Abstract
We performed, for first time, ab initio calculations for the ReO2-terminated ReO3 (001) surface and analyzed systematic trends in the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces using first-principles calculations. According to the ab initio calculation results, all ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surface upper-layer atoms relax inwards towards the crystal bulk, all second-layer atoms relax upwards and all third-layer atoms, again, relax inwards. The ReO2-terminated ReO3 and ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surface band gaps at the Γ–Γ point are always reduced in comparison to their bulk band gap values. The Zr–O chemical bond populations in the SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskite bulk are always smaller than those near the ZrO2-terminated (001) surfaces. In contrast, the Re–O chemical bond population in the ReO3 bulk (0.212e) is larger than that near the ReO2-terminated ReO3 (001) surface (0.170e). Nevertheless, the Re–O chemical bond population between the Re atom located on the ReO2-terminated ReO3 (001) surface upper layer and the O atom located on the ReO2-terminated ReO3 (001) surface second layer (0.262e) is the largest.
1. Introduction
Forefront (001) surfaces as well as (001) interface phenomena—which occur in the ABO3 perovskite oxides and ReO3—are hot topics in modern solid state physics due to their desirable atomic and electronic processes [1,2,3,4,5,6,7,8,9]. During the last quarter century, due to their great technological importance, as well as a comprehensive fundamental interest, the SrZrO3, BaZrO3, CaZrO3 and PbZrO3 (001) surfaces have been extensively investigated both theoretically and experimentally [10,11,12,13,14,15,16,17,18,19,20,21,22]. SrZrO3, BaZrO3, PbZrO3 and CaZrO3 matrices are so-called ABO3 perovskites, where A = Sr; Ba; Pb; or Ca and B = Zr. ABO3 perovskites have a large number of industrially important applications, for example, as actuators, capacitors and charge storage devices, etc. [23,24,25,26,27]. For many of those ABO3 perovskite applications, surface quality and structure play important roles. For example, recent studies have shown that the catalytic properties of ABO3 perovskite oxides are largely related to oxygen vacancies, which alter their electronic and crystal structures as well as surface chemistry [28,29,30,31,32].
Rhenium trioxide, ReO3, is often referred to as a covalent metal since it has very high electrical conductivity [33]. The electrical conductivity of ReO3 is similar to that of silver or copper [33]. Despite the great technological interest, there have been very few ab initio calculations and experimental studies performed on ReO3 polar (001) surfaces [34,35,36,37]. It is worth noting that there have been no ab initio studies performed, to the best of our knowledge, on the atomic relaxation of the ReO2-terminated polar ReO3 (001) surface. ReO3 related materials, such as LiReO3 and Li2ReO3, are prospective battery cathode materials [38]. The predictive power of first-principle calculations allows for the theoretical design of new materials for advanced technology applications. An excellent example is the theoretical prediction of the average voltages for a four-volt battery cathodes from first-principles calculations by Ceder and his coworkers [39,40]. Moreover, recently, based on first-principles calculations, it was shown that a five-volt battery was possible using Li2CoMn3O8 as the cathode material [41,42].
In the classical cubic unit cells of SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskites, which contain five atoms, the A type atom is located at the cube-corner position with the coordinates (0, 0, 0). The B type atom is located at the body-center position with the coordinates (½, ½, ½). Finally, the three O atoms are located at face-centered positions equal to (½, ½, 0), (½, 0, ½) and (0, ½, ½). The ABO3 perovskite’s A atom is always considerably larger than its B atom. All cubic ABO3 perovskites belong to the Pm3m space group, for which the space group number is 221. ReO3 forms the crystallizes in the cubic ABO3 perovskite structure—Pm3m space group and space group number 221—with the only difference being the unoccupied A-cation site [37].
The goal of the work reported in this paper is to perform, for the first time, ab initio calculations for polar ReO2-terminated ReO3 (001) surfaces. The ab initio calculation results for the polar ReO2-terminated ReO3 (001) surface were compared with the calculation results for neutral ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces. The calculation results for all five materials were carefully analyzed and systematic trends common for all ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces were elucidated and are reported herein.
2. Computational Method
We performed ab initio calculations for the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk and ReO2 or ZrO2-terminated (001) surfaces, respectively, using the hybrid exchange–correlation functionals B3PW [43] or B3LYP [44] as well as the widely recognized CRYSTAL computer code [45]. The SrTiO3 [46], CaF2 [47] and MgF2 [48] bulk Γ–Γ band gaps that were calculated using different exchange–correlation functionals are provided in Table 1. The experimentally measured SrTiO3 [49], CaF2 [50] and MgF2 [51,52] bulk band gaps at the Γ-point are listed in Table 1 for the purpose of comparison. It is well known that the local-density approximations (LDA) and generalized-gradient approximations (GGA) used in density functional theory (DFT) systematically underestimate the band gap in complex oxide materials, such as ABO3 perovskites and insulators by a factor of almost two (Table 1). In contrast, it is well known that the Hartree–Fock (HF) method systematically overestimates the band gap of solids. With the aim of generating a reliable basis for further ABO3 perovskites and ReO3 bulk and (001) surface calculations, which require a precise description of the Γ–Γ band gap, we performed the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk and (001) surface calculations by means of the hybrid exchange–correlation functionals B3PW or B3LYP, which utilize 20% of the HF method and 80% of the DFT Hamiltonian method when implemented in the CRYSTAL computer package [45].

Table 1.
SrTiO3, CaF2 and MgF2 bulk Γ–Γ band gaps calculated using different exchange–correlation functionals. Experimental bulk band gap data at the Γ-point are listed for comparison.
For example, as can be seen from Table 1, the ab initio HF calculations strongly overestimate the experimental SrTiO3 bulk band gap at the Γ-point—by 3.29 times. In contrast, the DFT-based PWGGA and PBE exchange–correlation functionals considerably underestimate the experimental SrTiO3 band gap at the Γ-point—by 1.62 and 1.60 times, respectively. Finally, the B3PW and B3LYP hybrid-exchange correlation functionals only slightly overestimate the experimental SrTiO3 band gap at the Γ-point—by 1.06 and 1.04 times, respectively. For predominantly this reason, the B3PW and B3LYP hybrid exchange–correlation functionals were used in all subsequent ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk and (001) surface ab initio calculations performed by means of the CRYSTAL computer code [45].
The key power of the CRYSTAL computer code, which is important for the study of neutral SrZrO3, BaZrO3, PbZrO3 and CaZrO3 as well as polar ReO3 (001) surfaces, is its use of the 2D isolated slab model, without artificial repetition along the z-axis. The reciprocal space integration, in the ab initio calculations, were performed by sampling the Brillouin zone with an 8 × 8 × 1 times extended Pack–Monkhorst mesh for the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces and 8 × 8 × 8 mesh for the bulk of those materials. In order to achieve highly accurate calculations, large enough tolerances of 7, 8, 7, 7 and 14 were chosen for the Coulomb overlap, Coulomb penetration, exchange overlap, first-exchange pseudo-overlap and second-exchange pseudo-overlap, respectively [45].
In order to calculate the neutral BO2-terminated ABO3 perovskite (001) surfaces (Figure 1), we used symmetrical slabs consisting of nine, neutral, alternating BO2 or AO layers perpendicular to the [001] crystal direction [11,14,16,41]. The slabs were rotated to make them perpendicular to the Oz axis. The CRYSTAL computer package [45] made it possible to avoid artificial periodicity along the Oz direction and to perform calculations for stand-alone 2D slabs. Taking into account the classical ionic charges for A(+2e), for B(+4e) and for O(−2e), both the AO and BO2 layers have a formal ionic charge equal to zero. The nine-layer slab, used in ABO3 perovskite (001) surface calculations, was terminated on both sides by BO2 planes and thereby consisted of a 23-atom supercell (Figure 1). The calculated BO2-terminated ABO3 perovskite (001) slabs were non-stoichiometric, with an empirical unit cell of A4B5O14 [11,14,16,41].
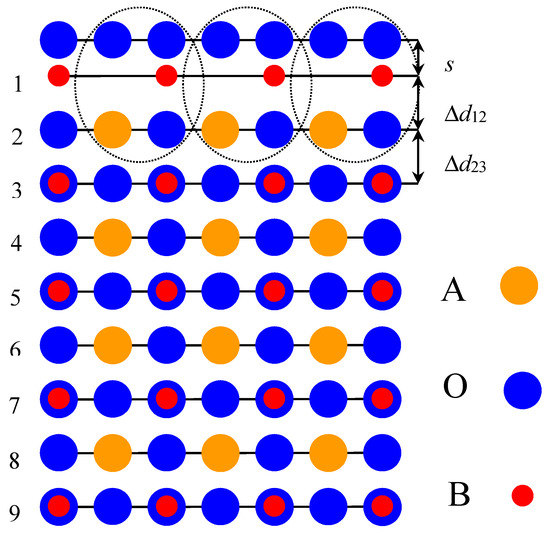
Figure 1.
Side-view of the nine-layer BO2-terminated ABO3 perovskite (001) surface.
In contrast to neutral ABO3 perovskite (001) surfaces, it is much more difficult to calculate polar, ReO2-terminated ReO3 (001) surfaces, which consist of charged ReO2 and O layers, taking into account the classical ionic charges of Re(+6e) and O(−2e) (Figure 2). Furthermore, for the ReO2-terminated polar ReO3 (001) surface calculations, we used symmetrical nine-layer slabs, which consisted of polar, alternating ReO2 and O layers and contained 19 atoms, with an empirical unit cell of B5O14. The nine-layer ReO2-terminated ReO3 surface, taking into account formal ionic charges (ReO2(+2e)–O(−2e)–ReO2(+2e)–O(−2e)–ReO2(+2e)–O(−2e)–ReO2(+2e)–O(−2e)–ReO2(+2e)), has a positive charge equal to +2e. In all calculations performed by CRYSTAL computer code, the unit cell should be neutral. In order to make the calculations feasible, for polar ReO2-terminated ReO3 (001) surfaces, instead of ionic basis sets—as in case of ABO3 perovskites—the basis sets for neutral Re and O atoms were used. For example, for the O atom, the basis sets developed by Piskunov et al. [46] were used, and two electrons were removed from the O2− ion to obtain the basis set for the neutral O atom [5,53,54]. For the Re atom, we used the basis set developed by Cora [45]. Using the atomic basis sets for Re and O atoms, we obtained the polar ReO2-terminated ReO3 (001) surface with a formal charge equal to 0, and thereby, such calculations are feasible with the CRYSTAL computer code. As we know from previous studies, on, for example, with polar CaTiO3 and SrTiO3 (111) surfaces [55,56,57,58,59], a very strong electron redistribution is observed, which deletes the polarity effects. It is evident that it is impossible to calculate the asymmetric slabs with different terminations, such as ReO2–O–ReO2––O–ReO2–O–ReO2–O, since, in the case for the asymmetric slab, it has a large dipole moment perpendicular to the ReO2-terminated ReO3 crystal (001) surface.
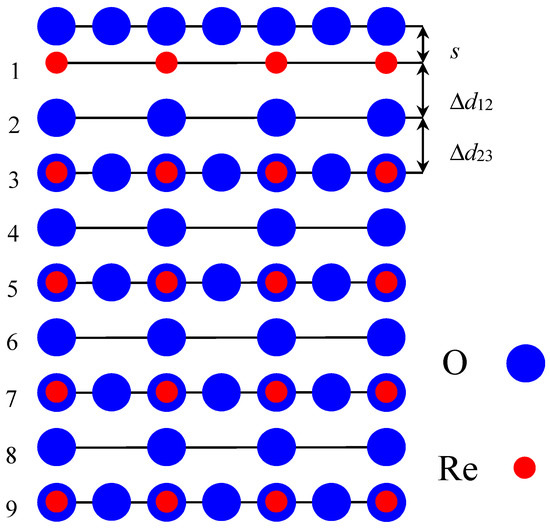
Figure 2.
Side-view of the nine-layer ReO2-terminated ReO3 polar (001) surface.
To correctly describe the chemical bonding as well as covalency effects for both ReO3, SrZrO3, BaZrO3, PbZrO3, CaZrO3 bulk and their (001) surfaces, we used a standard Mulliken population analysis as it is implemented in the CRYSTAL computer code [45]. Namely, the Mulliken population analysis was used for the chemical bond populations P, effective atomic charges q, as well as another local properties of the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 electronic structure, such as the bond orders, atomic covalences and full valences [60,61,62].
3. Numeric Results of ReO3, SrZrO3, BaZrO3, PbZrO3, CaZrO3 Bulk and (001) Surface Calculations
3.1. Ab Initio Calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 Bulk Properties
In order to begin the calculations, by means of the B3LYP or B3PW functional, the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk lattice constants were calculated and compared with actual experimental data (Table 2). As shown in Table 2, the B3LYP calculated ReO3 bulk lattice constant (3.758 Å) is only overestimated by 0.29% with respect to the experimental value of 3.747 Å [63]. The, by means of hybrid exchange–correlation functionals, calculated SrZrO3, BaZrO3 and PbZrO3 bulk lattice constants are overestimated with respect to the experimentally measured bulk lattice constants by 0.99%, 0.83% and 1.41%, respectively [64,65,66]. The theoretical ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 [67] bulk lattice constants were used in all subsequent (001) surface calculations.
Table 2.
B3LYP or B3PW calculated ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk lattice constants (in Å). The experimental bulk lattice constants are listed for the purpose of comparison.
The calculated effective charge is +2.382e for the Re atom in the ReO3 bulk matrix (Table 3). The calculated Zr effective charges in the SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskites (+2.174e, +2.134e, +2.111e and +2.144e, respectively) are similar to each other and strongly different from the Zr formal ionic charge (+4e). The calculated O effective charge in the ReO3 bulk is equal to −0.794e. Ab initio calculated O effective charges in the SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskites are equal to −1.351e, −1.316e, −1.160e and −1.310e, respectively. Therefore, the SrZrO3, BaZrO3 and CaZrO3 O effective charges are similar, but the O effective charge in the PbZrO3 crystal is considerably smaller, only −1.160e (Table 3). The chemical bond population between Re and O atoms in ReO3 is equal to +0.212e. The chemical bond population between Zr and O atoms in SrZrO3, BaZrO3, PbZrO3 and CaZrO3 matrices are equal to +0.092e, 0.108e, 0.106e, 0.086e, respectively. Large chemical bond population values between B and O atoms in ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 crystals indicate that the chemical bonding in these materials is covalent.
Table 3.
By means of the hybrid exchange–correlation functionals B3LYP or B3PW calculated effective atomic charges Q and bond populations P in ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3.
By means of the B3LYP or B3PW hybrid exchange–correlation functionals, the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk band gaps at the Γ–Γ point were calculated for the cubic phase of these crystals. It is worth mentioning that the hybrid exchange–correlation functionals, such as B3LYP or B3PW are in excellent agreement with the experimentally obtained band gaps of related ABO3 perovskites and their (001) surfaces [5,16,47,48,68], whereas the density functional theory, consistently underestimates the band gap of complex oxide materials. From another side, it is well known that the Hartree–Fock method considerably overestimates the band gap of complex oxide materials. The B3LYP-calculated bulk band gap for ReO3 at the Γ-point is equal to 5.76 eV (Table 4). To the best of our knowledge, there are no reported experimental data for the ReO3 bulk band gap at the Γ-point. The calculated optical band gap for BaZrO3 at the Γ-point (4.93 eV) is only underestimated by 6.98% regarding the experimental value of 5.3 eV [69]. The ab initio calculated optical band gaps at the Γ-point for SrZrO3, PbZrO3 and CaZrO3 perovskite cubic phases are 5.31, 5.63 and 5.40 eV, respectively. Unfortunately, it is not possible to compare the ab initio calculation results for the band gaps at the Γ-point for SrZrO3, PbZrO3 and CaZrO3 perovskites with experimental results, since there are, currently, no reports of the band gaps of SrZrO3, PbZrO3 and CaZrO3 perovskite cubic phases in the literature.
Table 4.
B3LYP or B3PW calculated ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk band gaps at the Γ–Γ point for the cubic phase. The ab initio calculation results are compared with the available experimental data.
3.2. Ab Initio Calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) Surfaces
B3LYP or B3PW ab initio calculations for the upper three-layer atom relaxation for the neutral ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 as well as polar ReO2-terminated ReO3 (001) surfaces (Table 5) were performed. It is worth noting that the ReO3 material has the cubic ABO3 perovskite structure and symmetry with the space group number 221, but with the only difference being the A atom vacancy (Figure 2). For the cases of SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskite ZrO2-terminated as well as ReO3 crystal ReO2-terminated (001) surfaces, according to the ab initio calculations, all upper-layer atoms relax towards the bulk (Table 5). The ReO2-terminated ReO3 (001) surface upper-layer Re atom displacement magnitude (3.19% of a0) is slightly larger than the ab initio calculated ABO3 perovskite ZrO2-terminated (001) surface Zr atom relaxation magnitudes, which are in the range of 1.30% of a0 for the CaZrO3 to 2.37% of a0 for the PbZrO3 perovskite (Table 5). In contrast, all SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskite second-layer ZrO2-terminated (001) surface atoms relax in the outward direction. The only exception to this systematic trend is the second-layer ReO2-terminated ReO3 (001) surface O atom inward relaxation towards the bulk; however, this has a small relaxation magnitude, equal to −0.32% of a0. All the ab initio calculated third-layer atoms for the ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 as well as ReO2-terminated ReO3 (001) surfaces, again, as in the case of the upper-layer atoms, relax inwards, towards the crystal bulk (Table 5). Nevertheless, the relaxation magnitudes of all first-layer atoms for the ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskite as well as ReO2-terminated ReO3 (001) surfaces are much larger than the relevant relaxation magnitudes of the respective third-layer atoms (Table 5).
Table 5.
ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 upper three-layer atom relaxation (in percent of the crystal bulk lattice constant) for the BO2-terminated (001) surfaces calculated by the B3LYP exchange–correlation functional for ReO3, SrZrO3, PbZrO3 and CaZrO3 perovskites as well as by the B3PW method for BaZrO3.
To compare ab initio calculation results for ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces with the available experimental results, the calculated surface rumplings s (the relative displacement of oxygen with respect to the metal in the upper surface layer) as well as the changes in interlayer distances, Δd12 and Δd23, are shown in Table 6. The calculations of the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surface interlayer distances rely on the positions of the metal ions (Figure 1), which are well known to be much stronger electron scatterers than oxygen ions [70]. As can be seen from Table 6, all the calculated ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces show the reduction of the interlayer distance Δd12 and expansion of Δd23. For all ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces, the reduction in the interlayer distance, Δd12, is larger than the expansion of the respective interlayer distance, Δd23. The ab initio calculated surface rumpling, s, is positive and largest between all calculated surface rumplings (+2.02) for the ReO2-terminated ReO3 (001) surface. The calculated surface rumplings, s, for the ZrO2-terminated BaZrO3 and PbZrO3 (001) surfaces (+0.09 and +0.38, respectively) are also positive, but much smaller than for the ReO2-terminated ReO3 (001) surface (+2.02). In contrast, the calculated surface rumplings, s, for the ZrO2-terminated SrZrO3 and CaZrO3 (001) surfaces are negative (−0.72 and −1.01, respectively).
Table 6.
B3PW and B3LYP calculated surface rumplings, s, as well as relative displacements, Δdij, between the 3 near-surface planes for the BO2-terminated ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces as a percent of the bulk crystal lattice constant.
Unfortunately, to the best of our knowledge, there are no experimental data available for the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surface rumpling, s, as well as interlayer distances, Δd12 and Δd23. However, such experimental data exist for the related ABO3 perovskite, SrTiO3 (Table 7). To compare the calculated and experimental SrTiO3 (001) surface structures, the calculated surface rumpling, s, as well as the changes in interlayer distances, Δdij, are detailed in Table 7. From Table 7, it can be seen that the agreement is fairly good for all theoretical calculation methods, which all give the same sign for the surface rumpling, s, as well as the changes in the interlayer distances, Δdij. For example, the calculated surface rumpling, s, for the SrO-terminated surface is much larger than for the TiO2-terminated SrTiO3 (001) surface for all theoretical methods [25,71,72,73,74,75]. From Table 7, it can be seen that both the calculated SrO and TiO2-terminated SrTiO3 (001) surfaces always exhibit a reduction in the interlayer distance, Δd12 and an expansion of Δd23. The theoretically calculated surface rumpling amplitudes, s, for both SrTiO3 (001) surface terminations are in fair agreement with the LEED [70], RHEED [76], MEIS [77] and SXRD [78] experiments (Table 7). Nevertheless, the calculated changes in interlayer distances disagree with the LEED experiments [70] for the TiO2-terminated SrTiO3 (001) surface, which show an increase in Δd12 and reduction in Δd23 (Table 7). In contrast, all ab initio as well as classical shell model calculations show a reduction in the interlayer distance, Δd12 and an expansion of Δd23 (Table 7). Nevertheless, as can be seen from Table 7, unfortunately, the different experiments contradict each other with respect to the sign of Δd12 and Δd23 for the SrO-terminated SrTiO3 (001) surface and for the sign of Δd23 for the TiO2-terminated SrTiO3 (001) surface (Table 7).
Table 7.
Calculated and experimental surface rumpling s and relative displacements Δdij (in percent of the bulk lattice constant) for the upper-three surface layers of SrO and TiO2-terminated SrTiO3 (001) slabs.
The ab initio calculated atomic displacements, the Mulliken static charges as well as bond populations between nearest atoms are reported in Table 8. The most important effect, as can be seen from Table 8, is strengthening of the Zr–O chemical bond near the ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surface in comparison to the bulk [79,80,81,82]. In contrast, for the ReO2-terminated ReO3 (001) surface, the chemical bond population between the Re and O atoms in the upper surface layer 0.170e (Table 8) is slightly smaller than the Re–O chemical bond population in the ReO3 bulk (0.212e). Nevertheless, the chemical bond population between the upper-layer Re atom and the second-layer O atom (0.262e) for the ReO2-terminated ReO3 (001) surface is considerably larger than the Re–O chemical bond population in the ReO3 crystal bulk (0.212e). It is worth noticing, that the Re and O effective charges in the ReO3 crystal bulk (+2.382e for Re and −0.794e for O) are much smaller than those expected in the ionic model (+6e for Re and −2e for O). Moreover, the Re–O chemical bond in the ReO3 bulk is considerably populated (+0.212e). It is interesting to note that the Re–O chemical bond population for the ReO2-terminated ReO3 (001) surface third layer (+0.208e) (Table 8) is already highly similar to the Re–O chemical bond population in the ReO3 bulk matrix (0.212e). The Re effective charge in the ReO2-terminated ReO3 (001) surface third layer (+2.341e) is almost as high as the Re effective charge value (+2.382e) in the ReO3 bulk crystal. In contrast, the Re effective charge on the ReO2-terminated ReO3 (001) surface upper layer, where the surface effect is strong, (+2.258e) is much smaller than the Re effective charge in the ReO3 crystal bulk (+2.382e).
Table 8.
Ab initio calculated absolute magnitudes of atomic shifts D (in Å), the effective atomic charges Q (in e) and nearest atom Me–O bond populations P (in e) for the ReO2 and ZrO2-terminated ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces.
As can be seen from the ab initio calculation results, detailed in Table 9, the Zr–O chemical bond populations for all four calculated perovskites SrZrO3, BaZrO3, PbZrO3 and CaZrO3 are larger near their ZrO2-terminated (001) surfaces than in the bulk. However, the opposite is true for the ReO3 crystal. The Re–O chemical bond population in the ReO3 bulk (0.212e) is larger than it is near the ReO2-terminated ReO3 (001) surface (0.170e). However, it is worth noting that the Re–O chemical bond population between the ReO2-terminated (001) surface upper-layer Re atom and the second-layer O atom (0.262e) is considerably larger than the Re–O chemical bond population in the ReO3 crystal bulk (0.212e).
Table 9.
Ab initio calculated B–O chemical bond populations for ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk as well as for BO2-terminated (001) surfaces (in e).
The, by means of the hybrid exchange–correlation functionals, calculated bulk band gaps at the Γ–Γ point for ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 crystals are equal to 5.76, 5.31, 4.93, 5.63 and 5.40 eV, respectively (Table 10). In most cases, there are no experimental data available for the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk band gaps in the cubic phase. However, the calculated BaZrO3 band gap at the Γ–Γ point (4.93 eV) is in fair agreement with the experimental data (5.3 eV) [69]. According to the performed ab initio calculations, the systematic trend is reduction of the ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk band gaps near their ReO2 or ZrO2-terminated (001) surfaces, respectively. Namely, the calculated band gap values at the Γ–Γ point for ReO2-terminated ReO3 and ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 terminated (001) surfaces of 0.22, 4.91, 4.48, 4.60 and 5.22 eV, respectively, were always smaller with respect to the bulk band gap value (Table 10).
Table 10.
Ab initio calculated optical band gaps at the Γ–Γ point for ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 bulk as well as their ReO2 or ZrO2-terminated (001) surfaces.
4. Conclusions
For the ab initio calculated ReO2-terminated ReO3 as well as ZrO2-terminated SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces, the systematic trend was that all upper-layer surface atoms relaxed inwards, towards the bulk, all second-layer surface atoms relaxed upwards, and again, all third-layer surface atoms relaxed inwards. As a result of the performed relaxation, all five material surfaces exhibited a reduction in the interlayer distance, Δd12 and expansion of Δd23.
For all the ab initio calculated materials, the changes in the interlayer distances between the first and second layer were larger than the respective changes in the interlayer distances between the second and third layer.
According to the performed ab initio calculations, the SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskite BO2-terminated as well as ReO2-terminated ReO3 (001) surface band gaps were always smaller with respect to their bulk band gap values.
The Zr–O chemical bond population in SrZrO3, BaZrO3, PbZrO3 and CaZrO3 perovskite bulk was always smaller than that near the ZrO2-terminated (001) surface. In contrast, the Re–O chemical bond population in the ReO3 bulk (0.212e) was larger than that near the ReO2-terminated ReO3 (001) surface (0.170e). The Re–O chemical bond population between the Re atom located on the ReO2-terminated ReO3 (001) surface upper layer as well as the O atom located on the ReO2-terminated ReO3 (001) surface second layer was the largest (0.262e).
Author Contributions
All authors equally contributed to the performed ab initio calculations as well as to the preparation of the manuscript. Namely, mostly R.I.E. and A.I.P. wrote the Introduction. Mostly J.P., J.G. and R.J. wrote the section Computational Method. All authors equally wrote the sections Numeric Results and Conclusions. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Latvian Government ERAF Grant number 1.1.1.1/18/A/073.
Acknowledgments
We greatly acknowledge the financial support via the ERAF Project No. 1.1.1.1/18/A/073.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Mastrikov, Y.A.; Merkle, R.; Kotomin, E.A.; Kuklja, M.M.; Maier, J. Surface termination effects on the oxygen reduction reaction rate at fuel cell cathodes. J. Mater. Chem. A 1998, 6, 11929–11940. [Google Scholar] [CrossRef]
- Enterkin, J.A.; Subramanian, A.K.; Russell, B.C.; Castell, M.R.; Poeppelmeier, K.R.; Marks, L.D. A homologous series of structures on the surface of SrTiO3 (110). Nat. Mater. 2010, 9, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Piskunov, S.; Eglitis, R.I. First principles hybrid DFT calculations of BaTiO3/SrTiO3 (001) interface. Solid State Ion. 2015, 274, 29–33. [Google Scholar] [CrossRef]
- Saeed, S.W.; Norby, T.; Bjornheim, T.S. Charge-Carrier enrichment at BaZrO3/SrTiO3 interfaces. J. Phys. Chem. C 2019, 123, 20808–20816. [Google Scholar] [CrossRef]
- Eglitis, R.I. Comparative ab initio calculations of SrTiO3 and CaTiO3 polar (111) surfaces. Phys. Status Solidi B 2015, 252, 635–642. [Google Scholar] [CrossRef]
- Zhao, X.H.; Selloni, A. Structure and stability of NaTaO3 (001) and KTaO3 (001) surfaces. Phys. Rev. Mater. 2019, 3, 015801. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Kleperis, J.; Purans, J.; Popov, A.I.; Jia, R. Ab initio calculations of CaZrO3 (011) surfaces: Systematic trends in polar (011) surface calculations of ABO3 perovskites. J. Mater. Sci. 2020, 55, 203–217. [Google Scholar] [CrossRef]
- Watanabe, Y. Ferroelectricity of stress-free and strained pure SrTiO3 revealed by ab initio calculations with hybrid and density functionals. Phys. Rev. B 2019, 99, 064107. [Google Scholar] [CrossRef]
- Piskunov, S.; Eglitis, R.I. Comparative ab initio calculations of SrTiO3/BaTiO3 and SrZrO3/PbZrO3 (001) heterostructures. Nucl. Instrum. Methods B 2016, 374, 20–23. [Google Scholar] [CrossRef]
- Meng, J.; Lan, Z.Y.; Lin, Q.Y.; Chen, T.; Chen, X.; Wei, X.; Lu, Y.H.; Li, J.X.; Zhang, Z. Cubic-like BaZrO3 nanocrystals with exposed {001}/{011} facets and tuned electronic band structure for enhanced photocatalytic hydrogen production. J. Mater. Sci. 2019, 54, 1967–1976. [Google Scholar] [CrossRef]
- Eglitis, R.I. First-principles calculations of BaZrO3 (001) and (011) surfaces. J. Phys. Condens. Matter 2007, 19, 356004. [Google Scholar] [CrossRef]
- Sambrano, J.R.; Longo, V.M.; Longo, E.; Taft, C.A. Electronic and structural properties of the (001) SrZrO3 surface. J. Mol. Struct. THEOCHEM 2007, 813, 49–56. [Google Scholar] [CrossRef]
- Kim, J.S.; Yang, J.H.; Kim, B.K.; Kim, Y.C. Proton conduction at BaO-terminated (001) BaZrO3 surface using density functional theory. Solid State Ion. 2015, 275, 19–22. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Rohlfing, M. First-principles calculations of the atomic and electronic structure of SrZrO3 and PbZrO3 (001) and (011) surfaces. J. Phys. Condens. Matter 2010, 22, 415901. [Google Scholar] [CrossRef] [PubMed]
- Brik, M.G.; Ma, C.G.; Krasnenko, V. First-principles calculations of the structural and electronic properties of the cubic CaZrO3 (001) surfaces. Surf. Sci. 2013, 608, 146–153. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I. Systematic trends in (001) surface ab initio calculations of ABO3 perovskites. J. Saudi Chem. Soc. 2018, 22, 459–468. [Google Scholar] [CrossRef]
- Guo, X.; Ge, J.; Ponchel, F.; Remiens, D.; Chen, Y.; Dong, X.; Wang, G. Effect of Sn substitution on the energy storage properties of high (001)-oriented PbZrO3 thin films. Thin Solid Films 2017, 632, 93–96. [Google Scholar] [CrossRef]
- Kotomin, E.A.; Piskunov, S.; Zhukovskii, Y.F.; Eglitis, R.I.; Gopejenko, A.; Ellis, D.E. The electronic properties of an oxygen vacancy at ZrO2-terminated (001) surfaces of a cubic PbZrO3: Computer simulations from the first principles. Phys. Chem. Chem. Phys. 2008, 10, 4258–4263. [Google Scholar] [CrossRef]
- Sung, H.J.; Mochizuki, Y.; Oba, F. Surface reconstruction and band alignment of nonmetallic A(II)B(IV)O3 perovskites. Phys. Rev. Mater. 2020, 4, 044606. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Piskunov, S. First principles calculations of SrZrO3 bulk and ZrO2-terminated (001) surface F centers. Comput. Condens. Matter 2016, 7, 1–6. [Google Scholar] [CrossRef]
- Nguyen, M.D.; Trinh, T.T.; Dang, H.T.; Hung, N.V. Understanding the effects of electric-field-induced phase transition and polarization loop behavior on the energy storage performance of antiferroelectric PbZrO3 thin films. Thin Solid Films 2020, 697, 137794. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Popov, A.I.; Jia, R. Systematic trends in YAlO3, SrTiO3, BaTiO3, BaZrO3 (001) and (111) surface ab initio calculations. Int. J. Mod. Phys. B 2019, 33, 1950390. [Google Scholar] [CrossRef]
- Hwang, H.Y.; Iwasa, Y.; Kawasaki, M.; Keimer, B.; Nagaosa, N.; Tokura, Y. Emergent phenomena at oxide interfaces. Nat. Mater. 2012, 11, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.N.; Machala, M.L.; Bluhm, H.; Chuech, W.C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 2015, 6, 6097. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Vanderbilt, D. First-principles calculations of atomic and electronic structure of SrTiO3 (001) and (011) surfaces. Phys. Rev. B 2008, 77, 195408. [Google Scholar] [CrossRef]
- Nahas, Y.; Akbarzadeh, A.; Prokharenko, S.; Prosandeev, S.; Walter, R.; Kornev, I.; Iniguez, J.; Bellaiche, L. Microscopic origins of the large piezoelectricity of leadfree (Ba,Ca)(Zr,Ti)O3. Nat. Commun. 2017, 8, 15944. [Google Scholar] [CrossRef]
- Jia, W.; Vikhnin, V.S.; Liu, H.; Kapphan, S.; Eglitis, R.; Usvyat, D. Critical effects in optical response due to charge transfer vibronic excitions and their structure in perovskite-like systems. J. Lumin. 1999, 83, 109–113. [Google Scholar] [CrossRef]
- Ji, Q.; Bi, L.; Zhang, J.; Cao, H.; Zhao, X.S. The role of oxygen vacancies of ABO3 perovskite oxides in the oxygen reduction reaction. Energy Environ. Sci. 2020, 13, 1408–1428. [Google Scholar] [CrossRef]
- Wrana, D.; Rodenbucher, C.; Belza, W.; Szot, K.; Krok, F. In situ study of redox processes on the surface of SrTiO3 single crystals. Appl. Surf. Sci. 2018, 432, 46–52. [Google Scholar] [CrossRef]
- Wojtyniak, M.; Balin, K.; Szade, J.; Szot, K. Inhomogeneity and segregation effect in the surface layer of Fe-doped SrTiO3 single crystals. Crystals 2020, 10, 33. [Google Scholar] [CrossRef]
- Rodenbucher, C.; Meuffels, P.; Speier, W.; Ermich, M.; Wrana, D.; Krok, F.; Szot, K. Stability and decomposition of perovskite-type titanates upon high temperature reduction. Phys. Status Solidi Rapid Res. Lett. 2017, 11, 1700222. [Google Scholar] [CrossRef]
- Eglitis, R.; Kruchinin, S.P. Ab initio calculations of ABO3 perovskite (001), (011) and (111) nano-surfaces, interfaces and defects. Mod. Phys. Lett. B 2020, 34, 2040057. [Google Scholar] [CrossRef]
- Evarestov, R.A.; Kalinko, A.; Kuzmin, A.; Losev, M.; Purans, J. First-principles LCAO calculations on 5d transition metal oxides: Electronic and phonon properties. Integr. Ferroelectr. 2009, 108, 1–10. [Google Scholar] [CrossRef]
- Ling, S.; Mei, D.; Gutowski, M. Reactivity of hydrogen and methanol on (001) surfaces of WO3, ReO3, WO3/ReO3 and ReO3/WO3. Catal. Today 2011, 165, 41–48. [Google Scholar] [CrossRef]
- Tsukada, M.; Tsukada, N.; Minami, F. Theory of the electronic structure of ReO3 (001) surface and the surface oxygen vacancy. J. Phys. Soc. Jpn. 1980, 49, 1115–1122. [Google Scholar] [CrossRef]
- Ge, Q.; Gutowski, M. A comparative study of methanol adsorption and dissociation over WO3 (001) and ReO3 (001). Top. Catal. 2015, 58, 655–664. [Google Scholar] [CrossRef]
- Evans, H.A.; Wu, Y.; Seshadri, R.; Cheetham, A.K. Perovskite-related ReO3-type structures. Nat. Rev. Mater. 2020, 5, 196–213. [Google Scholar] [CrossRef]
- Bashian, N.H.; Zhou, S.; Zuba, M.; Ganose, A.M.; Stiles, J.W.; Ee, A.; Ashby, D.S.; Scanlon, D.O.; Piper, L.F.J.; Dunn, B.; et al. Correlated polyhedral rotation in the absence of polarons during electrochemical insertion of lithium in ReO3. ACS Energy Lett. 2018, 3, 2513–2519. [Google Scholar] [CrossRef]
- Ceder, G.; Chiang, Y.M.; Sadoway, D.R.; Aydinol, M.K.; Jang, Y.I.; Huang, B. Identification of cathode materials for lithium batteries guided by first-principles calculations. Nature 1998, 392, 694–696. [Google Scholar] [CrossRef]
- Ceder, G. Predicting properties from scratch. Science 1998, 280, 1099–1100. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Borstel, G. Towards a practical rechargeable 5 V Li ion battery. Phys. Status Solidi A 2005, 202, R13–R15. [Google Scholar] [CrossRef]
- Eglitis, R. Ab initio calculations of Li2(Co,Mn)O8 solid solutions for rechargeable batteries. Int. J. Mod. Phys. B 2019, 33, 1950151. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Saunders, V.R.; Dovesi, R.; Roetti, C.; Causa, N.; Harrison, N.M.; Orlando, R.; Zicovich-Wilson, C.M. CRYSTAL-2009 User Manual; University of Torino: Torino, Italy, 2009. [Google Scholar]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Shi, H.; Eglitis, R.I.; Borstel, G. Ab initio calculations of the CaF2 electronic structure and F centers. Phys. Rev. B 2005, 72, 045109. [Google Scholar] [CrossRef]
- Vassilyeva, A.F.; Eglitis, R.I.; Kotomin, E.A.; Dauletbekova, A.K. Ab initio calculations of MgF2 (001) and (011) surface structure. Phys. B Condens. Matter 2010, 405, 2125–2127. [Google Scholar] [CrossRef]
- van Benthem, K.; Elsasser, C.; French, R.H. Bulk electronic structure of SrTiO3: Experiment and theory. J. Appl. Phys. 2001, 90, 6156–6164. [Google Scholar] [CrossRef]
- Rubloff, G.W. Far-Ultraviolet Reflectance Spectra and the electronic structure of ionic crystals. Phys. Rev. B 1972, 5, 662–684. [Google Scholar] [CrossRef]
- Thomas, J.; Stephan, G.; Lemonnier, J.C.; Nisar, M.; Robin, S. Optical anisotropy of MgF2 in its UV absorption region. Phys. Status Solidi B 1973, 56, 163–170. [Google Scholar] [CrossRef]
- Lisitsyn, V.M.; Lisitsyna, L.A.; Popov, A.I.; Kotomin, E.A.; Abuova, F.U.; Akilbekov, A.; Maier, J. Stabilization of primary mobile radiation defects in MgF2 crystals. Nucl. Instrum. Methods B 2016, 374, 24–28. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio calculations of the atomic and electronic structure of BaZrO3 (111) surfaces. Solid State Ion. 2013, 230, 43–47. [Google Scholar] [CrossRef]
- Eglitis, R.I. Comparative first-principles calculations of SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001), (011) and (111) surfaces. Ferroelectrics 2015, 483, 53–67. [Google Scholar] [CrossRef]
- Liu, W.; Wang, C.; Cui, J.; Man, Z.Y. Ab initio calculations of the CaTiO3 (111) polar surfaces. Solid State Commun. 2009, 149, 1871–1876. [Google Scholar] [CrossRef]
- Pojani, A.; Finocchi, F.; Noguera, C. Polarity of the SrTiO3 (111) and (110) surfaces. Surf. Sci. 1999, 442, 179–198. [Google Scholar] [CrossRef]
- Pojani, A.; Finocchi, F.; Noguera, C. A theoretical study of the unreconstructed polar (111) face of SrTiO3. Appl. Surf. Sci. 1999, 142, 177–181. [Google Scholar] [CrossRef]
- Noguera, C. Polar oxide surfaces. J. Phys. Condens. Matter 2000, 12, R367–R410. [Google Scholar] [CrossRef]
- Tasker, P.W. The stability of ionic crystal surfaces. J. Phys. C Solid State Phys. 1979, 12, 4977–4984. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence: Relations to Mulliken’s population analysis. Int. J. Quantum Chem. 1984, 26, 151–154. [Google Scholar] [CrossRef]
- Bochicchio, R.C.; Reale, H.F. On the nature of crystalline bonding-extension of statistical population analysis to 2-dimensional and 3-dimensional crystalline systems. J. Phys. B 1993, 26, 4871–4883. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio calculations of SrTiO3, BaTiO3, PbTiO3, CaTiO3, SrZrO3, PbZrO3 and BaZrO3 (001), (011) and (111) surfaces as well as F centers, polarons, KTN solid solutions and Nb impurities therein. Int. J. Mod. Phys. B 2014, 28, 1430009. [Google Scholar] [CrossRef]
- Schirber, J.E.; Morosin, B. “Compressibility Collapse” transition in ReO3. Phys. Rev. Lett. 1979, 42, 1485–1487. [Google Scholar] [CrossRef]
- Kennedy, B.J.; Howard, C.J.; Chakoumakos, B.C. High-temperature phase transitions in SrZrO3. Phys. Rev. B 1999, 59, 4023–4027. [Google Scholar] [CrossRef]
- Mathews, M.D.; Mirza, E.B.; Momin, A.C. High-temperature X-ray diffractometric studies of CaZrO3, SrZrO3 and BaZrO3. J. Mater. Sci. Lett. 1991, 10, 305–306. [Google Scholar] [CrossRef]
- Aoyagi, S.; Kuroiwa, Y.; Sawada, A.; Tanaka, H.; Nishibori, E.; Takata, M.; Sakata, M. Direct observation of covalency between O and disordered Pb in cubic PbZrO3. J. Phys. Soc. Jpn. 2002, 71, 2353–2356. [Google Scholar] [CrossRef]
- Eglitis, R.I. Theoretical modelling of the energy surface (001) and topology of CaZrO3 perovskite. Ferroelectrics 2008, 483, 75–85. [Google Scholar] [CrossRef]
- Shi, H.; Chang, L.; Jia, R.; Eglitis, R.I. Ab initio calculations of the charge transfer and aggregation of F centers in CaF2. J. Phys. Chem. C 2012, 116, 4832–4839. [Google Scholar] [CrossRef]
- Robertson, J. Band offsets of wide-band-gap oxides and implications for future electronic devices. J. Vac. Sci. Technol. B 2000, 18, 1785–1791. [Google Scholar] [CrossRef]
- Bickel, N.; Schmidt, G.; Heinz, K.; Muller, K. Ferroelectric relaxation of the SrTiO3 (100) surface. Phys. Rev. Lett. 1989, 62, 2009–2011. [Google Scholar] [CrossRef]
- Piskunov, S.; Kotomin, E.A.; Heifets, E.; Maier, J.; Eglitis, R.I.; Borstel, G. Hybrid DFT calculations of the atomic and electronic structure for ABO3 perovskite (001) surfaces. Surf. Sci. 2005, 575, 75–88. [Google Scholar] [CrossRef]
- Heifets, E.; Kotomin, E.A.; Maier, J. Semi-empirical simulations of surface relaxation for perovskite titanates. Surf. Sci. 2000, 462, 19–35. [Google Scholar] [CrossRef]
- Heifets, E.; Eglitis, R.I.; Kotomin, E.A.; Maier, J.; Borstel, G. Ab initio modeling of surface structure for SrTiO3 perovskite. Phys. Rev. B 2001, 64, 235417. [Google Scholar] [CrossRef]
- Padilla, J.; Vanderbilt, D. Ab initio study of SrTiO3 surfaces. Surf. Sci. 1998, 418, 64–70. [Google Scholar] [CrossRef]
- Cheng, C.; Kunc, K.; Lee, M.H. Structural relaxation and longitudinal dipole moment of SrTiO3 (001) (1 × 1) surfaces. Phys. Rev. B 2000, 62, 10409. [Google Scholar] [CrossRef]
- Hikita, T.; Hanada, T.; Kudo, M.; Kawai, M. Structure and electronic state of the TiO2 and SrO-terminated SrTiO3 (100) surface. Surf. Sci. 1993, 287–288, 377–381. [Google Scholar] [CrossRef]
- Ikeda, A.; Nishimura, T.; Morishita, T.; Kido, Y. Surface relaxation and rumpling of TiO2-terminated SrTiO3 (001) determined by medium energy ion scattering. Surf. Sci. 1999, 433–435, 520–524. [Google Scholar] [CrossRef]
- Charlton, G.; Brennan, S.; Muryn, C.A.; McGrath, R.; Norman, D.; Thornton, G. Surface relaxation of SrTiO3 (001). Surf. Sci. 2000, 457, L376–L380. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I. Comparative ab initio calculations for ABO3 perovskite (001), (011) and (111) surfaces as well as YAlO3 (001) surfaces and F centers. J. Nano Electron. Phys. 2019, 11, 01001. [Google Scholar] [CrossRef]
- Borstel, G.; Eglitis, R.I.; Kotomin, E.A.; Heifets, E. Modelling of defects and surfaces in perovskite ferroelectrics. Phys. Status Solidi B 2003, 236, 253–264. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Vanderbilt, D. Ab initio calculations of BaTiO3 and PbTiO3 (001) and (011) surface structures. Phys. Rev. B 2007, 76, 155439. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Vanderbilt, D. Ab initio calculations of the atomic and electronic structure of CaTiO3 (001) and (011) surfaces. Phys. Rev. B 2008, 78, 155420. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).