
A Robust Supramolecular Heterosynthon Assembled by a Hydrogen Bond and a Chalcogen Bond


Abstract
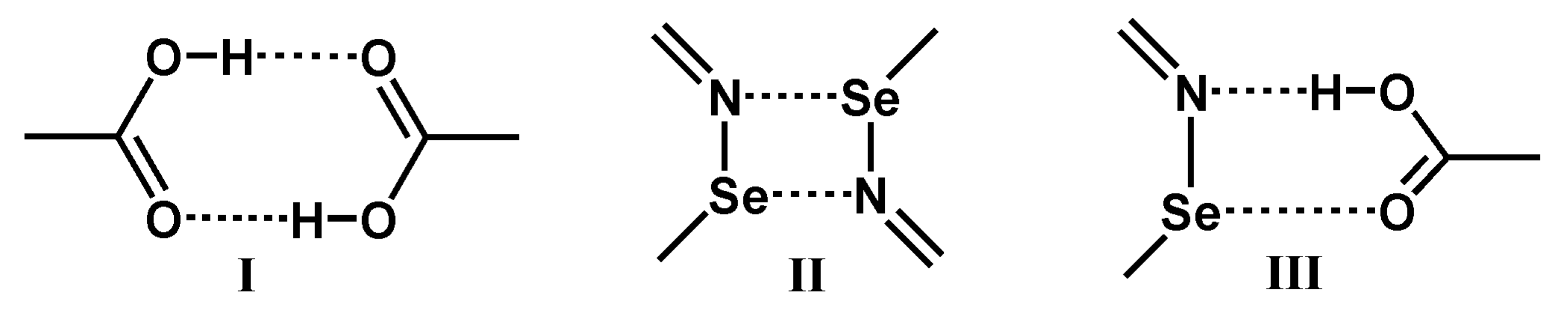
:1. Introduction
2. Materials and Methods
2.1. Cocrystal Synthesis
2.2. X-ray Crystallography
2.3. Computational Details
3. Results and Discussion
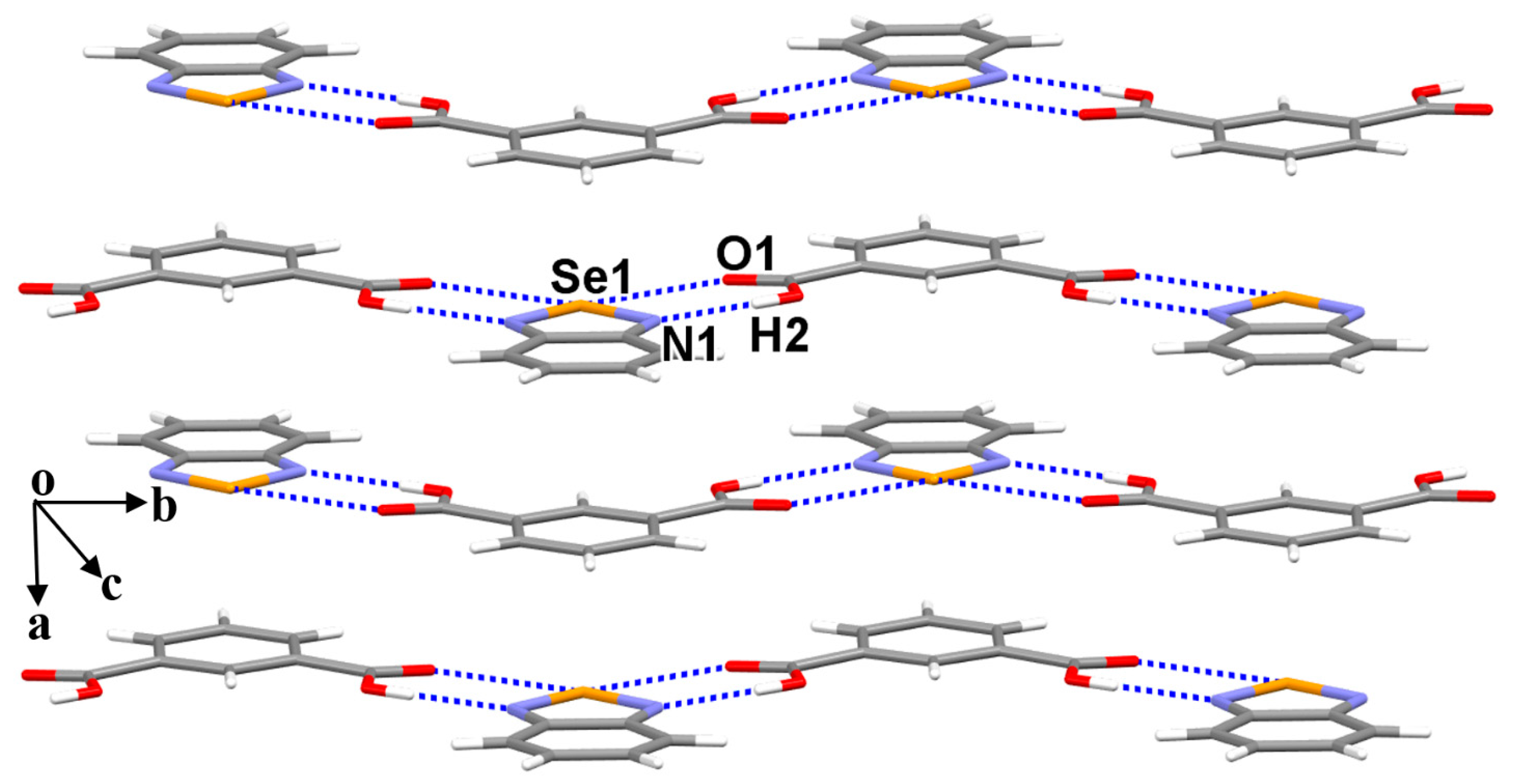
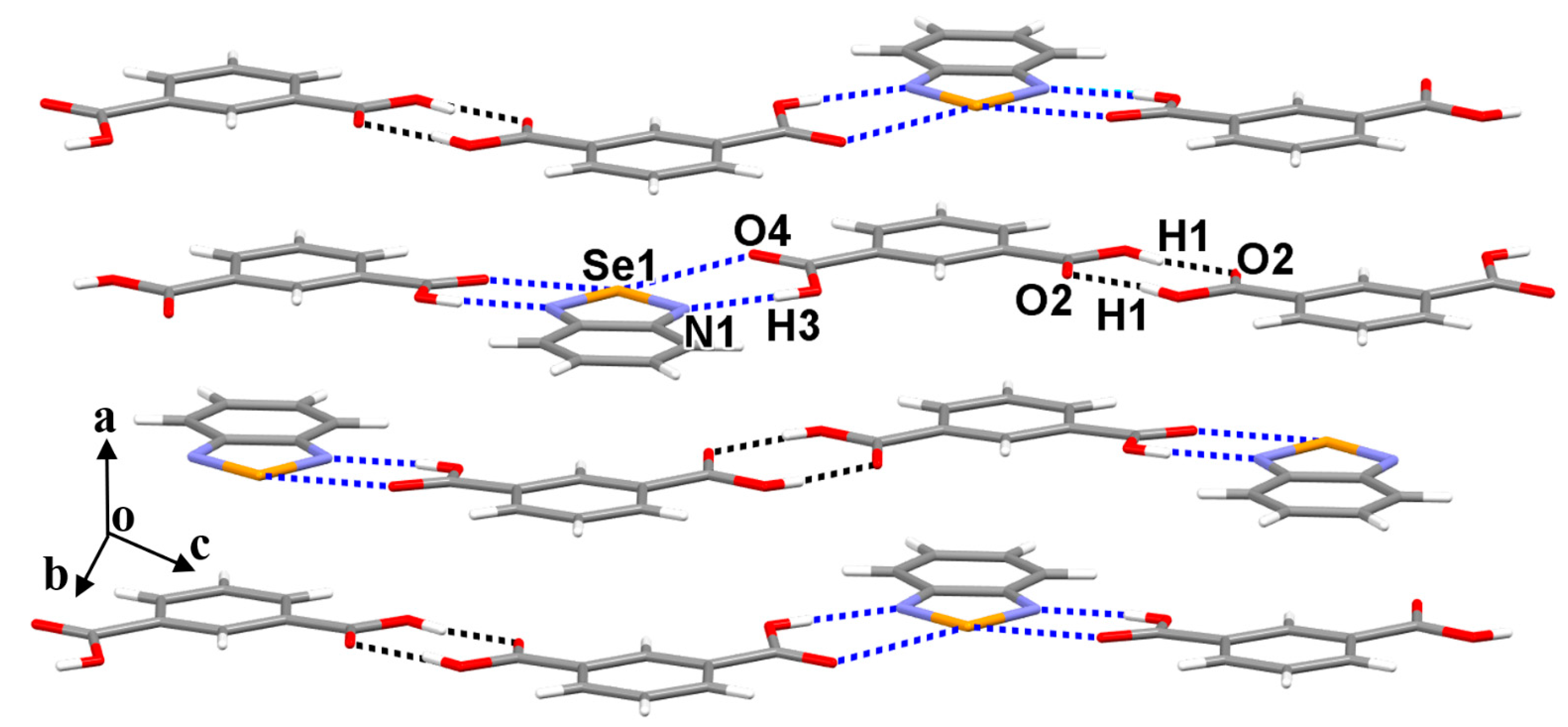
3.1. The Crystal Structures of Cocrystals 1 and 2
3.2. The π···π Stacking Interactions in the Crystal Structures
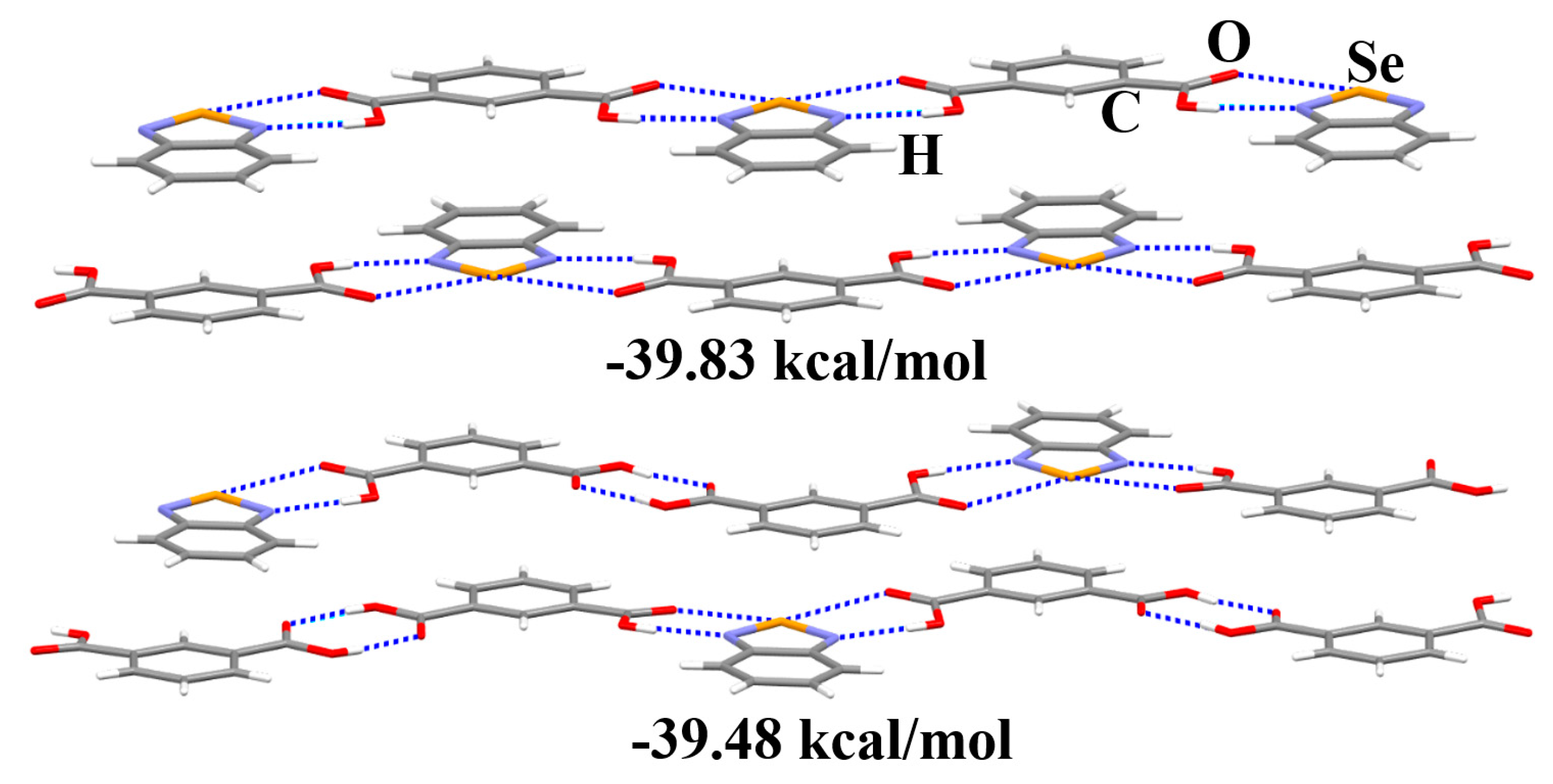
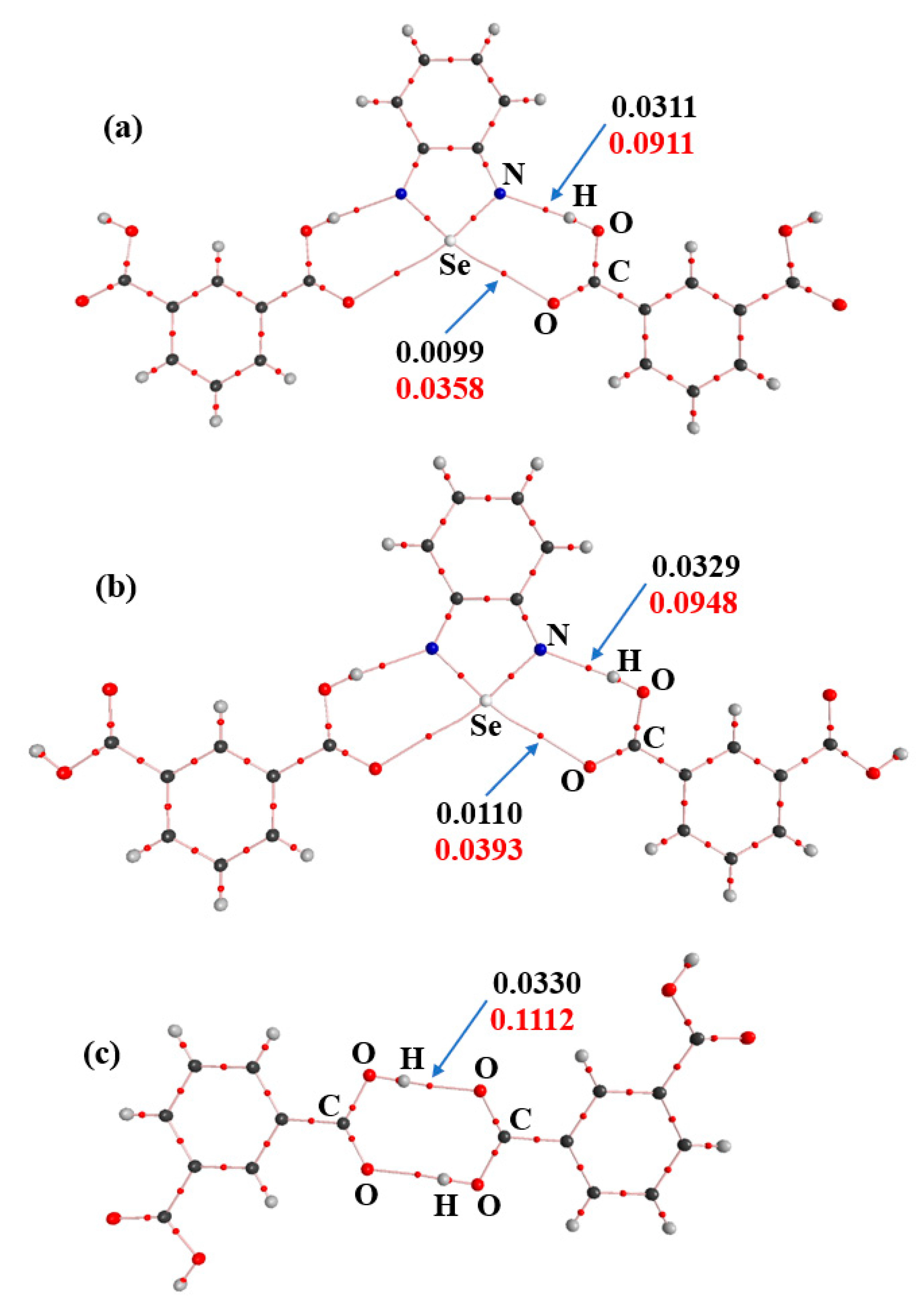
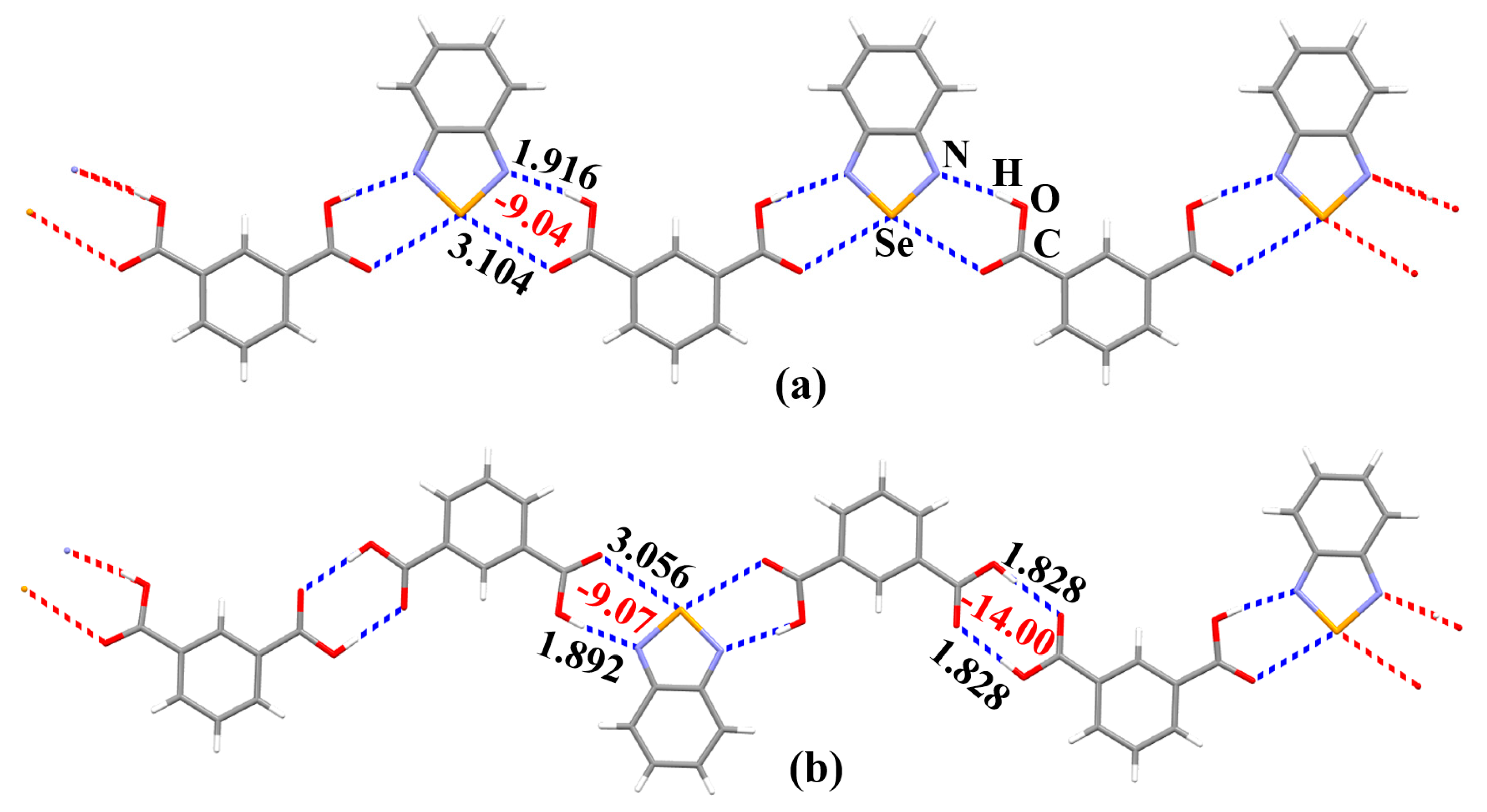
3.3. The Nature of the Supramolecular Synthons in the Crystal Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 31, 2311–2327. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: The Design of Organic Solids; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Nangia, A.; Lynch, V.M. Molecular Complexes of Homologous Alkanedicarboxylic Acids with Isonicotinamide: X-ray Crystal Structures, Hydrogen Bond Synthons, and Melting Point Alternation. Cryst. Growth Des. 2003, 3, 783–790. [Google Scholar] [CrossRef]
- Sanphui, P.; Bolla, G.; Das, U.; Mukherjee, A.K.; Nangia, A. Acemetacin Polymorphs: A Rare Case of Carboxylic Acid Catemer and Dimer Synthons. CrystEngComm 2013, 15, 34–38. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Eichstaedt, K.; Wasilewska, A.; Wicher, B.; Gdaniec, M.; Połoński, T. Supramolecular Synthesis Based on a Combination of Se···N Secondary Bonding Interactions with Hydrogen and Halogen Bonds. Cryst. Growth Des. 2016, 16, 1282–1293. [Google Scholar] [CrossRef]
- Rahman, F.-U.; Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef] [PubMed]
- Biot, N.; Bonifazi, D. Chalcogen-Bond Driven Molecular Recognition at Work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular Aggregation Patterns Featuring Se···N Secondary Bonding Interactions in Mono-Nuclear Selenium Compounds: A Comparison with their Congeners. Coord. Chem. Rev. 2021, 443, 214031. [Google Scholar] [CrossRef]
- Walsh, R.D.B.; Bradner, M.W.; Fleischman, S.; Morales, L.A.; Moulton, B.; Rodríguez-Hornedo, N.; Zaworotko, M.J. Crystal Engineering of the Composition of Pharmaceutical Phases. Chem. Commun. 2003, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical Cocrystals: Along the Path to Improved Medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef]
- Gong, Y.; Zhao, L.; Peng, Q.; Fan, D.; Yuan, W.Z.; Zhang, Y.; Tang, B.Z. Crystallization-Induced Dual Emission from Metal- and Heavy Atom-Free Aromatic Acids and Esters. Chem. Sci. 2015, 6, 4438–4444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem, Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The Benzene···Naphthalene Complex: A more Challenging System than the Benzene Dimer for newly Developed Computational Methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef]
- Li, M.M.; Wang, Y.B.; Zhang, Y.; Wang, W. The Nature of the Noncovalent Interactions between Benzene and C60 Fullerene. J. Phys. Chem. A 2016, 120, 5766–5772. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly Accurate Benchmark Calculations of the Interaction Energies in the Complexes C6H6···C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Biegler-König, F.; Schönbohm, J.; Bayles, D. AIM2000–A Program to Analyze and Visualize Atoms in Molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar]
- Derissen, J.L. Isophthalic Acid. Acta Cryst. 1974, B30, 2764–2765. [Google Scholar] [CrossRef]
- Pitoňák, M.; Neogrády, P.; Řezáč, J.; Jurečka, P.; Urban, M.; Hobza, P. Benzene Dimer: High-Level Wave Function and Density Functional Theory Calculations. J. Chem. Theory Comput. 2008, 4, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Popelier, P.L.A. Characterization of C–H–O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Wang, W.; Wong, N.-B.; Zheng, W.; Tian, A. Theoretical Study on the Blueshifting Halogen Bond. J. Phys. Chem. A 2004, 108, 1799–1805. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Lipkowski, P. Characteristics of X–H···π Interactions: Ab Initio and QTAIM Studies. J. Phys. Chem. A 2011, 115, 4765–4773. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. QTAIM Characteristics of Halogen Bond and Related Interactions. J. Phys. Chem. A 2012, 116, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathi, R.; Subramanian, V. Characterization of Hydrogen Bonding: From van der Waals Interactions to Covalency. In Hydrogen Bonding-New Insights; Grabowski, S.J., Ed.; Springer: New York, NY, USA, 2006; pp. 1–50. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocrystal | 1 | 2 |
|---|---|---|
| CCDC No. | 2090234 | 2090225 |
| Empirical formula | C14H10N2O4Se | C22H16N2O8Se |
| Formula weight | 349.20 | 515.33 |
| Crystal size/mm3 | 0.27 × 0.15 × 0.14 | 0.27 × 0.25 × 0.22 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/m | C2/c |
| a/Å | 6.9731(4) | 10.4027(7) |
| b/Å | 12.4491(5) | 14.8477(7) |
| c/Å | 8.2841(3) | 13.9101(8) |
| α/° | 90 | 90 |
| β/° | 107.827(5) | 101.425(6) |
| γ/° | 90 | 90 |
| V/Å3 | 684.60(6) | 2105.9(2) |
| Z | 2 | 4 |
| ρcalc/g·cm−3 | 1.694 | 1.625 |
| T/K | 292 | 290 |
| 2θ range for data collection/° | 6.546–56.698 | 7.054–56.898 |
| Reflections collected | 9375 | 12212 |
| Independent reflections [Rint] | 1627 [0.0317] | 2387 [0.0539] |
| R1, wR2 (I > 2σ(I)) | 0.0318, 0.0696 | 0.0548, 0.0901 |
| R1, wR2 (all data) | 0.0406, 0.0726 | 0.0850, 0.0988 |
| Goodness-of-fit on F2 | 1.120 | 1.131 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, S.; Zhang, Y.; Shan, L.; Xu, M.; Wang, J.-G.; Zhang, Y.; Wang, W. A Robust Supramolecular Heterosynthon Assembled by a Hydrogen Bond and a Chalcogen Bond. Crystals 2021, 11, 1309. https://doi.org/10.3390/cryst11111309
Miao S, Zhang Y, Shan L, Xu M, Wang J-G, Zhang Y, Wang W. A Robust Supramolecular Heterosynthon Assembled by a Hydrogen Bond and a Chalcogen Bond. Crystals. 2021; 11(11):1309. https://doi.org/10.3390/cryst11111309
Chicago/Turabian StyleMiao, Shaobin, Yunfan Zhang, Linjie Shan, Mingyuan Xu, Jian-Ge Wang, Yu Zhang, and Weizhou Wang. 2021. "A Robust Supramolecular Heterosynthon Assembled by a Hydrogen Bond and a Chalcogen Bond" Crystals 11, no. 11: 1309. https://doi.org/10.3390/cryst11111309
APA StyleMiao, S., Zhang, Y., Shan, L., Xu, M., Wang, J.-G., Zhang, Y., & Wang, W. (2021). A Robust Supramolecular Heterosynthon Assembled by a Hydrogen Bond and a Chalcogen Bond. Crystals, 11(11), 1309. https://doi.org/10.3390/cryst11111309