine and N-(4-nitrobenzoyl) methylbenzylamine. Cryst. Growth Des. 2004, 4, 95–101. [Google Scholar] [CrossRef]
- Lineberry, A.M.; Benjamin, E.T.; Davis, R.E.; Kassel, W.S.; Wheeler, K.A. Structural studies of racemates and quasiracemates: Chloro, Bromo, and Methyl Adducts of 2-Phenoxypropionic Acid. Cryst. Growth Des. 2008, 8, 612–619. [Google Scholar] [CrossRef]
- Cross, J.T.; Rossi, N.A.; Serafin, M.; Wheeler, K.A. Tröger’s base quasiracemates and crystal packing tendencies. CrystEngComm 2014, 16, 7251–7258. [Google Scholar] [CrossRef]
- Spaniol, J.M.; Wheeler, K.A. Accessing Centnerszwer’s quasiracemate—Molecular shape controlled molecular recognition. RSC Adv. 2016, 6, 64921–64929. [Google Scholar] [CrossRef]
- Toda, F.; Tanaka, K.; Miyamoto, H.; Koshima, H.; Miyahara, I.; Hirotsu, K. Formation of racemic compound crystals by mixing of two enantiomericcrystals in the solid state. Liquid transport of molecules from crystal to crystal. J. Chem. Soc. Perkin Trans. 1997, 2, 1877–1886. [Google Scholar] [CrossRef] [Green Version]
- Fomulu, S.L.; Hendi, M.S.; Davis, R.E.; Wheeler, K.A. Structural studies of enantiomers, racemates, and quasiracemates. N-(2-Chlorobenzoyl)methylbenzylamine and N-(2-Bromobenzoyl)methylbenzylamine. Cryst. Growth Des. 2002, 2, 645–651. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Ranjan, S.; Devarapalli, R.; Kundu, S.; Saha, S.; Deolka, S.; Vangala, V.R.; Reddy, C.M. Isomorphism: Molecular similarity to crystal structure similarity in multicomponent forms of analgesic drugs tolfenamic and mefenamic acid. IUCrJ 2020, 7, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Nicholls, A.; McGaughey, G.B.; Sheridan, R.P.; Good, A.C.; Warren, G.; Mathieu, M.; Muchmore, S.W.; Brown, S.P.; Grant, J.A.; Haigh, J.A.; et al. Molecular shape and medicinal chemistry: A perspective. J. Med. Chem. 2010, 53, 3862–3886. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, E.; Ebejer, J.-P. Applying machine learning to ultrafast shape recognition in ligand-based virtual screening. Front Pharm. 2019, 10, 1675. [Google Scholar] [CrossRef]
- Spackman, P.R.; Yu, L.; Morton, C.J.; Parker, M.W.; Bond, C.S.; Spackman, M.A.; Jayatilaka, D.; Thomas, S.P. Bridging Crystal Engineering and drug discovery by utilizing intermolecular interactions and molecular shapes in crystals. Angew. Chem. Int. Ed. 2019, 58, 16780–16784. [Google Scholar] [CrossRef]
- Seddon, M.P.; Cosgrove, D.A.; Packer, M.J.; Gillet, V.J. Alignment-free molecular shape comparison using spectral geometry: The framework. J. Chem. Inf. Model. 2019, 59, 98–116. [Google Scholar] [CrossRef] [Green Version]
- Ebalunode, J.O.; Ouyang, Z.; Liang, J.; Zheng, W. Novel approach to structure-based pharmacophore search using computational geometry and shape matching techniques. J. Chem. Inf. Model 2008, 48, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.S.; Grant, J.A.; Mosyak, L.; Nicholls, A. A Shape-Based 3-D Scaffold Hopping Method and Its Application to a Bacterial Protein−Protein Interaction. J. Med. Chem. 2005, 48, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Sahlstrom, K.D.; Ekelem, F.I.; Wheeler, K.A. Amino acid hydrogen oxalate quasiracemates—Hydrocarbon side chains. CrystEngComm 2021, 23, 8053–8060. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Nueno, V.I.; Venkatraman, V.; Mavridis, L.; Clark, T.; Ritchie, D.W. using spherical harmonic surface property representations for ligand-based virtual screening. Mol. Inform. 2011, 30, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. FieldScreen: Virtual screening using molecular fields. Application to the dud data Set. J. Chem. Inf. Model. 2008, 48, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Zissimos, A.M. Fast Calculation of van der Waals Volume as a sum of atomic and bond contributions and its application to drug compounds. J. Org. Chem. 2003, 68, 7368–7373. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandt, A.K.; Boyle, D.J.; Butler, J.P.; Gillingham, A.R.; Penner, S.E.; Spaniol, J.M.; Stockdill, A.K.; Vanderwall, M.M.; Yeraly, A.; Schepens, D.R.; et al. Molecular Recognition and Shape Studies of 3- and 4-Substituted Diarylamide Quasiracemates. Crystals 2021, 11, 1596. https://doi.org/10.3390/cryst11121596
Brandt AK, Boyle DJ, Butler JP, Gillingham AR, Penner SE, Spaniol JM, Stockdill AK, Vanderwall MM, Yeraly A, Schepens DR, et al. Molecular Recognition and Shape Studies of 3- and 4-Substituted Diarylamide Quasiracemates. Crystals. 2021; 11(12):1596. https://doi.org/10.3390/cryst11121596
Chicago/Turabian StyleBrandt, Ali K., Derek J. Boyle, Jacob P. Butler, Abigail R. Gillingham, Scott E. Penner, Jacqueline M. Spaniol, Alaina K. Stockdill, Morgan M. Vanderwall, Almat Yeraly, Diana R. Schepens, and et al. 2021. "Molecular Recognition and Shape Studies of 3- and 4-Substituted Diarylamide Quasiracemates" Crystals 11, no. 12: 1596. https://doi.org/10.3390/cryst11121596
APA StyleBrandt, A. K., Boyle, D. J., Butler, J. P., Gillingham, A. R., Penner, S. E., Spaniol, J. M., Stockdill, A. K., Vanderwall, M. M., Yeraly, A., Schepens, D. R., & Wheeler, K. A. (2021). Molecular Recognition and Shape Studies of 3- and 4-Substituted Diarylamide Quasiracemates. Crystals, 11(12), 1596. https://doi.org/10.3390/cryst11121596