Abstract
Crystallographic phasing recovers the phase information that is lost during a diffraction experiment. Molecular replacement is a commonly used phasing method for crystal structures in the protein data bank. In one form it uses a protein sequence to search a structure database to find suitable templates for phasing. However, sequence information is not always available, such as when proteins are crystallized with unknown binding partner proteins or when the crystal is of a contaminant. The recent development of AlphaFold published the predicted protein structures for every protein from twenty distinct species. In this work, we tested whether AlphaFold-predicted E. coli protein structures were accurate enough to enable sequence-independent phasing of diffraction data from two crystallization contaminants of unknown sequence. Using each of more than 4000 predicted structures as a search model, robust molecular replacement solutions were obtained, which allowed the identification and structure determination of YncE and YadF. Our results demonstrate the general utility of the AlphaFold-predicted structure database with respect to sequence-independent crystallographic phasing.
1. Introduction
Crystallographic phasing requires the retrieval of phase information that is lost during diffraction experiments. When there are no homology models, such phase information is recovered experimentally using isomorphous replacement preferably with their anomalous signals [1,2]. With the accumulation of experimentally determined structures, molecular replacement [3,4] has become a routine method for crystallographic phasing. Indeed, 71% of deposited crystal structures in the PDB database (www.pdb.org accessed on 12 August 2021) were determined using molecular replacement.
Molecular replacement exploits the similarity between known structures and the structure to be determined. Programs such as MOLREP [5], PHASER [6], and AMoRe [7] have been developed to accomplish this. When protein sequence information is known, molecular replacement pipelines may be used to automate the process as implemented in MrBUMP [8], BALBES [9], and MRage [10]. Using ab initio modelling software such as ROSETTA [11], predicted structures may also be used for molecular replacement as implemented in AMPLE [12]. However, there are cases in which the sequence information is unknown. Examples include the crystallization of contaminant proteins or unknown protein-binding partner proteins [13]. Under such scenarios, MoRDa (a non-redundant, annotated PDB database) [14], ContaMiner/ContaBase (a collection of previously reported contaminant protein structures) [13], and a sequence-independent molecular replacement based on available databases (SIMBAD) pipeline [15] may be used for sequence-independent molecular replacement using database-searching approaches. Among these tools, SIMBAD searches contaminant and MoRDa databases for a protein sequence-independent molecular replacement [15].
Machine learning has been extensively used for protein structure predictions with the recent development of the revolutionary attention-based AlphaFold [16,17] and RoseTTAFold algorithms [18]. Both methods have enabled accurate prediction of protein structures approaching the fidelity of their crystal structures. In collaboration with an European Molecular Biology Laboratory (EMBL) team, AlphaFold released more than 350,000 predicted structures representing the full protein complement of twenty species including humans and predominant model systems including yeast, Arabidopsis, and E. coli (https://alphafold.ebi.ac.uk accessed on 20 July 2021) [19]. The AlphaFold-predicted structures may serve as a valuable new resource to support crystallographic phasing. It is therefore possible to use these structural databases for a protein sequence-independent molecular replacement for phasing of diffraction data. This database approach may be of particular use for phasing proteins crystallized inadvertently, proteolysis products, and structures with significant conformational changes. In cases in which a protein crystallizes with an unexpected binding partner, the AlphaFold database could be also used to identify the identity of the unknown protein without the need for using mass spectrometry or protein sequencing.
For X-ray crystallography, many proteins are expressed in E. coli and purified using affinity columns. Often, in addition to protein of interest, E. coli contaminant proteins may bind either to the affinity resin or the protein of interest and may be co-purified and inadvertently crystallized. Although crystallization of a contaminant protein is relatively rare, many contaminant structures have been identified as reported in the ContaBase database [13]. For new contaminant proteins it may take some effort to identify it through experimental phasing, mass spectrometry, protein sequencing, or using database searches. Because AlphaFold has generated a complete database of predicted structures for all folded protein sequences in E. coli, we sought to test whether this resource could enable crystallographic phasing in the absence of protein sequence information.
In recent crystallization work on two plant proteins that were over-expressed in E. coli, we unexpectedly crystallized two contaminants and collected diffraction data to about 2.3–2.5 Å resolution. For one of them, we could not solve its structure using existing methods. In this work, we used the two contaminant data sets for sequence-independent molecular replacement. Using a relatively straightforward workflow, we showed that predicted AlphaFold structures can be used to phase both structures without any protein sequence information. Our work highlights the broad utility of the AlphaFold-predicted structure database for crystallographic analysis.
2. Materials and Methods
2.1. Sample Preparation for YncE/P76116
E. coli contaminant protein YncE was co-purified while we worked on the expression of a plant Δ6 desaturase over-expressed in BL21-Gold (DE3) cells (Novagen). The desaturase protein was over-expressed at 30 °C for 4 h by addition of 0.2 mM IPTG to the cell culture with an A600 of 0.6. Harvested cells were re-suspended in resuspension buffer (30 mM MES, 33 mM HEPES, 33 mM NaOAc, pH 7.5) supplemented with 2 mM MgCl2 and 0.1 mg/mL DNase. The cells were lysed using a French press, and cell debris was removed by centrifugation at 25,000× g for 30 min at 4 °C. The clarified extract was loaded onto a Poros 20 HS column (Perceptive Biosystems, Framingham, MA, USA), washed with five column volumes of resuspension buffer, and eluted with a linear gradient of 0–1.2 M NaCl in the resuspension buffer. Desaturase fractions were pooled and concentrated, subjected to a size-exclusion HPLC column (TSKgel G3000SW column, Tosoh Bioscience, South San Francisco, CA, USA), and eluted with 20 mM HEPES, pH 7.0, and 100 mM NaCl. The protein fractions were pooled and concentrated to 15 mg/mL for crystallization.
Crystals were grown using the hanging drop vapor diffusion method consisting of 0.6 µL of protein mixed with an equal volume of reservoir solution containing 0.2 M Li2SO4, 0.1 M MES, pH 6.0, and 20% PEG 4000. Plate-shaped crystals were flash-frozen with liquid nitrogen. Cryo-protectant was not added prior to freezing.
2.2. Sample Preparation for YadF/P61517
E. coli. contaminant protein YadF was co-purified with the production of Arabidopsis Metacaspase 4 (AtMC4) in BL21 (DE3) pLysS cells (Novagen). Cells were lysed using a homogenizer, and the soluble fraction of AtMC4 was collected for a three-step purification by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (HisTrap FF column, GE Healthcare, Inc., Chicago, IL, USA), ion exchange chromatography (HiTrap Q HP column, GE Healthcare, Inc.), and gel filtration (Superdex 200 10/300 GL column, GE Healthcare, Inc.). Purified AtMC4 was then mixed and incubated with the excess molar amount of the inhibitor PPACK (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). This mixture was further purified by gel filtration, and the inhibitor-bound complex was concentrated to 8–10 mg/mL for crystallization.
Crystals were grown using the hanging drop vapor diffusion method. One µL of inhibitor-bound AtMC4 was mixed with an equal volume of precipitant that contains 100 mM sodium cacodylate, pH 6.8, and 1.8 M ammonium sulfate. For cryo-crystallography, crystals were transferred into the precipitant supplemented with 10% glycerol and were flash-cooled into liquid nitrogen for cryogenic data collection.
2.3. Diffraction Data Collection and Reduction
Diffraction data were collected at the NSLS-II beamline FMX (17ID-2) at 100 K [20]. The beamline is equipped with an Eiger 16M detector. For YncE, we collected data at an X-ray wavelength of 0.979 Å. A total of 1800 frames were collected from a single YncE crystal with a rotation angle of 0.2°. For YadF, we collected data at an X-ray wavelength of 1.891 Å. A total of ~1500 frames were collected from four YadF crystals with a rotation angle of 0.3°.
Single-crystal data sets were indexed and integrated independently using DIALS [21] and then scaled and merged using CCP4 programs POINTLESS and AIMLESS [22,23] with the outlier rejection as implemented in PyMDA [24,25]. For the YncE data, we rejected 700 radiation-damaged frames. For the YadF data, we rejected 948 radiation-damaged frames using a decay value of 1.0 as defined by frame_cutoff = (Min(SmRmerge) × (1+decay)), where Min(SmRmerge) is the lowest SmRmerge (reported in AIMLESS log file) within a single-crystal data set; and decay is a rejection ratio [24]. The data collection and data processing statistics for the two data sets are shown in Table 1.

Table 1.
Data collection and refinement statistics.
2.4. AlphaFold Structures for Database-Driven Molecular Replacement
Figure 1 shows the workflow of using AlphaFold-predicted E. coli structure database for sequence-independent molecular replacement. From these twenty AlphaFold-predicted structure databases (https://alphafold.ebi.ac.uk/download accessed on 20 July 2021), we downloaded all 4363 E. coli protein structures. Among these structures, we removed those with less than 50 residues from further use. Then, we set up a molecular replacement search using the remaining 4175 structures. For each structure, we performed molecular replacement in MOLREP [5] with both rotation and translation searches with a high-resolution data cut-off at 3.0 Å resolution. To speed up the searches, in MOLREP we turned off the pack and score function and searched for the 10 highest rotation and translation peaks. We performed the searches in parallel by submitting the jobs to a custom-built Linux cluster using the batch-queuing system SGE (Sun Grid Engine, Oracle Corporation, Austin, TX, USA). The first rotation and translation peak heights for each structure were extracted from MOLREP log files, written to a file, and sorted. The structures displaying the highest rotation and translation peaks were used to narrow the molecular replacement search. For YncE, we removed 34 disordered residues from its N-terminus and used MOLREP for multi-copy molecular replacement [26]. For YadF, we tried molecular replacement in different space groups to identify the one with the highest translation peak height.
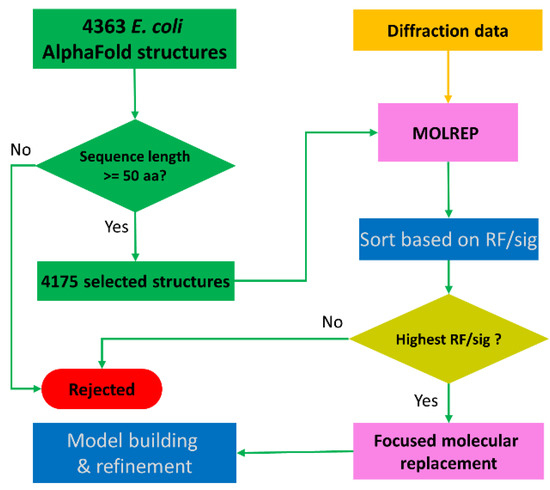
Figure 1.
Schematic workflow of sequence-independent crystallographic phasing using AlphaFold-predicted E. coli structures. A total number of 4363 AlphaFold-predicted structures were downloaded from the AlphaFold structure database. After filtering based on protein sequence length, 4175 structures were selected for molecular replacement using MOLREP. The output candidate solutions were sorted based on RF/sig, and the AlphaFold structure with the highest RF/sig peak height was selected for focused molecular replacement and downstream model building and refinement.
2.5. Model Building and Structure Refinement
Iterative model building and refinement were performed in COOT [27] and PHENIX.REFINE [28,29], respectively. For the YncE data, Bijvoet pairs were averaged for structure refinement. For the YadF data, Bijvoet pairs were treated as two different reflections in structure refinement, and the resultant Fourier coefficients were used for calculation of Bijvoet-difference Fourier maps. We also used anomalous signals for a f″ refinement [30] to find anomalous scattering elements in the YadF structure. For the f” refinement, the occupancies for the potassium and zinc ions were first estimated so that their refined individual B factors are close to the average B factors from their interacting protein and water atoms. We then refined f″ in PHENIX.REFINE starting with f″ values of zero for sulfur, potassium, and zinc. The stereochemistry of the refined structures was validated with PROCHECK [31] and MolProbity [32] for quality assurance. The refinement statistics for the two data sets are shown in Table 1.
3. Results
3.1. AlphaFold Structures for Phasing YncE
During our work on the purification and crystallization of a plant desaturase, we co-purified YncE under crystallization conditions of 0.2 M Li2SO4, 0.1 M MES, pH 6.0, and 20% PEG 4000. We collected diffraction data and processed the data to dmin 2.5 Å in space group P21 with unit dimensions a = 53.2 Å, b = 147.3 Å, c = 96.9 Å, and β = 104.4°. Although the expected sequence identity for the desaturase to its homologous structures in PDB was beyond 80%, we were unable to solve the structure using the PDBs as search models, suggesting that this crystallized protein could be a contaminant. We used CCP4 online servers to search for contaminants but did not find a clear solution. To identify the contaminant, we also tried to repeat the crystallization and used mass spectrometry to identify the contaminant.
With only the diffraction data available, we hypothesized that the contaminant protein must originate from E. coli. With the release of the AlphaFold-predicted E. coli structures, we reasoned that the crystallized contaminant should be represented in the AlphaFold structure database. We proceeded with the workflow described in Figure 1 to search for a monomer. All AlphaFold structures give their highest rotation and translation peaks beyond zero, while a single structure, YncE (UNIPROT entry P76116), showed the highest RF/sig and TF/sig of 12.43 and 13.08, respectively (Figure 2a).
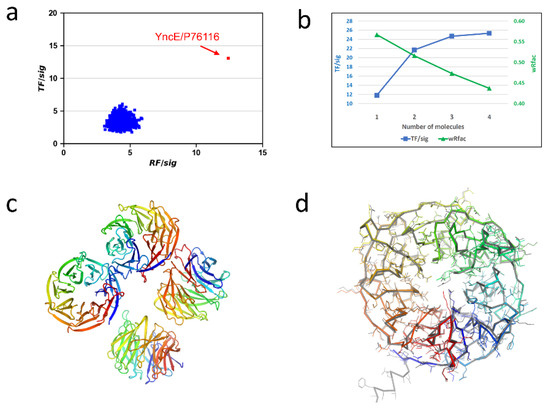
Figure 2.
Alphafold structures for phasing E. coli YncE. (a) Histogram of rotation and translation peaks. (b) Progressive molecular replacement while searching for four molecules in a.u. (c) Refined YncE structure. (d) Comparison of the refined structure with the AlphaFold structure. The AlphaFold-predicted structure is shown in gray.
Unit-cell content and self-rotation function analyses suggested the presence of multiple copies of YncE in the asymmetric unit (a.u.). We therefore performed focused molecular replacement searches for multiple copies using MOLREP. Visualization of the AlphaFold-predicted YncE structure indicated that it has a long N-terminal extension consisting of 34 poorly predicted/disordered residues. To assure that such a long extension would not affect the packing analysis in MOLREP, we removed the N-terminal 34 residues and used the truncated model for a search of two to five molecules. We obtained the best results while searching for four molecules in a.u. and observed that both TF/sig and wRfac improved with an increasing number of molecules (Figure 2b). With the four-molecule search, the final TF/sig and wRfac were 25.35 and 0.437, respectively, strongly indicating a correct solution for protein identification and structure determination.
The refined YncE structure has four molecules, each containing residues from 32 to 342 and forming a seven-bladed β-propeller structure (Figure 2c). Except the N-terminal extension, the structure is very similar to the AlphaFold-predicted structure with an RMSD of 0.39 Å for 321 aligned Cα atoms (Figure 2d). However, we found that many side chains have different conformations, perhaps due to crystal contacts or disordered conformations.
In the UNIPROT entry for P76116/YncE, two PDBs (3VGZ and 3VH0) were reported: one crystallized in C2221 lattice and the other crystallized in I41 lattice [33]. Our P21-form structure is a new contaminant structure. The P21-form structure has an RMSD of 0.44 Å with the C2221-form structure and 0.37 Å with the I41-form structure, indicating that all three structures are very similar although being crystallized in different space groups. Table 2 summarizes the detailed crystallographic comparison of the YncE structure determined in three different lattices.
Table 2.
Comparison of YncE/P76116 structure with PDB structures listed under UNIPROT entry P76116.
3.2. AlphaFold Structures for Phasing YadF
E. coli YadF is another contaminant protein that was co-purified with an Arabidopsis metacaspase 4 (AtMC4). AtMC4 is a cysteine protease, and we have previously determined its structure in an apo form [34]. To obtain a complex structure of AtMC4 with a protease inhibitor PPACK, we attempted to crystallize the complex for structural analysis. Crystals with dimensions of about 20–30 µm were obtained under the crystallization conditions of 0.1 M sodium cacodylate, pH 6.8, and 1.8 M (NH4)2SO4. We collected diffraction data from four crystals at a relatively longer X-ray wavelength of 1.891 Å. The processed data at dmin 2.3 Å had a tetragonal lattice with unit-cell dimensions of a = 67.5 Å and c = 85.3 Å. However, we were unable to solve its structure using the AtMC4 structures of either the full length or its truncations. Therefore, we suspected that this could be another E. coli contaminant and may be suitable for structure determination through searching the AlphaFold-predicted structure database.
Using the same workflow described above for YncE, we performed molecular replacement searches using MOLREP for each of the 4175 structures. Figure 3a shows the histogram plot for RF/sig and TF/sig. Although there are four targets with the highest translation peaks beyond 10 (UNIPROT entries P0CF69, P75971, P0CF68, and P61517), P61517/YadF is the only target with the highest rotation peak at 9.04, suggesting it is a possible solution for downstream model building and refinement. YadF has 220 residues, and the unit-cell content analysis suggested a single molecule in a.u. with an estimated solvent content of 43%. The initial refinement in PHENIX.REFINE yielded an R/free R of 0.30/0.39, suggesting larger structural differences relative to the AlphaFold-predicted structure. Therefore, we rebuilt the model using ARP/WARP [35]. ARP/WARP produced a nearly complete model of 208 residues in one chain with an R/free R of 0.194/0.252, indicating a correct identification and structure determination using the AlphaFold structure database.
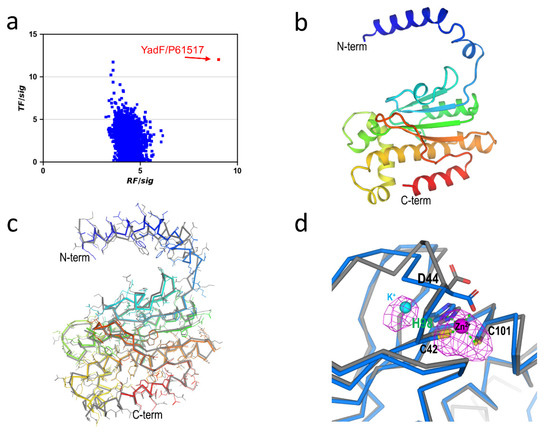
Figure 3.
AlphaFold structures for phasing E. coli. YadF. (a) Histogram of rotation and translation peaks. (b) Refined YadF structure. (c) Comparison with the AlphaFold structure. The AlphaFold-predicted structure is shown in gray. (d) Active-site structure. Residues interacting with the zinc site are shown as sticks. Bijvoet difference Fourier map for anomalous scatterers were shown as magenta isomeshes contoured at 3σ. As a comparison, the AlphaFold-predicted structure is shown in gray.
The refined structure has 211 residues, and its structure is shown in Figure 3b. The structure has an N-terminal α-helix domain and a C-terminal mixed αβ domain. Compared with the AlphaFold-predicted structure, the RMSD was 1.18 Å for 206 aligned Cα atoms. Most structural differences were on the N-terminal helix and the loop connecting it to the αβ domain (Figure 3c).
YadF is a carbonic anhydrase whose activity is zinc-dependent [36]. We had collected data at an X-ray wavelength of 1.891 Å at which the theoretical anomalous signal f″ was 0.98 e. Therefore, we used an f″ refinement to characterize zinc anomalous signals [30]. With an estimated occupancy of 1.0 for the zinc site, the refined f″ was 0.94 e, clearly validating the specialization of the zinc site. Zinc is coordinated with two cysteine residues (Cys42 and Cys101), His98, and Asp44. Figure 3d shows the Bijvoet difference Fourier densities for the active site. The Bijvoet densities cover zinc as well as two sulfur atoms. Surprisingly, next to the zinc/sulfur densities, we observed an extra electron density next to His98. To identify the type of anomalous scatterers associated with this density, we performed the f″ refinement with a candidate ion of Zn2+, Ca2+, K+, or Na+. Through the f” refinements, the only reasonable fit for this anomalous scatterer was K+ with an occupancy of 0.6 and a B-factor of 33.5 Å2. However, we did not include K+ either in protein purification or crystallization. Its exact origin and potential functional role will therefore be the subject of further investigation.
The AlphaFold-predicted structure does not contain any ions, neither Zn2+ nor K+. Structural superimposition of the AlphaFold structure with the ion-bound YadF structure indicates conformational changes of Asp44 (Figure 3d). Interestingly, the same residue has been proposed to undergo conformational change so that substrate CO2 can approach Zn2+ to form a CO-Zn2+ species [36]. Thus, it is possible that the AlphaFold-predicted structure might resemble an intermediate state of YadF, at least for the active site structure.
Under UNIPROT entry P61517, there are four reported PDBs (1I6O, 1I6P, 1T75, and 2ESF) [36,37], which were all determined in tetragonal lattices but with different crystallization conditions. Our structure had an RMSD between 0.35 and 0.79 Å compared to these structures. Table 3 summarizes detailed crystallographic comparison of YadF under different crystallization conditions.
Table 3.
Comparison of YadF/P61517 structures with PDB structures listed under UNIPROT entry P61517.
4. Discussion
4.1. AlphaFold-Predicted Structure Database
Crystallizing protein contaminants is a relatively common problem. In this work, we demonstrated that AlphaFold-predicted E. coli structures can be useful for molecular replacement to identify unknown crystallized contaminant proteins and to determine their structures. In our tests, we did not modify the predicted structures for the initial molecular replacement searches even though these predicted structures may contain unstructured extensions and poorly predicted regions such as we found with the N-terminal long extension in YncE.
For the two contaminant structures that we determined using AlphaFold-predicted structure database, YncE is a new contaminant. Although there are two crystal structures (PDB entries 3VGZ and 3VH0), we did not obtain a clear solution while trying database search approaches using the CCP4 online server. As a comparison, for YadF, in addition to using AlphaFold structure database, we were able to find a solution using its unit cell dimensions to search the PDB database, and PDBs 1I6P and 4ZNZ were identified. Apparently PDB 4ZNZ had already been reported as a crystallization contaminant [38] that was crystallized in a different condition (Table 3). We note that the YadF structure in this work has a larger RMSD with the AlphaFold-predicted structure (1.2 Å) than with other crystal structures (0.35–0.79 Å) with the largest structural differences located at the N-terminal helix (Figure 3c). In the YadF crystal structures, this helix is stabilized by forming a dimer with its symmetry mate [36]. In contrast, the AlphaFold-predicted structure is a monomer, and the N-terminal helix is thus more flexible.
Phasing with an E. coli structure database has multiple advantageous over using the PDB database. First, the predicted structures contain only single-chain structures, which may be used directly for rotation searches without further processing, i.e., removing non-protein components or splitting a protein complex into individual components. Second, the predicted structure is based on the entire encoded protein sequences. Consequently, using such a database provides a higher probability of finding a promising structure template for phasing. Although in this work we only used E. coli structures for identification and determination of contaminant structures, the AlphaFold databases contain 350,000 predicted protein structures from 20 species [19]; and those databases may be well suited for phasing contaminant structures from proteins expressed in mammalian cells, yeast, Arabidopsis, etc. Third, AlphaFold structures may be used to identify and phase unexpected proteolytic fragments or unexpected binding partner proteins.
Using a domain-structure database and modelled structure for phasing has been previously implemented in MoRDa and AMPLE, respectively [12,14]. However, due to the limited number of structural domains and the uncertainty associated with the modelling, database-based phasing has not been routine and is normally used as a method of last resort after exhausting other phasing strategy options. As AlphaFold-predicted structures approach the accuracy of experimental structures, molecular replacement using AlphaFold structures could have more routine applications even for de novo phasing of proteins for which there is no homologous structure. The AlphaFold algorithm uses an artificial intelligence model that was extensively trained with available PDB and sequence databases [16]. Hence the AlphaFold-predicted structures could be biased toward known structures. Accordingly, additional protein structures with novel folds are needed to improve the prediction accuracy of AlphaFold. Based on our findings, we speculate that an increasing number of crystal structures will be phased using AlphaFold-predicted structural workflows.
4.2. Combining AlphaFold Phasing with Anomalous Signals
Perhaps due to the existence of prior crystal structures for both YncE and YadF, AlphaFold-predicted structures are quite accurate, with RMSD values of 0.39 Å and 1.18 Å, relative to their refined structures (Figure 2d and Figure 3c). When there are only remote or no homologous structures, AlphaFold-predicted structures may be insufficient for phasing solely through molecular replacement. We propose that molecular replacement with anomalous signals, e.g., MR-SAD [39], might be a highly productive strategy.
For YadF, we collected long-wavelength data at 1.891 Å, which allowed the characterization of anomalous scatterers of zinc, potassium, and sulfur atoms within the structure. To determine whether anomalous signals would enhance AlphaFold-based crystallographic phasing, we tested MR-SAD [39] using the PHASER_EP pipeline [6]. With the initial phases from the AlphaFold structure, PHASER_EP identified seven anomalous scatterers with a figure-of-merit of 0.467. The MR-SAD map was of high quality; the pipeline could build 201 residues in eight fragments, with the longest fragment representing 71 residues. Subsequently, ARP/wARP built the same model as starting from the AlphaFold structure without using anomalous signals. For phasing YadF, anomalous signals did not help much because ARP/wARP overcame the model errors (for example, the N-terminal helix—Figure 3c) through automated model building. In cases where the model is not accurate enough or the diffraction data are not of sufficient resolution, MR-SAD may help to solve structures that are otherwise very challenging or even currently considered unsolvable. Most proteins contain intrinsic sulfur atoms that are native anomalous scatterers of long-wavelength X-rays. Thus, to optimize the use of AlphaFold-predicted structures for phasing a de novo structure, it might be advantageous to collect long-wavelength native-SAD data, preferably using a helium flight path if available. That would enable the anomalous signals from sulfur atoms to be used for AlphaFold-based phasing using MR-SAD.
5. Conclusions
Using the AlphaFold-predicted E. coli structure database, we identified the proteins and determined structures for two crystallization contaminants without protein sequence information. The molecular replacement solutions and the structural comparison of refined structures with those AlphaFold-predicted structures suggest that the predicted structures are of sufficiently high accuracy to enable crystallographic phasing and will likely be integrated into other structure determination pipelines.
Author Contributions
Conceptualization, Q.L.; formal analysis, L.C, P.Z., S.M. and Q.L.; investigation, P.Z., J.C., C.P. and B.A.; writing of original draft preparation, Q.L.; writing of review and editing, S.M., J.S. and Q.L.; visualization, Q.L.; supervision, Q.L. and J.S.; project administration, Q.L.; L.C. and P.Z. contributed equally to this article. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by Brookhaven National Laboratory LDRD 22-008 and NIH grant GM107462. P.Z. and Q.L. were supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, as part of the Quantitative Plant Science Initiative at BNL. J.C. and J.S. were supported by Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, United States Department of Energy Grant DOE KC0304000.
Institutional Review Board Statement
Not applicable. Our study did not involve humans or animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
Atomic coordinates and structure factor files were deposited in the RCSB Protein Data Bank (PDB) under the accession codes 7SEU (YncE) and 7SEV (YadF).
Acknowledgments
The diffraction data were collected at FMX beamline as part of the Center for BioMolecular Structure (CBMS), which is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893) and by the DOE Office of Biological and Environmental Research (KP1607011). NSLS-II is supported in part by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Program under contract number DE-SC0012704 (KC0401040).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, Q.; Hendrickson, W.A. Crystallographic phasing from weak anomalous signals. Curr. Opin. Struc. Biol. 2015, 34, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, W.A. Anomalous diffraction in crystallographic phase evaluation. Q. Rev. Biophys. 2014, 47, 49–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.; Mccoy, A. An introduction to molecular replacement. Acta Crystallogr. Sect. D Struct. Biol. 2008, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G. The Molecular Replacement Method. Acta Cryst. A 1990, 46, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Cryst. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Mccoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Navaza, J. Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 1367–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, R.M.; McNicholas, S.J.; Thomas, J.M.H.; Simpkin, A.J.; Simkovic, F.; Uski, V.; Ballard, C.C.; Winn, M.D.; Wilson, K.S.; Rigden, D.J. Recent developments in MrBUMP: Better search-model preparation, graphical interaction with search models, and solution improvement and assessment. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. BALBES: A molecular-replacement pipeline. Acta Crystallogr. Sect. D Struct. Biol. 2008, 64, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunkoczi, G.; Echols, N.; McCoy, A.J.; Oeffner, R.D.; Adams, P.D.; Read, R.J. Phaser.MRage: Automated molecular replacement. Acta Crystallogr. Sect. D Struct. Biol. 2013, 69, 2276–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Baker, D. Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef] [Green Version]
- Bibby, J.; Keegan, R.M.; Mayans, O.; Winn, M.D.; Rigden, D.J. AMPLE: A cluster-and-truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1622–1631, Erratum in 2014, 70, 1174–1174. [Google Scholar] [CrossRef] [Green Version]
- Hungler, A.; Momin, A.; Diederichs, K.; Arold, S.T. ContaMiner and ContaBase: A webserver and database for early identification of unwantedly crystallized protein contaminants. J. Appl. Crystallogr. 2016, 49, 2252–2258. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.; Lebedev, A. MoRDa, an automatic molecular replacement pipeline. Acta Crystallogr. A 2015, 71, S19. [Google Scholar] [CrossRef] [Green Version]
- Simpkin, A.J.; Simkovic, F.; Thomas, J.M.H.; Savko, M.; Lebedev, A.; Uski, V.; Ballard, C.; Wojdyr, M.; Wu, R.; Sanishvili, R.; et al. SIMBAD: A sequence-independent molecular-replacement pipeline. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Bouatta, N.; Sorger, P.; AlQuraishi, M. Protein structure prediction by AlphaFold2: Are attention and symmetries all you need? Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Schneider, D.K.; Shi, W.; Andi, B.; Jakoncic, J.; Gao, Y.; Bhogadi, D.K.; Myers, S.F.; Martins, B.; Skinner, J.M.; Aishima, J. FMX–the Frontier Microfocusing Macromolecular Crystallography Beamline at the National Synchrotron Light Source II. J. Synchrotron. Radiat. 2021, 28, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Waterman, D.G.; Winter, G.; Gildea, R.J.; Parkhurst, J.M.; Brewster, A.S.; Sauter, N.K.; Evans, G. Diffraction-geometry refinement in the DIALS framework. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 558–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.; Axford, D.; Waterman, D.; Owen, R.L. Macromolecular microcrystallography. Crystallogr. Rev. 2011, 17, 105–142. [Google Scholar] [CrossRef]
- Takemaru, L.; Guo, G.R.; Zhu, P.; Hendrickson, W.A.; McSweeney, S.; Liu, Q. PyMDA: Microcrystal data assembly using Python. J. Appl. Crystallogr. 2020, 53, 277–281. [Google Scholar] [CrossRef]
- Guo, G.R.; Fuchs, M.R.; Shi, W.X.; Skinner, J.; Rerman, E.; Ogata, C.M.; Hendrickson, W.A.; McSweeney, S.; Liu, Q. Sample manipulation and data assembly for robust microcrystal synchrotron crystallography. IUCrJ 2018, 5, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.; Teplyakov, A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. Sect. D Struct. Biol. 2000, 56, 1622–1624. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Echols, N.; Morshed, N.; Afonine, P.V.; McCoy, A.J.; Miller, M.D.; Read, R.J.; Richardson, J.S.; Terwilliger, T.C.; Adams, P.D. Automated identification of elemental ions in macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1104–1114. [Google Scholar] [CrossRef]
- Liu, Q.; Hendrickson, W.A. Robust structural analysis of native biological macromolecules from multi-crystal anomalous diffraction data. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1314–1332. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagawa, W.; Sagawa, T.; Niki, H.; Kurumizaka, H. Structural basis for the DNA-binding activity of the bacterial beta-propeller protein YncE. Acta Crystallogr. Sect. D Struct. Biol. 2011, 67, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Yu, X.H.; Wang, C.; Zhang, Q.F.; Liu, W.; McSweeney, S.; Shanklin, J.; Lam, E.; Liu, Q. Structural basis for Ca2+-dependent activation of a plant metacaspase. Nat. Commun. 2020, 11, 2249. [Google Scholar] [CrossRef] [PubMed]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Cronk, J.D.; Endrizzi, J.A.; Cronk, M.R.; O’neill, J.W.; Zhang, K.Y. Crystal structure of E. coli β–carbonic anhydrase, an enzyme with an unusual pH–dependent activity. Protein Sci. 2001, 10, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Cronk, J.D.; Rowlett, R.S.; Zhang, K.Y.; Tu, C.; Endrizzi, J.A.; Lee, J.; Gareiss, P.C.; Preiss, J.R. Identification of a novel noncatalytic bicarbonate binding site in eubacterial β-carbonic anhydrase. Biochemistry 2006, 45, 4351–4361. [Google Scholar] [CrossRef] [PubMed]
- Niedzialkowska, E.; Gasiorowska, O.; Handing, K.B.; Majorek, K.A.; Porebski, P.J.; Shabalin, I.G.; Zasadzinska, E.; Cymborowski, M.; Minor, W. Protein purification and crystallization artifacts: The tale usually not told. Protein Sci. 2016, 25, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Panjikar, S.; Parthasarathy, V.; Lamzin, V.S.; Weiss, M.S.; Tucker, P.A. On the combination of molecular replacement and single-wavelength anomalous diffraction phasing for automated structure determination. Acta Crystallogr. Sect. D Struct. Biol. 2009, 65, 1089–1097. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).