
Low Frequency Vibrations and Diffusion in Disordered Polymers Bearing an Intrinsic Microporosity as Revealed by Neutron Scattering

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
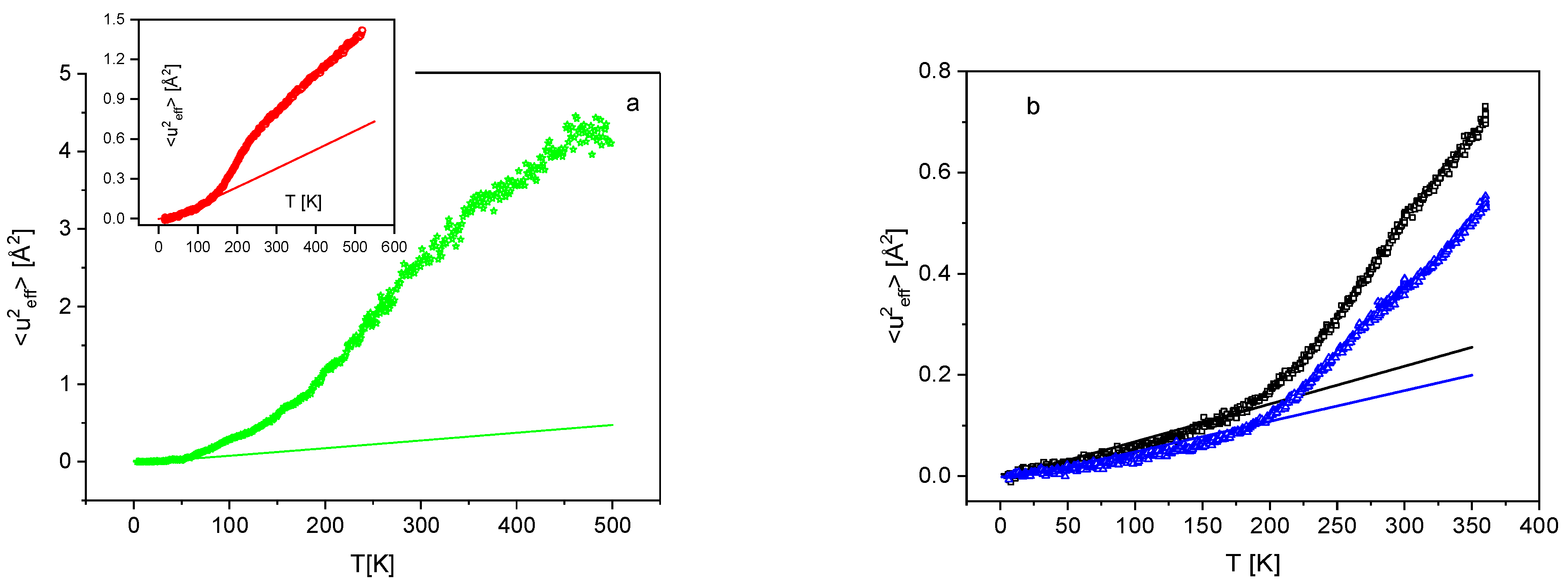
3. Results and Discussion
3.1. Low Frequency Density of States
3.2. Microscopic Diffusion
3.3. Microscopic Dynamics from the Vibrational Density of States
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijmans, J.G.; Baker, R.W. The solution-diffusion model: A review. J. Membr. Sci. 1995, 107, 1–21. [Google Scholar] [CrossRef]
- Baker, R.W. Membrane Technology and Applications; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Yampolskii, Y. Polymeric gas separation membranes. Macromolecules 2012, 45, 3298–3311. [Google Scholar] [CrossRef]
- Robeson, L.M. The upper bound revisited. J. Membr. Sci. 2008, 320, 390–400. [Google Scholar] [CrossRef]
- Swaidan, R.; Ghanem, B.; Pinnau, I. Fine-tuned intrinsically ultramicroporous polymers redefine the permeability/selectivity upper bounds of membrane-based air and hydrogen separations. ACS Macro Lett. 2015, 4, 947–951. [Google Scholar] [CrossRef]
- Comesaña-Gándara, B.; Chen, J.; Bezzu, C.G.; Carta, M.; Rose, I.; Ferrari, M.-C.; Esposito, E.; Fuoco, A.; Jansen, J.C.; McKeown, N.B. Redefining the Robeson upper bounds for CO2/CH4 and CO2/N2 separations using a series of ultrapermeable benzotriptycene-based polymers of intrinsic microporosity. Energy Environm. Sci. 2019, 12, 2733–2740. [Google Scholar] [CrossRef]
- Freeman, B.D. Basis of permeability/selectivity tradeoff relations in polymeric gas separation membranes. Macromolecules 1999, 32, 375–380. [Google Scholar] [CrossRef]
- Robeson, L.M.; Freeman, B.D.; Paul, D.R.; Rowe, B.W. An empirical correlation of gas permeability and permselectivity in polymers and its theoretical basis. J. Membr. Sci. 2009, 341, 178–185. [Google Scholar] [CrossRef]
- Higashimura, T.; Masuda, T.; Okada, M. Gas permeability of polyacetylenes with bulky substituents. Polym. Bull. 1983, 10, 114–117. [Google Scholar] [CrossRef]
- Masuda, T.; Kawasaki, M.; Okano, Y.; Higashimura, T. Polymerization of methylpentynes by transition metal catalysts: Monomer structure, reactivity, and polymer properties. Polym. J. 1982, 14, 371–377. [Google Scholar] [CrossRef]
- Morisato, A.; Pinnau, I. Synthesis and gas permeation properties of poly(4-methyl-2-pentyne). J. Membr. Sci. 1996, 121, 243–250. [Google Scholar] [CrossRef]
- Budd, P.M.; Msayib, K.J.; Tattershall, C.E.; Ghanem, B.S.; Reynolds, K.J.; McKeown, N.B.; Fritsch, D. Gas separation membranes from polymers of intrinsic microporosity. J. Membr. Sci. 2005, 251, 263–269. [Google Scholar] [CrossRef]
- Carta, M.; Malpass-Evans, R.; Croad, M.; Rogan, Y.; Jansen, J.C.; Bernardo, P.; Bazzarelli, F.; McKeown, N.B. An efficient polymer molecular sieve for membrane gas separations. Science 2013, 339, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, D.; Heuchel, M.; Yampolskii, Y.; Khotimskii, V.; Shantarovich, V. Free volume distributions in ultrahigh and lower free volume polymers: Comparison between molecular modeling and positron lifetime studies. Macromolecules 2002, 35, 2129–2140. [Google Scholar] [CrossRef]
- Heuchel, M.; Fritsch, D.; Budd, P.M.; McKeown, N.B.; Hofmann, D. Atomistic packing model and free volume distribution of a polymer with intrinsic microporosity (PIM-1). J. Membr. Sci. 2008, 318, 84–99. [Google Scholar] [CrossRef]
- Alentiev, D.A.; Bermeshev, M. Design and synthesis of porous organic polymeric materials from norbornene derivatives. Polym. Rev. 2021, 38. [Google Scholar] [CrossRef]
- Wang, X.; Wilson, T.J.; Alentiev, D.; Gringolts, M.; Finkelshtein, E.; Bermeshev, M.; Long, B.K. Substituted polynorbornene membranes: A modular template for targeted gas separations. Polym. Chem. 2021, 12, 2947–2977. [Google Scholar] [CrossRef]
- Yampolskii, Y.P. A current position of polyacetylenes among other highly permeable membrane materials. Polym. Rev. 2017, 57, 200–212. [Google Scholar] [CrossRef]
- Chapala, P.P.; Bermeshev, M.V.; Starannikova, L.E.; Belov, N.A.; Ryzhikh, V.E.; Shantarovich, V.P.; Lakhin, V.G.; Gavrilova, N.N.; Yampolskii, Y.P.; Finkelshtein, E.S. A novel, highly gas-permeable polymer representing a new class of silicon-containing polynorbornens as efficient membrane materials. Macromolecules 2015, 48, 8055–8061. [Google Scholar] [CrossRef]
- Gringolts, M.; Bermeshev, M.; Yampolskii, Y.; Starannikova, L.; Shantarovich, V.; Finkelshtein, E. New high permeable addition poly(tricyclononenes) with Si(CH3)3 side groups. synthesis, gas permeation parameters, and free volume. Macromolecules 2010, 43, 7165–7172. [Google Scholar] [CrossRef]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry, and Use; John Wiley & Sons: New York, NY, USA, 1974. [Google Scholar]
- Teplyakov, V.; Meares, P. Correlation aspects of the selective gas permeabilities of polymeric materials and membranes. Gas Sep. Purif. 1990, 4, 66–74. [Google Scholar]
- Robeson, L.M.; Smith, Z.P.; Freeman, B.D.; Paul, D.R. Contributions of diffusion and solubility selectivity to the upper bound analysis for glassy gas separation membranes. J. Membr. Sci. 2014, 453, 71–83. [Google Scholar] [CrossRef]
- Robeson, L.M.; Dose, M.E.; Freeman, B.D.; Paul, D.R. Analysis of the transport properties of thermally rearranged (TR) polymers and polymers of intrinsic microporosity (PIM) relative to upper bound performance. J. Membr. Sci. 2017, 525, 18–24. [Google Scholar] [CrossRef]
- Fuoco, A.; Rizzuto, C.; Tocci, E.; Monteleone, M.; Esposito, E.; Budd, P.M.; Carta, M.; Comesañ-Gándara, B.; McKeown, N.B.; Jansen, J.C. The origin of size-selective gas transport through polymers of intrinsic microporosity. J. Mater. Chem. A 2019, 7, 20121–20126. [Google Scholar] [CrossRef]
- Malpass-Evans, R.; Rose, I.; Fuoco, A.; Bernardo, P.; Clarizia, G.; McKeown, N.B.; Jansen, J.C.; Carta, M. Effect of the bridgehead methyl substituts on the gas permeabiliby of Tröger‘s-base derived polymers of intrinsic microporosity. Membranes 2020, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Mazo, M.; Balabaev, M.; Alentiev, A.; Yampolskii, Y. Molecular dynamics simulation of nanostructure of high free volume polymers with SiMe3 side groups. Macomolecules 2018, 51, 1398–1408. [Google Scholar] [CrossRef]
- Inoue, R.; Kanaya, T.; Masuda, T.; Nishida, K.; Yamamuro, O. Relationship between the local dynamics and gas permeability of para-substituted poly(1-chloro-2-phenylacetylenes). Macromolecules 2012, 45, 6008–6014. [Google Scholar] [CrossRef]
- Kanaya, T.; Teraguchi, M.; Masuda, T.; Kaji, K. Local mobility of substituted polyacetylenes measured by quasielastic neutron scattering and its relationship with gas permeability. Polymer 1999, 40, 7157–7161. [Google Scholar] [CrossRef]
- Kanaya, T.; Kawaguchi, T.; Kaji, K.; Sakaguchi, T.; Kwak, G.; Masuda, T. Role of local dynamics in the gas permeability of glassy substituted polyacetylenes. A quasielastic neutron scattering study. Macromolecules 2002, 35, 5559–5564. [Google Scholar] [CrossRef]
- Zorn, R.; Lohstroh, W.; Zamponi, M.; Harrison, W.J.; Budd, P.; Böhning, M.; Schönhals, A. Molecular mobility of a polymer of intrinsic microporosity revealed by quasielastic neutron scattering. Macromolecules 2020, 53, 6731–6739. [Google Scholar] [CrossRef]
- Schönhals, A.; Szymoniak, P.; Kolmangadi, M.A.; Böhning, M.; Zamponi, M.; Frick, B.; Appel, M.; Günther, G.; Russina, M.; Alenttiev, D.A.; et al. Microscopic dynamics of highly permeable super glassy polynorbornenes revealed by neutron scattering. J. Membr. Sci. 2021. [Google Scholar] [CrossRef]
- Shintani, H.; Tanaka, H. Universal link between the boson peak and transverse phonons in glass. Nat. Mat. 2008, 7, 870. [Google Scholar] [CrossRef] [PubMed]
- Zallen, R. The Physics of Amorphous Solids; John Wiley & Sons: New York, NY, USA, 1983. [Google Scholar]
- Angell, C.A.; Yue, Y.; Wang, L.-M.; Copley, J.R.; Borick, S.; Mossa, S. Potential energy, relaxation, vibrational dynamics and the boson peak of hyperquenched glasses. J. Phys. Cond. Matt. 2003, 15, S1051. [Google Scholar] [CrossRef]
- Malinovsky, V.K.; Novikov, V.N.; Sokolov, A.P. Investigation of structural correlations in disordered materials by Raman scattering measurements. J. Non-Cryst. Solids 1987, 90, 485–488. [Google Scholar] [CrossRef]
- Buchenau, U.; Galperin, Y.M.; Gurevich, V.L.; Parshin, A.D.; Ramos, M.A.; Schober, H.R. Interaction of soft modes and sound waves in glasses. Phys. Rev. B Condens Matter Mater. Phys. 1992, 46, 2798–2808. [Google Scholar] [CrossRef]
- Laird, B.B.; Schober, H.R. Localized low-frequency vibrational modes in a simple model glass. Phys. Rev. Lett. 1991, 66, 636–639. [Google Scholar] [CrossRef]
- Taraskin, S.N.; Loh, Y.L.; Natarajan, G.; Elliott, S.R. Origin of the boson peak in systems with lattice disorder. Phys. Rev. Lett. 2001, 86, 1255–1258. [Google Scholar] [CrossRef]
- Schirmacher, W.; Diezemann, G.; Ganter, C. Harmonic vibrational excitations in disordered solids and the “boson peak”. Phys. Rev. Lett. 1998, 81, 136–139. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, L.; Wang, Y.; Schirmacher, W.; Zhang, J. Disentangling boson peaks and Van Hove singularities in a model glass. Phys. Rev. B 2018, 98, 174207. [Google Scholar] [CrossRef]
- Milkus, R.; Zaccone, A. Local inversion-symmetry breaking controls the boson peak in glasses and crystals. Phys. Rev. B 2016, 93, 094204. [Google Scholar] [CrossRef]
- Schönhals, A.; Zorn, R.; Frick, B. Inelastic neutron spectroscopy as a tool to investigate nanoconfined polymer systems. Polymer 2016, 105, 393–406. [Google Scholar] [CrossRef]
- Zorn, R.; Mayorova, M.; Richter, D.; Schönhals, A.; Hartmann, L.; Kremer, F.; Frick, B. Effect of nanoscopic confinement on the microscopic dynamics of glass-forming liquids and polymers studied by inelastic neutron scattering. AIP Conf. Proc. 2008, 982, 79–84. [Google Scholar]
- Asthalter, T.; Bauer, M.; van Bürck, U.; Sergueev, I.; Franz, H.; Chumakov, A.I. Confined phonons in glasses. Eur. Phys. J. E: Soft Matter Biol. Phys. 2003, 12, S9–S12. [Google Scholar] [CrossRef]
- Schönhals, A.; Goering, H.; Schick, C.; Frick, B.; Zorn, R. Glass transition of polymers confined to nanoporous glasses. Colloid Polym. Sci. 2004, 282, 882–891. [Google Scholar] [CrossRef]
- Zorn, R. Boson peak in confined disordered systems. Phys. Rev. B: Condens. Matter Phys. 2010, 81, 054208. [Google Scholar] [CrossRef]
- Pérez-Castañeda, T.; Jiménez-Riobóo, R.J.; Ramos, M.A. Two-level systems and boson peak remain stable in 110-million-year-old amber glass. Phys. Rev. Lett. 2014, 112, 165901. [Google Scholar] [CrossRef]
- Pogna, E.; Chumakov, A.; Ferrante, C.; Ramos, M.; Scopigno, T. Tracking the connection between disorder and energy landscape in glasses using geologically hyperaged amber. J. Phys. Chem. Lett. 2019, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Castañeda, T.; Rodríguez-Tinoco, C.; Rodríguez-Viejo, J.; Ramos, M.A. Suppression of tunneling two level systems in ultrastable glasses of indomethacin. Proc. Natl. Acad. Sci. USA 2014, 111, 11275. [Google Scholar] [CrossRef]
- Singh, S.; Ediger, M.D.; De Pablo, J.J. Ultrastable glasses from in silico vapour deposition. Nat. Mater. 2013, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Monnier, X.; Colmenero, J.; Wolf, M.; Cangialosi, D. Reaching the ideal glass in polymer spheres: Thermodynamics and vibrational density of states. Phys. Rev. Lett. 2021, 126, 118004. [Google Scholar] [CrossRef] [PubMed]
- Zorn, R.; Szymoniak, P.; Kolmangadi, M.; Wolf, M.; Alentiev, D.; Bermeshev, M.; Böhning, M.; Schönhals, A. Low frequency vibrational density of state of highly permeable super glassy polynorbornenes—The Boson peak. Phys. Chem. Chem. Phys. 2020, 22, 8381–18387. [Google Scholar] [CrossRef]
- Rose, I.; Bezzu, C.G.; Carta, M.; Comesaña-Gándara, B.; Lasseuguette, E.; Ferrari, M.C.; Bernardo, P.; Clarizia, G.; Fuoco, A.; Jansen, J.C.; et al. Polymer ultrapermeability from the inefficient packing of 2D chains. Nat. Mater. 2017, 16, 932. [Google Scholar] [CrossRef]
- Bernardo, P.; Scorzafave, V.; Clarizia, G.; Tocci, E.; Jansen, J.C.; Borgogno, A.; Malpass-Evans, R.; McKeown, N.B.; Carta, M.; Tasselli, F. Thin film composite membranes based on a polymer of intrinsic microporosity derived from Tröger’s base: A combined experimental and computational investigation of the role of residual casting solvent. J. Membr. Sci. 2019, 569, 17–31. [Google Scholar] [CrossRef]
- Zorn, R.; Yin, H.; Lohstroh, W.; Harrison, W.; Budd, P.M.; Pauw, B.R.; Böhning, M.; Schönhals, A. Anomalies in the low frequency vibrational density of states for a polymer with intrinsic microporosity—The Boson peak of PIM-1. Phys. Chem. Chem. Phys. 2018, 20, 1355–1363. [Google Scholar] [CrossRef]
- Yin, H.; Yang, B.; Chua, Y.Z.; Szymoniak, P.; Carta, M.; Malpass-Evans, R.; McKeown, N.; Harrison, W.J.; Budd, P.M.; Schick, C.; et al. Effect of backbone rigidity on the glass transition of polymers of intrinsic microporosity probed by fast scanning calorimetry. ACS Macro Lett. 2019, 8, 1022–1028. [Google Scholar] [CrossRef]
- Copley, J.R.D.; Cook, J.C. The disk chopper spectrometer at NIST: A new instrument for quasielastic neutron scattering studies. Chem. Phys. 2003, 292, 477. [Google Scholar] [CrossRef]
- Azuah, R.T.; Kneller, L.R.; Qiu, Y.; Tregenna-Piggott, P.L.W.; Brown, C.M.; Copley, J.R.D.; Dimeo, R.M. DAVE: A comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. J. Res. Natl. Inst. Stan. Technol. 2009, 114, 341. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Dimeo, R.M.; Gehring, P.M.; Neumann, D.A. The high-flux backscattering spectrometer at the NIST Center for Neutron Research. Rev. Sci. Inst. 2003, 74, 2759–2777. [Google Scholar] [CrossRef]
- Bée, M. Quasielastic Neutron Scattering, Principles and Applications in Solid State Chemistry, Biology and Materials Science; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Lovesey, S.W. Theory of Neutron Scattering from Condensed Matter; Oxford University: New York, NY, USA, 1987; Volume 1, p. 121. [Google Scholar]
- Zorn, R.; Hartmann, L.; Frick, B.; Richter, D.; Kremer, F.J. Inelastic neutron scattering experiments on the dynamics of a glass-forming material in mesoscopic confinement. Non-Cryst. Solids 2002, 307, 547–554. [Google Scholar] [CrossRef]
- Schirmacher, W.; Schmid, B.; Tomaras, C.; Viliani, G.; Baldi, G.; Ruocco, G.; Scopigno, T. Vibrational excitations in systems with correlated disorder. Phys. Status Solidi C 2008, 5, 862–866. [Google Scholar] [CrossRef]
- Cicerone, M.T.; Soles, C.L. Fast dynamics and stabilization of proteins: Binary glasses of trehalose and glycerol. Biophys. J. 2004, 86, 3836–3845. [Google Scholar] [CrossRef]
- Krause, C.; Zorn, R.; Emmerling, F.; Falkenhagen, J.; Frick, B.; Huber, P.; Schönhals, A. Vibrational density of states of triphenylene based discotic liquid crystals: Dependence on the length of the alkyl chain. Phys. Chem. Chem. Phys. 2014, 16, 7324–7333. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Zorn, R.; Frick, B.; Schönhals, A. Thermal properties and vibrational density of states of a nanoconfined discotic liquid crystal. Colloid Polym. Sci. 2014, 292, 1949–1960. [Google Scholar] [CrossRef]
- DeGiuli, E.; Laversanne-Finot, A.; Düring, G.; Lerner, G.; Wyart, M. Effects of coordination and pressure on sound attenuation, boson peak and elasticity in amorphous solid. Soft Matter 2014, 10, 5628–5644. [Google Scholar] [CrossRef] [PubMed]
- Beltukov, Y.M.; Parshin, D.A. Boson peak in various random-matrix models. JETP Lett. 2016, 104, 552–556. [Google Scholar] [CrossRef]
- Conyuh, D.A.; Beltukov, Y.M. Random matrix approach to the boson peak and Ioffe-Regeln criterion in amorphous solids. Phys. Rev. B 2021, 103, 104204. [Google Scholar] [CrossRef]
- Sokolov, A.P.; Calemczuk, R.; Salce, B.; Quittmann, D.; Duval, E. Low- temperature anomalies in strong and fragil glass formers. Phys. Rev. Lett. 1997, 73, 2405. [Google Scholar] [CrossRef]
- Srivastava, S.; Das, S.P. Fragility and boson peak formation in a supercooled liquid. Phys. Lett. A 2001, 286, 76–79. [Google Scholar] [CrossRef][Green Version]
- Zorn, R. Multiple scattering correction of neutron scattering elastic scans. Nucl. Instr. Meth. A 2007, 572, 874–881. [Google Scholar] [CrossRef]
- Zorn, R. On the evaluation of neutron scattering elastic scan data. Nucl. Instr. Meth. A 2009, 603, 439–445. [Google Scholar] [CrossRef]
- Yin, H.; Chapala, P.; Bermeshev, M.; Pauw, B.R.; Schönhals, A.; Böhning, M. Influence of trimethylsilyl side groups on the molecular mobility and charge transport in highly permeable glassy polynorbornenes. ACS Appl. Polym. Sci. 2019, 1, 844–855. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A. Carbon-carbon torsional barriers driving the fast dynamics in glass-forming polymers. Phys. Rev. B 1998, 57, 1358–13513. [Google Scholar] [CrossRef]
- Golzar, K.; Modarress, H.; Amjad-Iranagh, S. Effect of pristine and functionalized single- and multi-walled carbon nanotubes on CO2 separation of mixed matrix membranes based on polymers of intrinsic microporosity (PIM-1): A molecular dynamics simulation study. J. Mol. Model. 2017, 23, 266. [Google Scholar] [CrossRef] [PubMed]
- Kupgan, G.; Abbot, L.J.; Hart, K.E.; Colina, C.M. Modeling amorphous microporous polymers for CO2 capture and separations. Chem. Rev. 2018, 11, 5488–5538. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PIM-EA-TB(CH3) | PIM-EA-TB(H2) | PIM-1 | PTCNSi1 | |
|---|---|---|---|---|
| BET surface area m2/g | 1030 | 845 | 720 | 610 |
| PIM-1 | PTCNSi1 | PIM-EA-TB(CH3) | |
|---|---|---|---|
| fCH3 | 0.523 | 0.423 | 0.271 |
| Δ<u2eff>CH3 | 0.552 Ǻ2 | 0.446 Ǻ2 | 0.286 Ǻ2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zorn, R.; Szymoniak, P.; Kolmangadi, M.A.; Malpass-Evans, R.; McKeown, N.B.; Tyagi, M.; Böhning, M.; Schönhals, A. Low Frequency Vibrations and Diffusion in Disordered Polymers Bearing an Intrinsic Microporosity as Revealed by Neutron Scattering. Crystals 2021, 11, 1482. https://doi.org/10.3390/cryst11121482
Zorn R, Szymoniak P, Kolmangadi MA, Malpass-Evans R, McKeown NB, Tyagi M, Böhning M, Schönhals A. Low Frequency Vibrations and Diffusion in Disordered Polymers Bearing an Intrinsic Microporosity as Revealed by Neutron Scattering. Crystals. 2021; 11(12):1482. https://doi.org/10.3390/cryst11121482
Chicago/Turabian StyleZorn, Reiner, Paulina Szymoniak, Mohamed A. Kolmangadi, Richard Malpass-Evans, Neil B. McKeown, Madhusudan Tyagi, Martin Böhning, and Andreas Schönhals. 2021. "Low Frequency Vibrations and Diffusion in Disordered Polymers Bearing an Intrinsic Microporosity as Revealed by Neutron Scattering" Crystals 11, no. 12: 1482. https://doi.org/10.3390/cryst11121482
APA StyleZorn, R., Szymoniak, P., Kolmangadi, M. A., Malpass-Evans, R., McKeown, N. B., Tyagi, M., Böhning, M., & Schönhals, A. (2021). Low Frequency Vibrations and Diffusion in Disordered Polymers Bearing an Intrinsic Microporosity as Revealed by Neutron Scattering. Crystals, 11(12), 1482. https://doi.org/10.3390/cryst11121482