
On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Spectral Characteristics of Compounds 3 and 4 (General Method)
2.2. XRD Experimental Part
2.3. Computational Details
3. Results and Discussion
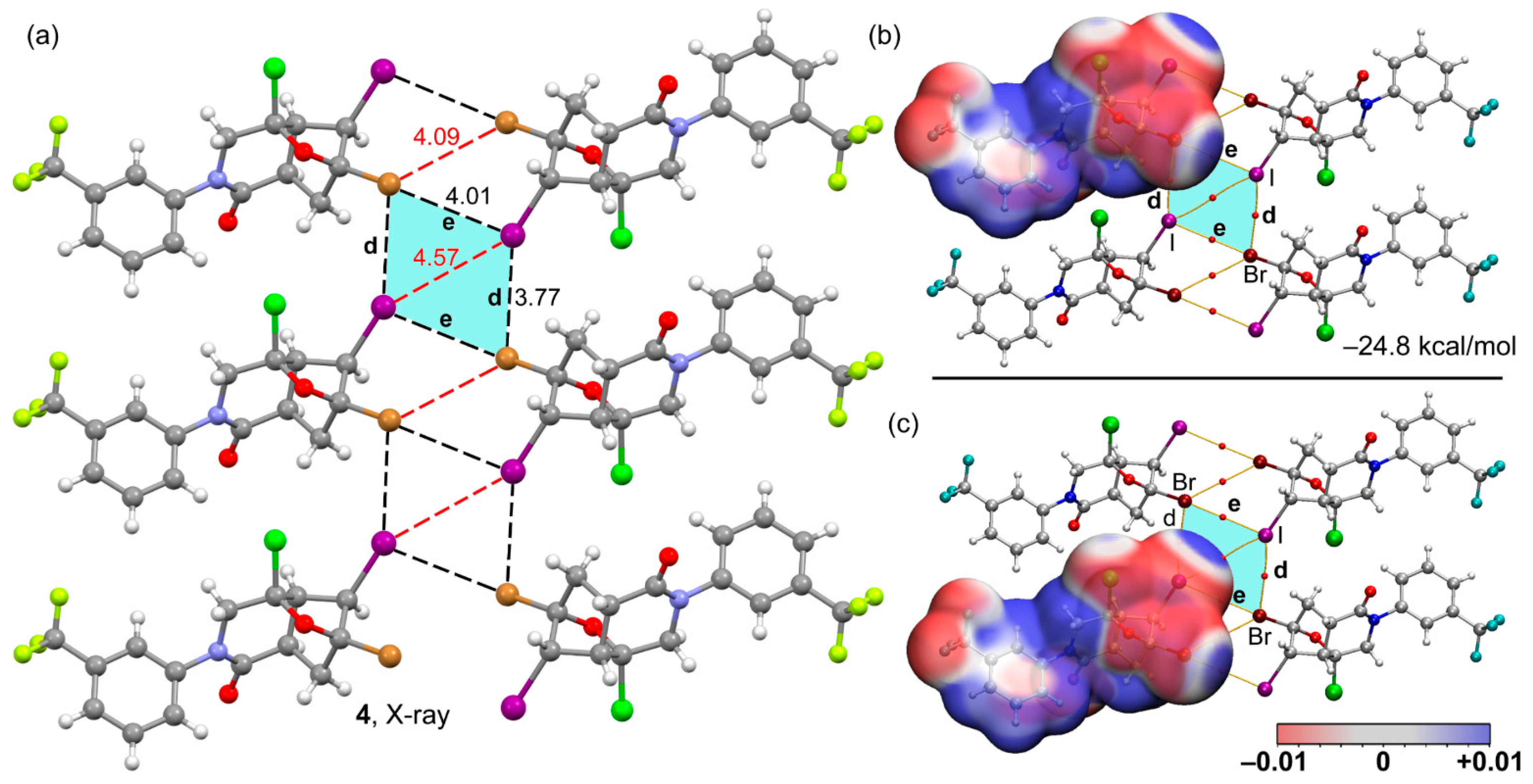
3.1. Structural Description of Compounds 3 and 4
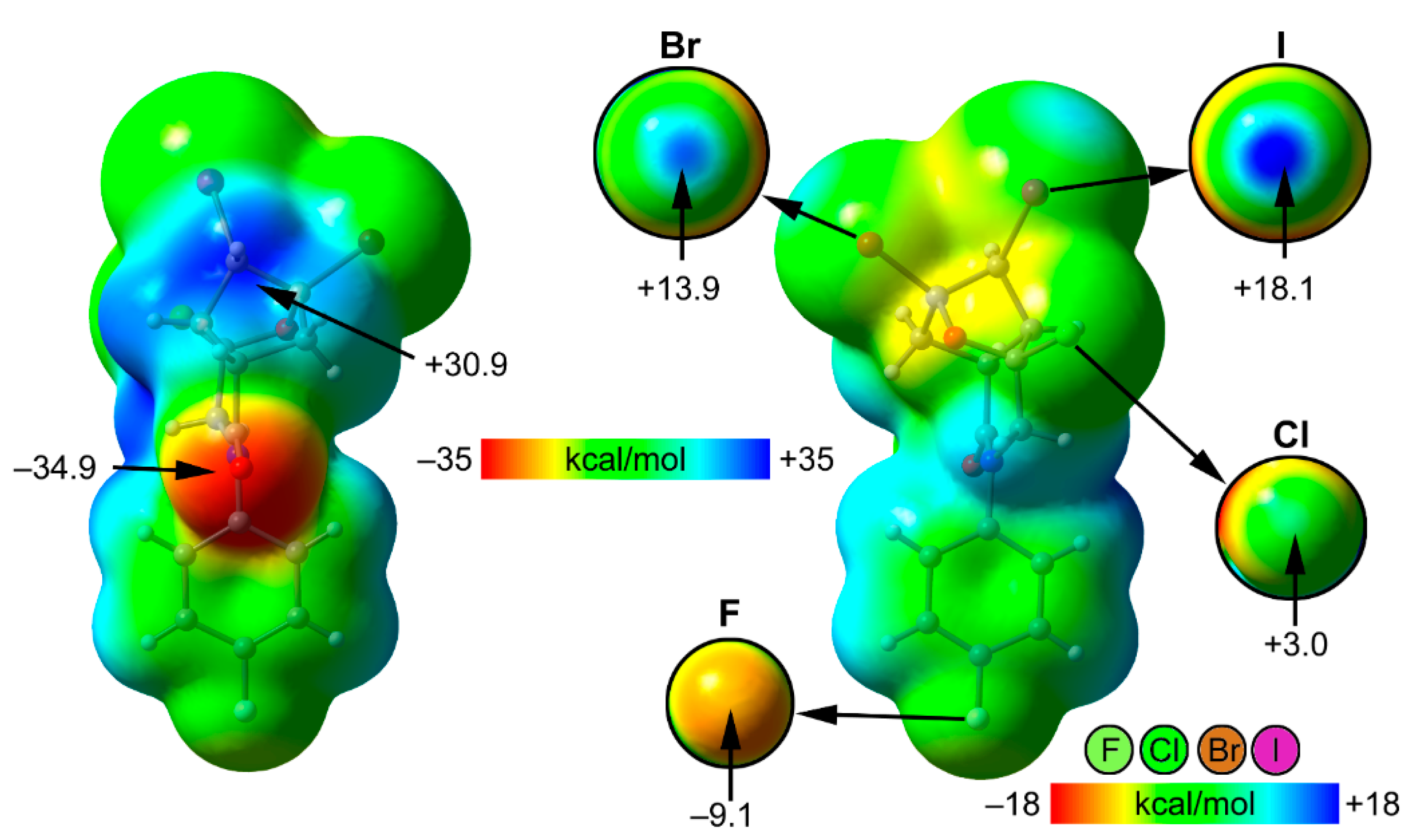
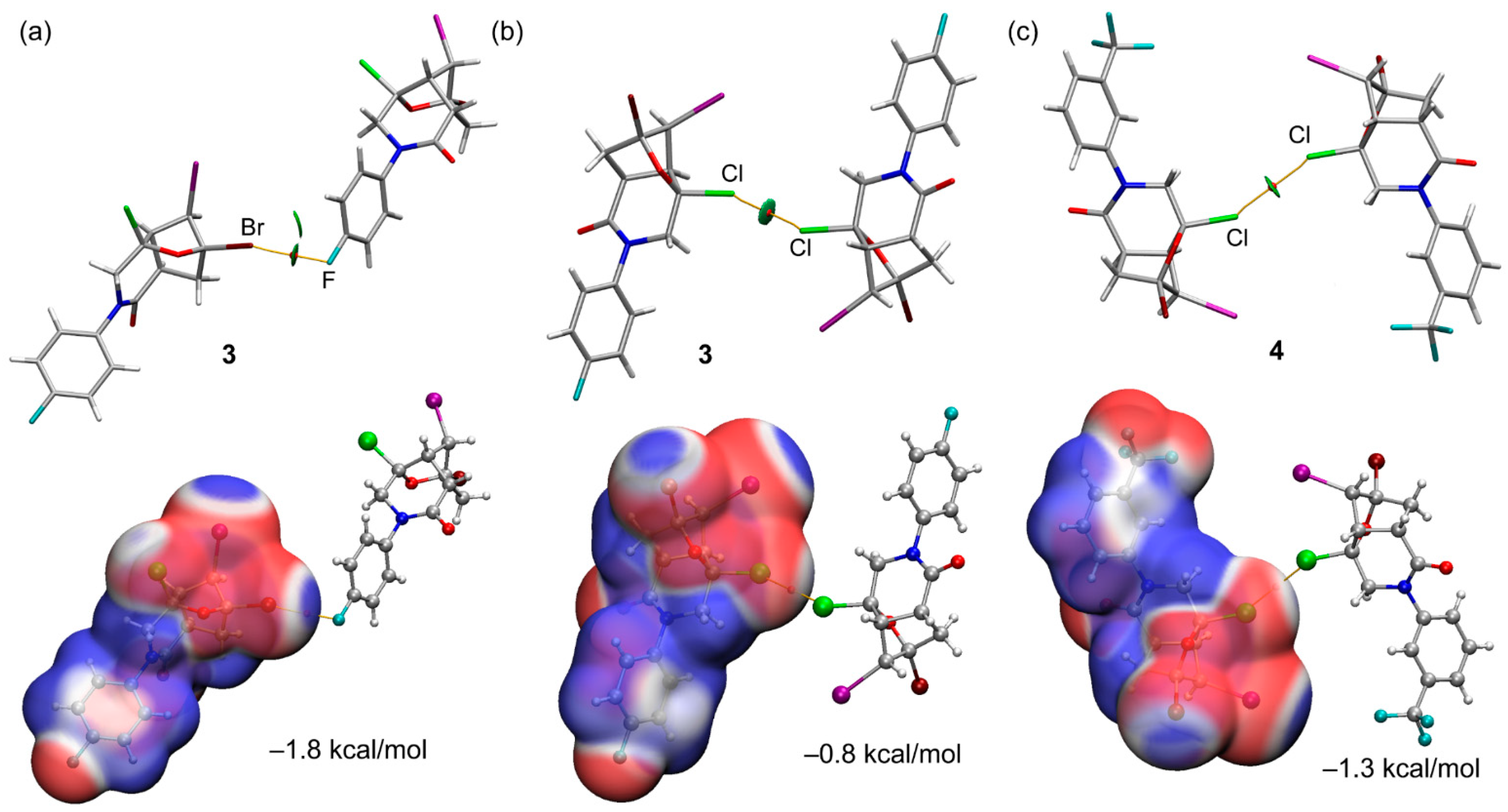
3.2. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. (Eds.) Non-Covalent Interactions in the Synthesis and Design of New Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Davis, H.J.; Phipps, R.J. Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem. Sci. 2017, 8, 864–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.T.; Wu, L.Z. (Eds.) Hydrogen Bonded Supramolecular Structures; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: Fundamentals and Applications (Structure and Bonding); Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Benito, M.; Roselló, Y.; Barceló-Oliver, M.; Frontera, A.; Molins, E. Uracil Derivatives for Halogen-Bonded Cocrystals. Int. J. Mol. Sci. 2021, 22, 10663. [Google Scholar] [CrossRef] [PubMed]
- Heinen, F.; Reinhard, D.L.; Engelage, E.; Huber, S.M. A Bidentate Iodine(III)-Based Halogen-Bond Donor as a Powerful Organocatalyst. Angew. Chem. Int. Ed. 2021, 60, 5069–5073. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef]
- Michalak, M.; Michalak, K.; Urbanczyk-Lipkowska, Z.; Wicha, J. Synthetic studies on dicyclopenta[a,d]cyclooctane terpenoids: Construction of the core structure of fusicoccins and ophiobolins on the route involving a Wagner-Meerwein rearrangement. J. Org. Chem. 2011, 76, 7497–7509. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Zaytsev, V.P.; Nikitina, E.V.; Khrustalev, V.N.; Gozun, S.V.; Boltukhina, E.V.; Varlamov, A.V. Skeletal Wagner-Meerwein rearrangement of perhydro-3a,6;4,5-diepoxyisoindoles. Tetrahedron 2011, 67, 9148–9163. [Google Scholar] [CrossRef]
- Morzycki, J.W.; Gryszkiewicz, A.; Jastrzebka, I. Some reactions of 16α,17α-oxido-steroids: A study related to the synthesis of the potent anti-tumor Saponin OSW-1 aglycone. Tetrahedron Lett. 2000, 41, 3751–3754. [Google Scholar] [CrossRef]
- Chen, Z.-M.; Zhang, Q.-W.; Chen, Z.-H.; Li, H.; Tu, Y.-Q.; Zhang, F.-M.; Tian, J.-M. Organocatalytic Asymmetric Halogenation/Semipinacol Rearrangement: Highly Efficient Synthesis of Chiral α-Oxa-Quaternary β-Haloketones. J. Am. Chem. Soc. 2011, 133, 8818–8821. [Google Scholar] [CrossRef]
- Li, H.; Zhang, F.-M.; Tu, Y.-Q.; Zhang, Q.-W.; Chen, Z.-M.; Chen, Z.-H.; Li, J. Enantioselective bromination/semipinacol rearrangement for the synthesis of β-bromoketones containing an all-α-carbon quaternary center. Chem. Sci. 2011, 2, 1839–1841. [Google Scholar] [CrossRef]
- Girdhar, N.K.; Ishar, M.P.S.; Kumar, R.; Singh, R.; Singh, G. Highly efficient Lewis acid catalyzed, one step conversions of 16α,17α-epoxy-3β-hydroxypregn-5-en-20-one to D-homosteroid and Δ13-steroids. Tetrahedron 2001, 57, 7199–7204. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Mertsalov, D.F.; Zaytsev, V.P.; Varlamov, A.V.; Gurbanov, A.V.; Dorovatovskii, P.V.; Timofeeva, T.V.; Khrustalev, V.N.; Mahmudov, K.T. Halogen bonding in Wagner-Meerwein rearrangement products. J. Mol. Liq. 2018, 249, 949–952. [Google Scholar] [CrossRef]
- Jung, M.E.; Street, L.J. Novel rearrangements of 7-oxanorbornene systems. Tetrahedron Lett. 1985, 26, 3639–3642. [Google Scholar] [CrossRef]
- Ciganek, E.; Calabrese, J.C. β-IodoKetones by Prevost Reaction of Vinyl Carbinol. J. Org. Chem. 1995, 60, 4439–4443. [Google Scholar] [CrossRef]
- Zubkov, F.I.; Nikitina, E.V.; Turchin, K.F.; Aleksandrov, G.G.; Safronova, A.A.; Borisov, R.S.; Varlamov, A.V. Wagner-Meerwein Skeletal Rearrangement of 3-Spiroannulated 6,8a-Epoxy- and 6,8a;7,8-Diepoxyisoquinolines (3-Aza-11-oxatricyclo[6.2.1.01,6]undec-9-enes). Isolation and Identification of 5-Aza-2-oxatricyclo[6.2.1.03,9]undec-3-enes. J. Org. Chem. 2004, 69, 432–438. [Google Scholar] [CrossRef]
- Zaytsev, V.P.; Mertsalov, D.F.; Trunova, A.M.; Khanova, A.V.; Nikitina, E.V.; Sinelshchikova, A.A.; Grigoriev, M.S. Iodine acetate as a mild selective agent for the Wagner–Meerwein rearrangement in 3a,6-epoxyisoindoles. Chem. Heterocycl. Compd. 2020, 56, 930–935. [Google Scholar] [CrossRef]
- Shellhamer, D.F.; Allen, J.L.; Allen, R.D.; Gleason, D.C.; Schlosser, C.O.N.; Powers, B.J.; Probst, J.W.; Rhodes, M.C.; Ryan, A.J.; Titterington, P.K.; et al. Ionic Reaction of Halogens with Terminal Alkenes: The Effect of Electron-Withdrawing Fluorine Substituents on the Bonding of Halonium Ions. J. Org. Chem. 2003, 68, 3932–3937. [Google Scholar] [CrossRef]
- Shellhamer, D.F.; Davenport, K.J.; Forberg, H.K.; Herrick, M.P.; Jones, R.N.; Rodriguez, S.J.; Sanabria, S.; Trager, N.N.; Weiss, R.J.; Heasley, V.L.; et al. Rearrangement of 3-Membered 1,1,2-Trifluorobromonium and Iodonium Ions and Comparison of Trifluorochloronium to Fluorocarbenium Ions. J. Org. Chem. 2008, 73, 4532–4538. [Google Scholar] [CrossRef]
- Fokin, A.A.; Schreiner, P.R.; Berger, R.; Robinson, G.H.; Wei, P.; Campana, C.F. Pseudotetrahedral Polyhalocubanes: Synthesis, Structures, and Parity Violating Energy Differences. J. Am. Chem. Soc. 2006, 128, 5332–5333. [Google Scholar] [CrossRef] [PubMed]
- SAINT-Plus, Version 7.68; Bruker AXS Inc.: Madison, WI, USA, 2012.
- Sheldrick, G.M. SADABS; Bruker AXS: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. On the importance of halogen-halogen interactions in the solid state of fullerene halides: A combined theoretical and crystallographic study. Crystals 2017, 7, 191. [Google Scholar] [CrossRef]
- Yogendra, S.; Hennersdorf, F.; Bauza, A.; Frontera, A.; Fischer, R.; Weigand, J.J. Selective and Reversible Fluoride Complexation from Water by a Cyclic Tri(phosphonio)methanide Dication. Angew. Chem. Int. Ed. 2017, 56, 7907–7911. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Zelenkov, L.E.; Ivanov, D.M.; Sadykov, E.K.; Bokach, N.A.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Semicoordination Bond Breaking and Halogen Bond Making Change the Supramolecular Architecture of Metal-Containing Aggregates. Cryst. Growth Des. 2020, 20, 6956–6965. [Google Scholar] [CrossRef]
- Torubaev, Y.; Skabitsky, I.V.; Rozhkov, A.V.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Highly Polar Stacking Interactions Wrap Inorganics in Organics: Lone-pair–π-Hole Interactions between the PdO4 Core and Electron-π deficient Arenes. Inorg. Chem. Front. 2021. [Google Scholar] [CrossRef]
- Suslonov, V.V.; Soldatova, N.S.; Ivanov, D.M.; Galmes, B.; Frontera, A.; Resnati, G.; Postnikov, P.S.; Kukushkin, V.Y.; Bokach, N.A. Diaryliodonium Tetrachloroplatinates(II): Recognition of a Trifurcated Metal-Involving μ3-I···(Cl, Cl, Pt) Halogen Bond. Cryst. Growth Des. 2021, 21, 5360–5372. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Petrov, A.A.; Galmes, B.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Ligand Steric Hindrances Switch Bridging (μ2-I)···O,O to Two-Center I···O Halogen-Bonding Mode in the Assembly of Diketonate Copper(II) Species. Cryst. Growth Des. 2021, 21, 4073–4082. [Google Scholar] [CrossRef]
- Baykov, S.V.; Geyl, K.K.; Ivanov, D.M.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. Azine Steric Hindrances Switch Halogen Bonding to N-Arylation upon Interplay with σ-Hole Donating Haloarenenitriles. Chem. Asian J. 2021, 16, 1445–1455. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Eliseeva, A.A.; Baykov, S.V.; Suslonov, V.V.; Galmes, B.; Frontera, A.; Kukushkin, V.Y.; Ivanov, D.M.; Bokach, N.A. Electron belt-to-σ-hole switch of noncovalently bound iodine(I) atoms in dithiocarbamate metal complexes. Inorg. Chem. Front. 2021, 8, 2505–2517. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Rozhkov, A.V.; Ananyev, I.V.; Frontera, A.; Kukushkin, V.Y. Bifurcated Halogen Bonding Involving Two Rhodium(I) Centers as an Integrated σ-Hole Acceptor. JACS Au 2021, 1, 354–361. [Google Scholar] [CrossRef]
- Efimenko, Z.M.; Eliseeva, A.A.; Ivanov, D.M.; Galmes, B.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Bifurcated μ2-I···(N,O) Halogen Bonding: The Case of (Nitrosoguanidinate)NiII Cocrystals with Iodine(I)-Based σ-Hole Donors. Cryst. Growth Des. 2021, 21, 588–596. [Google Scholar] [CrossRef]
- Soldatova, N.S.; Postnikov, P.S.; Suslonov, V.V.; Kissler, T.Y.; Ivanov, D.M.; Yusubov, M.S.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Diaryliodonium as a double σ-hole donor: The dichotomy of thiocyanate halogen bonding provides divergent solid state arylation by diaryliodonium cations. Org. Chem. Front. 2020, 7, 2230–2242. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl)I···dz2 [PtII] Halogen Bonds. Chem. Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef]
- Gomila, R.M.; Frontera, A. Metalloid Chalcogen–pnictogen σ-hole bonding competition in stibanyl telluranes. J. Organomet. Chem. 2021, 954–955, 122092. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 3 | 4 |
|---|---|---|
| CCDC number | 2118649 | 2118650 |
| Empirical formula | C14H11NO2FClBrI | C15H11NO2F3ClBrI |
| Formula weight | 486.50 | 536.51 |
| Temperature/K | 100(2) | 100(2) |
| Crystal system | orthorhombic | monoclinic |
| Space group | Pca21 | P21/c |
| a/Å | 19.310(2) | 12.2575(9) |
| b/Å | 7.2206(8) | 7.0718(5) |
| c/Å | 22.530(3) | 19.2040(13) |
| α/° | 90 | 90 |
| β/° | 90 | 90.781(4) |
| γ/° | 90 | 90 |
| Volume/Å3 | 3141.4(6) | 1664.5(2) |
| Z | 8 | 4 |
| ρcalcg/cm3 | 2.057 | 2.141 |
| μ/mm−1 | 4.765 | 4.524 |
| F(000) | 1856.0 | 1024.0 |
| Crystal size/mm3 | 0.16 × 0.12 × 0.08 | 0.4 × 0.18 × 0.14 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 8.378 to 54.998 | 6.968 to 70 |
| Index ranges | −25 ≤ h ≤ 25, −9 ≤ k ≤ 8, −29 ≤ l ≤ 29 | −19 ≤ h ≤ 19, −11 ≤ k ≤ 10, −30 ≤ l ≤ 30 |
| Reflections collected | 44,518 | 42,785 |
| Independent reflections | 7191 [Rint = 0.0816, Rsigma = 0.0605] | 7322 [Rint = 0.0282, Rsigma = 0.0217] |
| Data/restraints/parameters | 7191/1/380 | 7322/0/245 |
| Goodness-of-fit on F2 | 1.016 | 1.030 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0344, wR2 = 0.0575 | R1 = 0.0216, wR2 = 0.0443 |
| Final R indexes [all data] | R1 = 0.0503, wR2 = 0.0617 | R1 = 0.0310, wR2 = 0.0468 |
| Largest diff. peak/hole/e Å−3 | 0.93/−0.67 | 0.57/−1.00 |
| Bond | Length/Å | Length/Å | Length/Å |
|---|---|---|---|
| 3, molecule A | 3, molecule B | 4 | |
| I1-C5 | 2.146(8) | 2.138(7) | 2.1361(12) |
| Br1-C6 | 1.922(7) | 1.913(7) | 1.9150(12) |
| Cl1-C4 | 1.815(7) | 1.814(8) | 1.8202(12) |
| O1-C1 | 1.236(9) | 1.224(9) | 1.2262(16) |
| O8-C4 | 1.420(9) | 1.416(9) | 1.4177(15) |
| O8-C6 | 1.453(8) | 1.446(8) | 1.4457(16) |
| N2-C1 | 1.354(9) | 1.364(10) | 1.3576(17) |
| N2-C3 | 1.465(9) | 1.461(9) | 1.4621(16) |
| N2-C11 | 1.437(9) | 1.446(9) | 1.4369(17) |
| C1-C7A | 1.501(10) | 1.526(10) | 1.5111(18) |
| C3-C4 | 1.523(10) | 1.492(10) | 1.5159(18) |
| C4-C4A | 1.506(10) | 1.518(10) | 1.5189(18) |
| C4A-C5 | 1.534(10) | 1.535(10) | 1.5394(18) |
| C4A-C7A | 1.573(10) | 1.560(10) | 1.5612(18) |
| C5-C6 | 1.523(10) | 1.526(10) | 1.5261(18) |
| C6-C7 | 1.506(10) | 1.531(10) | 1.5219(18) |
| C7-C7A | 1.585(11) | 1.567(11) | 1.5755(18) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mertsalov, D.F.; Gomila, R.M.; Zaytsev, V.P.; Grigoriev, M.S.; Nikitina, E.V.; Zubkov, F.I.; Frontera, A. On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms. Crystals 2021, 11, 1406. https://doi.org/10.3390/cryst11111406
Mertsalov DF, Gomila RM, Zaytsev VP, Grigoriev MS, Nikitina EV, Zubkov FI, Frontera A. On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms. Crystals. 2021; 11(11):1406. https://doi.org/10.3390/cryst11111406
Chicago/Turabian StyleMertsalov, Dmitriy F., Rosa M. Gomila, Vladimir P. Zaytsev, Mikhail S. Grigoriev, Eugeniya V. Nikitina, Fedor I. Zubkov, and Antonio Frontera. 2021. "On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms" Crystals 11, no. 11: 1406. https://doi.org/10.3390/cryst11111406
APA StyleMertsalov, D. F., Gomila, R. M., Zaytsev, V. P., Grigoriev, M. S., Nikitina, E. V., Zubkov, F. I., & Frontera, A. (2021). On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms. Crystals, 11(11), 1406. https://doi.org/10.3390/cryst11111406