Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study



Abstract
1. Introduction
2. Results
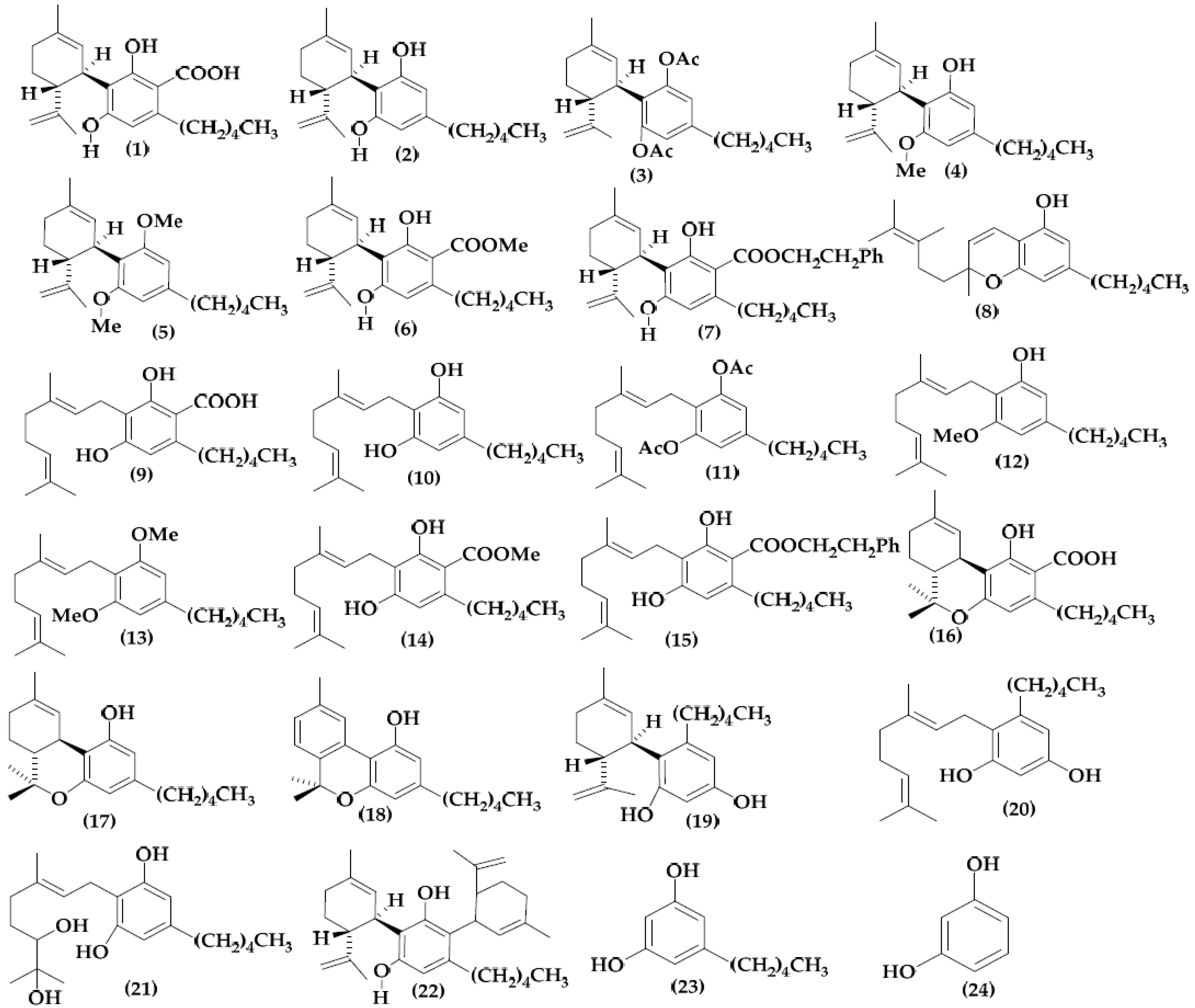
2.1. Molecular Structure and Data Collection
2.2. Molecular Descriptor Calculation
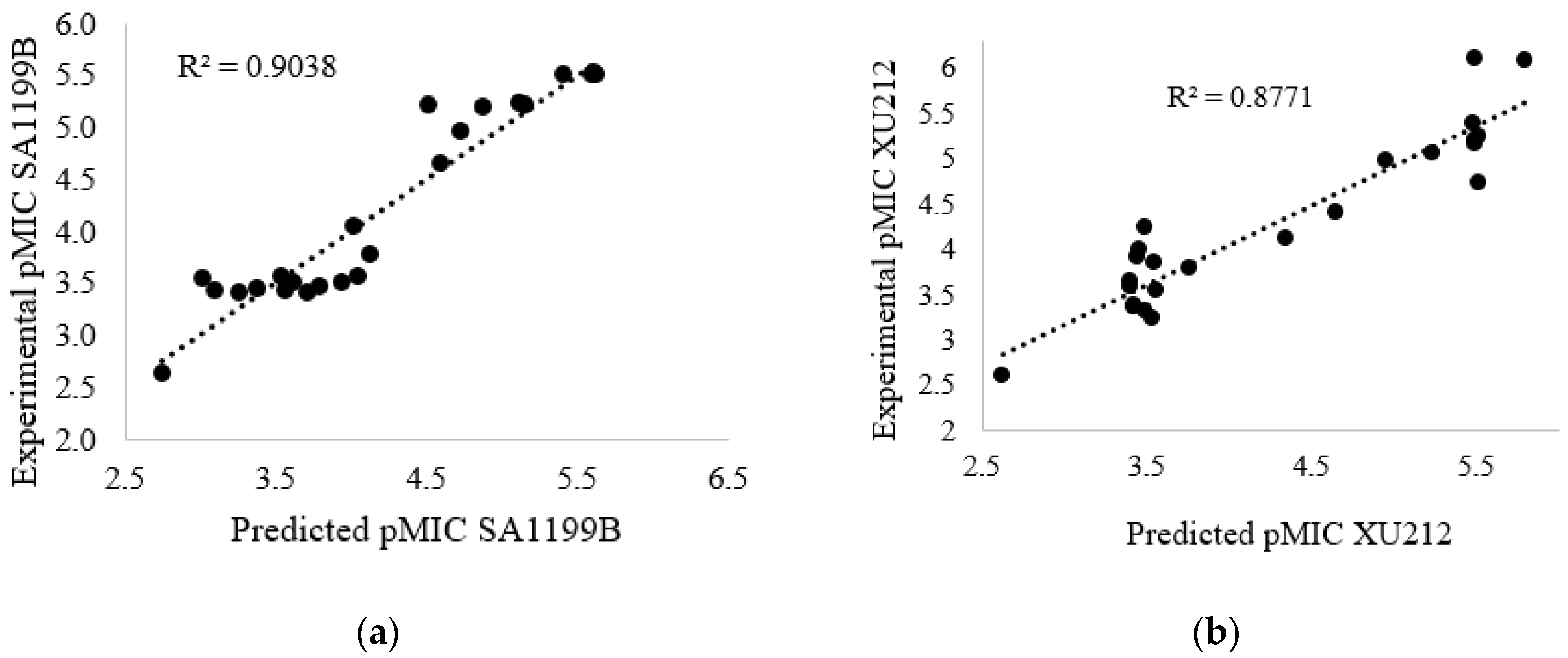
2.3. Quantitative Structure–Activity Relationships (QSAR):
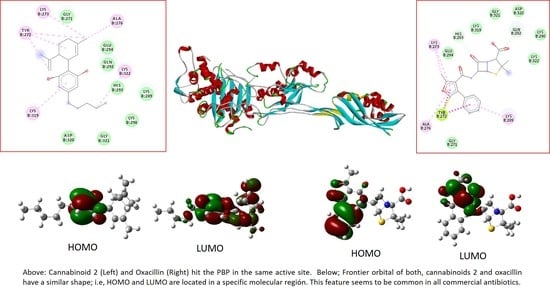
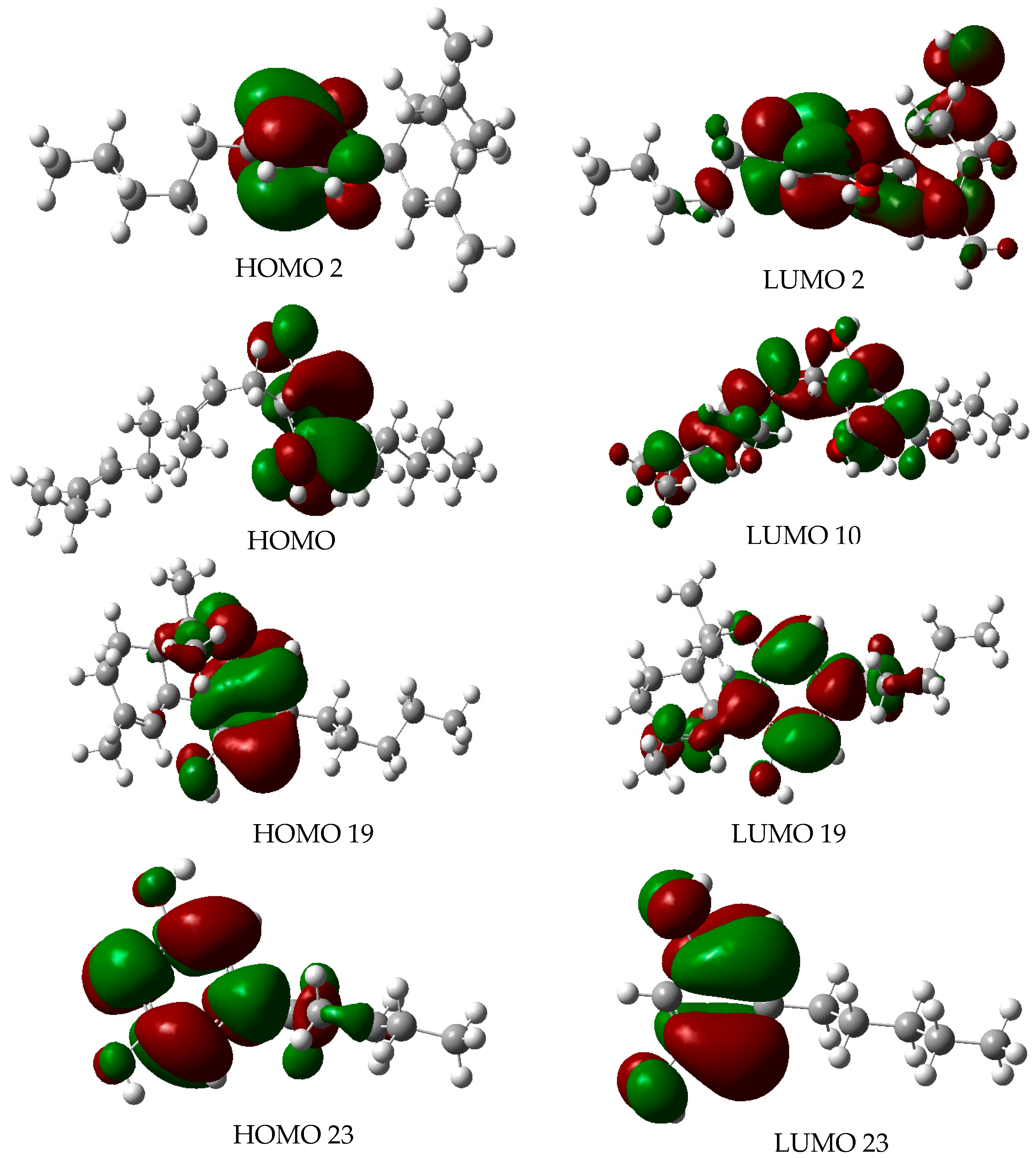
2.4. Molecular Orbitals
2.5. Drug-Likeness Properties
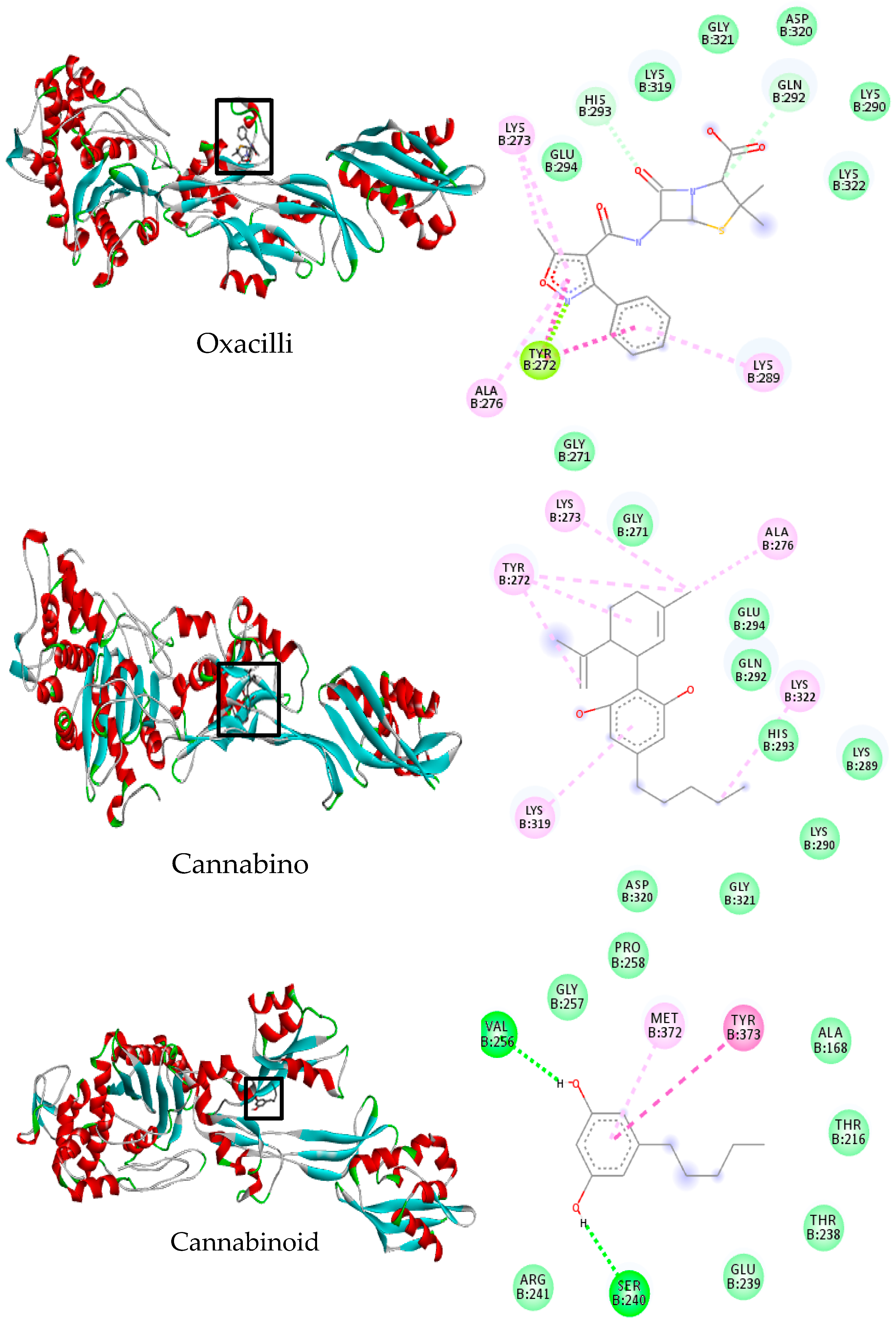
2.6. Molecular Docking
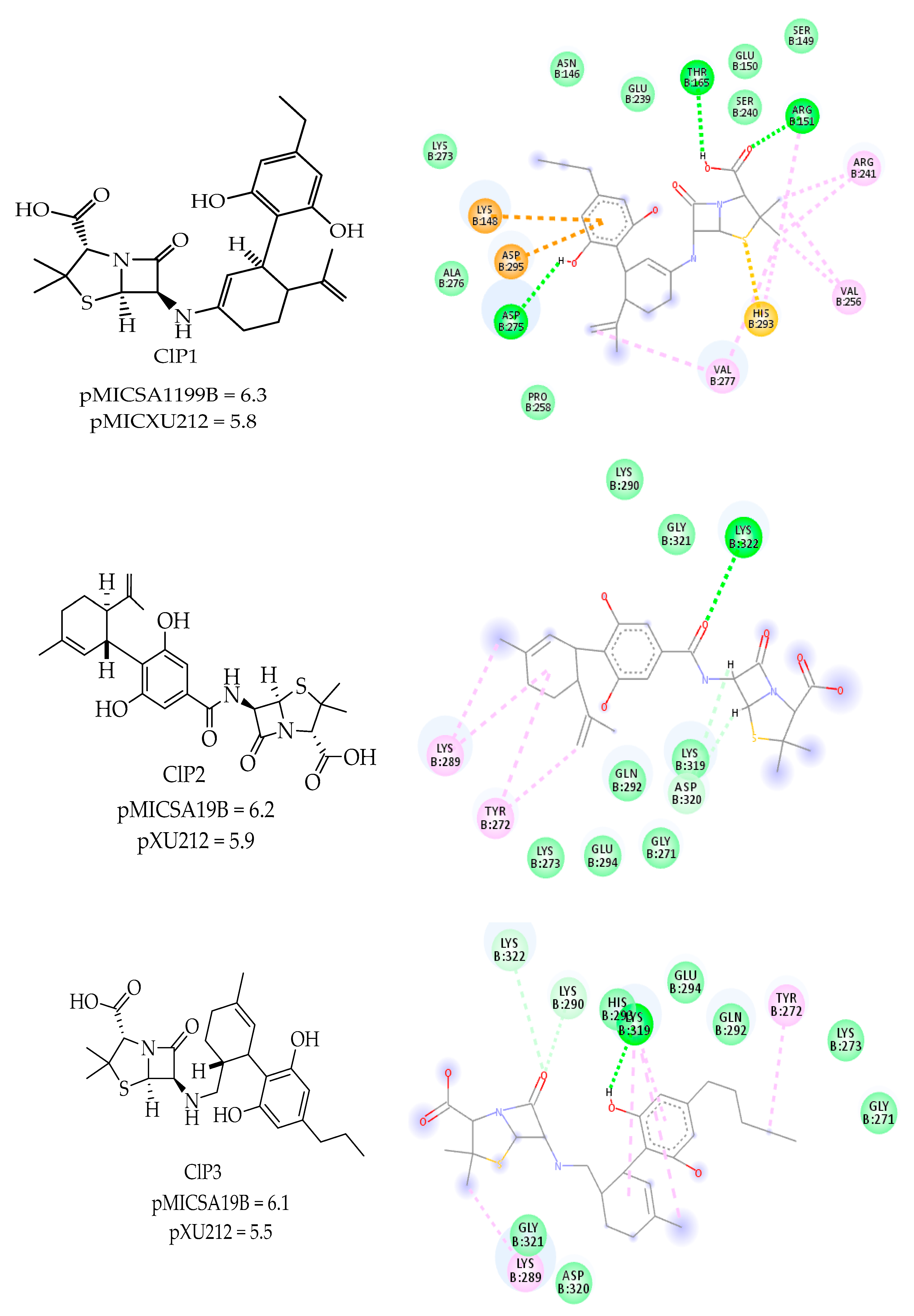
2.7. Predicted Compounds
3. Materials and Methods
3.1. Data Set Collection and Minimum Energy Structure Computation
3.2. Statistical Analysis
3.3. Drug-Likeness Property Prediction
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cosgrove, S.E.; Qi, Y.; Kaye, K.S.; Harbarth, S.; Karchmer, A.W.; Carmeli, Y. The Impact of Methicillin Resistance in Staphylococcus aureus Bacteremia on Patient Outcomes: Mortality, Length of Stay, and Hospital Charges. Infect. Control Hosp. Epidemiol. 2005, 26, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Cheesman, M.J.; Ilanko, A.; Blonk, B.; Cock, I.E. Developing new antimicrobial therapies: Are synergistic combinations of plant extracts/compounds with conventional antibiotics the solution? Pharmacogn. Rev. 2017, 11, 57–72. [Google Scholar]
- Gibbons, S. Anti-staphylococcal plant natural products. Nat. Prod. Rep. 2004, 21, 263–277. [Google Scholar] [CrossRef]
- Ballu, S.; Itteboina, R.; Sivan, S.K.; Manga, V. Rational design of methicillin resistance staphylococcus aureus inhibitors through 3D-QSAR, molecular docking and molecular dynamics simulations. Comput. Biol. Chem. 2018, 73, 95–104. [Google Scholar] [CrossRef]
- Dias, T.; Gaudêncio, S.; Pereira, F. A Computer-Driven Approach to Discover Natural Product Leads for Methicillin-Resistant Staphylococcus aureus Infection Therapy. Mar. Drugs 2018, 17, 16. [Google Scholar] [CrossRef]
- Uddin, R.; Lodhi, M.U.; Ul-Haq, Z. Combined Pharmacophore and 3D-QSAR Study on A Series of Staphylococcus aureus Sortase A inhibitors. Chem. Biol. Drug Des. 2012, 80, 300–314. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. Cannabinoids for treatment of alzheimer’s disease: Moving toward the clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Anand, P.; Whiteside, G.; Fowler, C.J.; Hohmann, A.G. Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res. Rev. 2009, 60, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Notcutt, W.G. Clinical Use of Cannabinoids for Symptom Control in Multiple Sclerosis. Neurotherapeutics 2015, 12, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.C.; Tsien, R.W.; Whalley, B.J.; Devinsky, O. Cannabinoids and Epilepsy. Neurotherapeutics 2015, 12, 747–768. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Hernández-Tiedra, S.; Dávila, D.; Lorente, M. The use of cannabinoids as anticancer agents. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.M.M.; Almagboul, A.Z.I.; Khogali, S.M.E.; Gergeir, U.M.A. Antimicrobial Activity of Cannabis sativa. J. Chin. Med. 2012, 3, 61–64. [Google Scholar] [CrossRef]
- Novak, J.; Zitterl-Eglseer, K.; Deans, S.G.; Franz, C.M. Essential oils of different cultivars ofCannabis sativa L. and their antimicrobial activity. Flavour Fragr. J. 2001, 16, 259–262. [Google Scholar] [CrossRef]
- Vemuri, V.K.; Makriyannis, A. Medicinal chemistry of cannabinoids. Clin. Pharmacol. Ther. 2015, 97, 553–558. [Google Scholar] [CrossRef]
- Stott, C.G.; Guy, G.W. Cannabinoids for the pharmaceutical industry. In Proceedings of the Euphytica; Springer: Berlin/Heidelberg, Germany, 2004; Volume 140, pp. 83–93. [Google Scholar]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial cannabinoids from Cannabis sativa: A structure-activity study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Aromatic Substitution. In Advanced Organic Chemistry; Springer US: Boston, MA, USA, 2007; pp. 771–831. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Structural Effects on Stability and Reactivity. In Advanced Organic Chemistry; Springer US: Boston, MA, USA, 2007; pp. 253–388. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. G16_C01. Gaussian 16 for Linux.
- Hydrogen-Bonding Capacity and Brain Penetration: Ingenta Connect. Available online: https://www.ingentaconnect.com/content/scs/chimia/1992/00000046/F0020007/art00003# (accessed on 13 May 2020).
- Torrent-Sucarrat, M.; De Proft, F.; Ayers, P.W.; Geerlings, P. On the applicability of local softness and hardness. Phys. Chem. Chem. Phys. 2010, 12, 1072–1080. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional Structure-Directed Quantitative Structure-Activity Relationships I. Partition Coefficients as a Measure of Hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [Google Scholar] [CrossRef]
- Labet, V.; Morell, C.; Cadet, J.; Eriksson, L.A.; Grand, A. Hydrolytic deamination of 5-methylcytosine in protic medium-A theoretical study. J. Phys. Chem. A 2009, 113, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Vazquez, J.; Gilbert, E.; Herrero, E.; Luque, F.J. Lipophilicity in drug design: An overview of lipophilicity descriptors in 3D-QSAR studies. Future Med. Chem. 2019, 11, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.L.L.; Ramos, A.L.D.; Filho, N.R.A.; Furtado, N.C.; Taft, C.A.; Aranda, D.A.G. Production of Biodiesel by a Two-Step Niobium Oxide Catalyzed Hydrolysis and Esterification. Lett. Org. Chem. 2010, 7, 571–578. [Google Scholar] [CrossRef]
- Lan, N.T.N.; Thu, N.T.N.; Barrail-Tran, A.; Duc, N.H.; Lan, N.N.; Laureillard, D.; Lien, T.T.X.; Borand, L.; Quillet, C.; Connolly, C.; et al. Randomised pharmacokinetic trial of rifabutin with lopinavir/ritonavir- antiretroviral therapy in patients with HIV-associated tuberculosis in Vietnam. PLoS ONE 2014, 9, e84866. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sun, X.; Zhu, F.; Ren, S.; Wang, S. OH-initiated transformation and hydrolysis of aspirin in AOPs system: DFT and experimental studies. Sci. Total Environ. 2017, 592, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Yamada, A. Molecular Orbital Principles of Oxygen-Redox Battery Electrodes. ACS Appl. Mater. Interfaces 2017, 9, 36463–36472. [Google Scholar] [CrossRef]
- Tarumi, M.; Matsuzaki, Y.; Suzuki, K. Theoretical study on the redox reaction mechanism of quinone compounds in industrial processes. Chem. Eng. Sci. 2019, 199, 381–387. [Google Scholar] [CrossRef]
- Winkler, D.A. The Role of Quantitative Structure±Activity Relationships (QSAR) in Biomolecular Discovery. Brief. Bioinform. 2002, 3, 73–86. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G. The Diels-Alder Reaction from the EDA-NOCV Perspective: A Re-Examination of the Frontier Molecular Orbital Model. Eur. J. Org. Chem. 2019, 2019, 478–485. [Google Scholar] [CrossRef]
- Grover, M.; Singh, B.; Bakshi, M.; Singh, S. Quantitative structure-property relationships in pharmaceutical research-Part 1. Pharm. Sci. Technol. Today 2000, 3, 28–35. [Google Scholar] [CrossRef]
- Malhotra, R.; Ravesh, A.; Singh, V. Synthesis, characterization, antimicrobial activities, and QSAR studies of organotin(IV) complexes. Phosphorussulfur Silicon Relat. Elem. 2017, 192, 73–80. [Google Scholar] [CrossRef]
- Kumer, A.; Paul, S. The Simulating Study of Homo, Lumo, Thermo Physical and Quantitative Structure of Activity Relationship (Qsar) of Some Anticancer Active Ionic Liquids. Eur. J. Environ. Res. 2019, 3, 1–10. [Google Scholar]
- Kumar, A.; Grewal, A.S.; Singh, V.; Narang, R.; Pandita, D.; Lather, V. Synthesis, Antimicrobial Activity and QSAR Studies of Some New Sparfloxacin Derivatives. Pharm. Chem. J. 2018, 52, 444–454. [Google Scholar] [CrossRef]
- Khodair, A.I.; Awad, M.K.; Gesson, J.P.; Elshaier, Y.A.M.M. New N-ribosides and N-mannosides of rhodanine derivatives with anticancer activity on leukemia cell line: Design, synthesis, DFT and molecular modelling studies. Carbohydr. Res. 2020, 487, 107894. [Google Scholar] [CrossRef]
- Suresh Kumar, S.; Athimoolam, S.; Sridhar, B. Structural, spectral, theoretical and anticancer studies on new co-crystal of the drug 5-fluorouracil. J. Mol. Struct. 2018, 1173, 951–958. [Google Scholar] [CrossRef]
- Kanagamani, K.; Muthukrishnan, P.; Ilayaraja, M.; Shankar, K.; Kathiresan, A. Synthesis, Characterisation and DFT Studies of Stigmasterol Mediated Silver Nanoparticles and Their Anticancer Activity. J. Inorg. Organomet. Polym. Mater. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Jeyaseelan, S.C.; Premkumar, R.; Kaviyarasu, K.; Franklin Benial, A.M. Spectroscopic, quantum chemical, molecular docking and in vitro anticancer activity studies on 5-Methoxyindole-3-carboxaldehyde. J. Mol. Struct. 2019, 1197, 134–146. [Google Scholar] [CrossRef]
- Sarkar, I.; Goswami, S.; Majumder, P. Quantitative structure–activity relationship (QSAR) study of some DNA-intercalating anticancer drugs. In Proceedings of the Lecture Notes in Electrical Engineering; Springer: Berlin/Heidelberg, Germany, 2020; Volume 575, pp. 357–366. [Google Scholar]
- Wang, J.; Yun, D.; Yao, J.; Fu, W.; Huang, F.; Chen, L.; Wei, T.; Yu, C.; Xu, H.; Zhou, X.; et al. Design, synthesis and QSAR study of novel isatin analogues inspired Michael acceptor as potential anticancer compounds. Eur. J. Med. Chem. 2018, 144, 493–503. [Google Scholar] [CrossRef]
- Baeten, A.; Tafazoli, M.; Kirsch-Volders, M.; Geerlings, P. Use of the HSAB principle in quantitative structure–activity relationships in toxicological research: Application to the genotoxicity of chlorinated hydrocarbons. Int. J. Quantum Chem. 1999, 74, 351–355. [Google Scholar] [CrossRef]
- Bradbury, S.P.; Mekenyan, O.G.; Ankley, G.T. The role of ligand flexibility in predicting biological activity: Structure-activity relationships for aryl hydrocarbon, estrogen, and androgen receptor binding affinity. Environ. Toxicol. Chem. 1998, 17, 15–25. [Google Scholar]
- Joshi, R.; Pandey, N.; Yadav, S.K.; Tilak, R.; Mishra, H.; Pokharia, S. Synthesis, spectroscopic characterization, DFT studies and antifungal activity of (E)-4-amino-5-[N’-(2-nitro-benzylidene)-hydrazino]-2,4-dihydro-[1,2,4]triazole-3-thione. J. Mol. Struct. 2018, 1164, 386–403. [Google Scholar] [CrossRef]
- Joshi, R.; Kumari, A.; Singh, K.; Mishra, H.; Pokharia, S. Triorganotin(IV) complexes of Schiff base derived from 1,2,4-triazole moiety: Synthesis, spectroscopic investigation, DFT studies, antifungal activity and molecular docking studies. J. Mol. Struct. 2020, 1206, 127639. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, A.; Huang, M.; Liu, M.; Pei, H.; Huang, L.; Yi, H.; Liu, W.; Hu, A. Design, synthesis, DFT study and antifungal activity of the derivatives of pyrazolecarboxamide containing thiazole or oxazole ring. Eur. J. Med. Chem. 2018, 149, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S.; Farah, M.A.; Al-Lohedan, H.A.; Al-Anazi, K.M. Comprehensive exploration of the anticancer activities of procaine and its binding with calf thymus DNA: A multi spectroscopic and molecular modelling study. RSC Adv. 2018, 8, 9083–9093. [Google Scholar] [CrossRef]
- Rachedi, K.O.; Ouk, T.S.; Bahadi, R.; Bouzina, A.; Djouad, S.E.; Bechlem, K.; Zerrouki, R.; Ben Hadda, T.; Almalki, F.; Berredjem, M. Synthesis, DFT and POM analyses of cytotoxicity activity of α-amidophosphonates derivatives: Identification of potential antiviral O,O-pharmacophore site. J. Mol. Struct. 2019, 1197, 196–203. [Google Scholar] [CrossRef]
- da Costa, R.M.; Bastos, J.K.; Costa, M.C.A.; Ferreira, M.M.C.; Mizuno, C.S.; Caramori, G.F.; Nagurniak, G.R.; Simão, M.R.; dos Santos, R.A.; Veneziani, R.C.S.; et al. In vitro cytotoxicity and structure-activity relationship approaches of ent-kaurenoic acid derivatives against human breast carcinoma cell line. Phytochemistry 2018, 156, 214–223. [Google Scholar] [CrossRef]
- Soffers, A.E.M.F.; Boersma, M.G.; Vaes, W.H.J.; Vervoort, J.; Tyrakowska, B.; Hermens, J.L.M.; Rietjens, I.M.C.M. Computer-modeling-based QSARs for analyzing experimental data on biotransformation and toxicity. In Proceedings of the Toxicology in Vitro; Pergamon: Bergama, Turkey, 2001; Volume 15, pp. 539–551. [Google Scholar]
- Lewis, D.F.V. Quantitative structure-activity relationships (QSARs) within the cytochrome P450 system: QSARs describing substrate binding, inhibition and induction of P450s. Inflammopharmacology 2003, 11, 43–73. [Google Scholar] [CrossRef]
- Strahan, J.; Popere, B.C.; Khomein, P.; Pointer, C.A.; Martin, S.M.; Oldacre, A.N.; Thayumanavan, S.; Young, E.R. Modulating absorption and charge transfer in bodipy-carbazole donor-acceptor dyads through molecular design. Dalton Trans. 2019, 48, 8488–8501. [Google Scholar] [CrossRef]
- Vikramaditya, T.; Saisudhakar, M.; Sumithra, K. Computational study on thermally activated delayed fluorescence of donor-linker-acceptor network molecules. RSC Adv. 2016, 6, 37203–37211. [Google Scholar] [CrossRef]
- Santos Silva, H.; Metz, S.; Hiorns, R.C.; Bégué, D. Targeting ideal acceptor-donor materials based on hexabenzocoronene. J. Mol. Struct. 2018, 1161, 442–452. [Google Scholar] [CrossRef]
- Wan, X.; Li, C.; Zhang, M.; Chen, Y. Acceptor–donor–acceptor type molecules for high performance organic photovoltaics–chemistry and mechanism. Chem. Soc. Rev. 2020, 49, 2828–2842. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, Z.; Li, S.; Luan, G.; Liang, D.; Tang, S.; Jin, R. Design of Acceptors with Suitable Frontier Molecular Orbitals to Match Donors via Substitutions on Perylene Diimide for Organic Solar Cells. Int. J. Mol. Sci. 2016, 17, 721. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, D.; Ma, X.; Ansari, R.; Kim, J.; Kieffer, J. Design principles for the energy level tuning in donor/acceptor conjugated polymers. Phys. Chem. Chem. Phys. 2019, 21, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.C.; Guo, E.Q.; Zhang, Y.L.; Ren, P.H.; Yang, W.J. Donor-acceptor-substituted anthracene-centered cruciforms: Synthesis, enhanced two-photon absorptions, and spatially separated frontier molecular orbitals. Chem. Mater. 2009, 21, 5125–5135. [Google Scholar] [CrossRef]
- Higashino, T.; Ishida, K.; Imahori, H. Modulation of Frontier Molecular Orbitals on Dithieno[3,4- b :3′,4′- d ]phosphole Derivatives by Donor-π-Acceptor Interaction. Chem. Lett. 2020, 49, 272–275. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, C.; Kwong, D.J.; Li, J.; Deng, X.; Fan, Z. A Dramatic Odd-Even Oscillating Behavior for the Current Rectification and Negative Differential Resistance in Carbon-Chain-Modified Donor-Acceptor Molecular Devices. Adv. Funct. Mater. 2013, 23, 2765–2774. [Google Scholar] [CrossRef]
- Sathya, A.; Prabhu, T.; Ramalingam, S. Structural, biological and pharmaceutical importance of antibiotic agent chloramphenicol. Heliyon 2020, 6, e03433. [Google Scholar] [CrossRef]
- Alnoman, R.B.; Parveen, S.; Hagar, M.; Ahmed, H.A.; Knight, J.G. A new chiral boron-dipyrromethene (BODIPY)-based fluorescent probe: Molecular docking, DFT, antibacterial and antioxidant approaches. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef]
- Marinescu, M.; Cinteza, L.O.; Marton, G.I.; Marutescu, L.G.; Chifiriuc, M.C.; Constantinescu, C. Density functional theory molecular modeling and antimicrobial behaviour of selected 1,2,3,4,5,6,7,8-octahydroacridine-N(10)-oxides. J. Mol. Struct. 2017, 1144, 14–23. [Google Scholar] [CrossRef]
- Celik, S.; Albayrak, A.T.; Akyuz, S.; Ozel, A.E.; Sigirci, B.D. Synthesis, antimicrobial activity, molecular docking and ADMET study of a caprolactam-glycine cluster. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Sobhani, S.; Pordel, M.; Beyramabadi, S.A. Design, synthesis, spectral, antibacterial activities and quantum chemical calculations of new Cu (II) complexes of heterocyclic ligands. J. Mol. Struct. 2019, 1175, 677–685. [Google Scholar] [CrossRef]
- Slassi, S.; Aarjane, M.; Yamni, K.; Amine, A. Synthesis, crystal structure, DFT calculations, Hirshfeld surfaces, and antibacterial activities of schiff base based on imidazole. J. Mol. Struct. 2019, 1197, 547–554. [Google Scholar] [CrossRef]
- Özbek, N.; Özdemir, Ü.Ö.; Altun, A.F.; Şahin, E. Sulfonamide-derived hydrazone compounds and their Pd (II) complexes: Synthesis, spectroscopic characterization, X-ray structure determination, in vitro antibacterial activity and computational studies. J. Mol. Struct. 2019, 1196, 707–719. [Google Scholar] [CrossRef]
- Khan, S.A.; Asiri, A.M.; Al-Ghamdi, N.S.M.; Asad, M.; Zayed, M.E.M.; Elroby, S.A.K.; Aqlan, F.M.; Wani, M.Y.; Sharma, K. Microwave assisted synthesis of chalcone and its polycyclic heterocyclic analogues as promising antibacterial agents: In vitro, in silico and DFT studies. J. Mol. Struct. 2019, 1190, 77–85. [Google Scholar] [CrossRef]
- Flores, M.C.; Márquez, E.A.; Mora, J.R. Molecular modeling studies of bromopyrrole alkaloids as potential antimalarial compounds: A DFT approach. Med. Chem. Res. 2018, 27, 844–856. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Iluz, N.; Maor, Y.; Keller, N.; Malik, Z. The synergistic effect of PDT and oxacillin on clinical isolates of Staphylococcus aureus. Lasers Surg. Med. 2018, 50, 535–551. [Google Scholar] [CrossRef]
- Lee, H.; Boyle-Vavra, S.; Ren, J.; Jarusiewicz, J.A.; Sharma, L.K.; Hoagland, D.T.; Yin, S.; Zhu, T.; Hevener, K.E.; Ojeda, I.; et al. Identification of small molecules exhibiting oxacillin synergy through a novel assay for inhibition of vraTSR expression in methicillin-resistant staphylococcus aureus. Antimicrob. Agents Chemother. 2019, 63, e02593-18. [Google Scholar] [CrossRef]
- Legrand, T.; Vodovar, D.; Tournier, N.; Khoudour, N.; Hulin, A. Simultaneous determination of eight β-lactam antibiotics, amoxicillin, cefazolin, cefepime, cefotaxime, ceftazidime, cloxacillin, oxacillin, and piperacillin, in human plasma by using ultra-high-performance liquid chromatography with ultraviolet detection. Antimicrob. Agents Chemother. 2016, 60, 4734–4742. [Google Scholar] [CrossRef]
- Nomura, R.; Nakaminami, H.; Takasao, K.; Muramatsu, S.; Kato, Y.; Wajima, T.; Noguchi, N. A class A β-lactamase produced by borderline oxacillin-resistant Staphylococcus aureus hydrolyzes oxacillin. J. Glob. Antimicrob. Resist. 2020, 22, 244–247. [Google Scholar] [CrossRef]
- de Sousa, J.N.; de Oliveira, A.B.M.; Ferreira, A.K.; Silva, E.; de Sousa, L.M.S.; França Rocha, M.C.; de, J.P.; Júnior, S.; William Kaatz, G.; da Silva Almeida, J.R.G.; et al. Modulation of the resistance to norfloxacin in Staphylococcus aureus by Bauhinia forficata link. Nat. Prod. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.C. Norfloxacin, a fluoroquinolone antibacterial agent. Classification, mechanism of action, and in vitro activity. Am. J. Med. 1987, 82, 3–17. [Google Scholar] [CrossRef]
- Barry, A.L.; Jones, R.N.; Thornsberry, C.; Ayers, L.W.; Gerlach, E.H.; Sommers, H.M. Antibacterial activities of ciprofloxacin, norfloxacin, oxolinic acid, cinoxacin, and nalidixic acid. Antimicrob. Agents Chemother. 1984, 25, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Lezama, J.; Márquez, E.; Escalante, L.; Córdova, T.; Chuchani, G. Theoretical study of neighboring carbonyl group participation in the elimination kinetics of chloroketones in the gas phase. J. Phys. Org. Chem. 2011, 24, 229–240. [Google Scholar] [CrossRef]
- Mora, J.; Cervantes, C.; Marquez, E. New Insight into the Chloroacetanilide Herbicide Degradation Mechanism through a Nucleophilic Attack of Hydrogen Sulfide. Int. J. Mol. Sci. 2018, 19, 2864. [Google Scholar] [CrossRef]
- Miao, J.; Hua, S.; Li, S. Assessment of density functionals on intramolecular dispersion interaction in large normal alkanes. Chem. Phys. Lett. 2012, 541, 7–11. [Google Scholar] [CrossRef]
- Eriksson, E.S.E.; Eriksson, L.A. Predictive power of long-range corrected functionals on the spectroscopic properties of tetrapyrrole derivatives for photodynamic therapy. Phys. Chem. Chem. Phys. 2011, 13, 7207–7217. [Google Scholar] [CrossRef]
- Zara, Z.; Iqbal, J.; Ayub, K.; Irfan, M.; Mahmood, A.; Khera, R.A.; Eliasson, B. A comparative study of DFT calculated and experimental UV/Visible spectra for thirty carboline and carbazole based compounds. J. Mol. Struct. 2017, 1149, 282–298. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, Å.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Mazzone, G.; Malaj, N.; Russo, N.; Toscano, M. Density functional study of the antioxidant activity of some recently synthesized resveratrol analogues. Food Chem. 2013, 141, 2017–2024. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Acevedo-Martínez, L.; Barigye, S.J.; Valdés-Martiní, J.R.; Contreras-Torres, E. QuBiLS-MIDAS: A parallel free-software for molecular descriptors computation based on multilinear algebraic maps. J. Comput. Chem. 2014, 35, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Basak, S.C. Beware of External Validation!-A Comparative Study of Several Validation Techniques used in QSAR Modelling. Curr. Comput. Aided Drug Des. 2018, 14, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K. Cross-validation as the objective function for variable-selection techniques. TrAC Trends Anal. Chem. 2003, 22, 395–406. [Google Scholar] [CrossRef]
- Kiralj, R.; Ferreira, M.M.C. Basic validation procedures for regression models in QSAR and QSPR studies: Theory and application. J. Braz. Chem. Soc. 2009, 20, 770–787. [Google Scholar] [CrossRef]
- Cawley, G.C. Leave-One-Out Cross-Validation Based Model Selection Criteria for Weighted LS-SVMs. In Proceedings of the 006 IEEE International Joint Conference on Neural Network Proceedings, Vancouver, BC, Canada, 16–21 July 2006; pp. 1661–1668. [Google Scholar]
- Shao, J. Linear model selection by cross-validation. J. Am. Stat. Assoc. 1993, 88, 486–494. [Google Scholar] [CrossRef]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Proudfoot, J.R. Drugs, leads, and drug-likeness: An analysis of some recently launched drugs. Bioorganic Med. Chem. Lett. 2002, 12, 1647–1650. [Google Scholar] [CrossRef]
- Vistoli, G.; Pedretti, A.; Testa, B. Assessing drug-likeness-what are we missing? Drug Discov. Today 2008, 13, 285–294. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 2 May 2020).
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA-Scientific Enterprise Software for Chemical Research, Material Science R&D. Available online: https://www.3dsbiovia.com/ (accessed on 2 May 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | pMIC SA-1199B | pMIC XU212 | Compound | pMIC SA-1199B | pMIC XU212 |
|---|---|---|---|---|---|
| 1 | 5.70 | 5.70 | 13 | 3.89 | 3.89 |
| 2 | 6.00 | 6.00 | 14 | 4.19 | 4.19 |
| 3 | 3.89 | 3.89 | 15 | 3.89 | 3.89 |
| 4 | 3.89 | 3.89 | 16 | 5.09 | 5.10 |
| 5 | 3.89 | 3.89 | 17 | 5.69 | 6.00 |
| 6 | 3.89 | 3.89 | 18 | 6.00 | 6.00 |
| 7 | 3.89 | 3.89 | 19 | 6.00 | 6.00 |
| 8 | 5.70 | 6.00 | 20 | 5.70 | 6.30 |
| 9 | 5.40 | 5.40 | 21 | 4.50 | 4.80 |
| 10 | 6.00 | 6.00 | 22 | 3.89 | 3.89 |
| 11 | 3.89 | 3.89 | 23 | 4.19 | 4.19 |
| 12 | 3.89 | 3.89 | 24 | 3.59 | 3.59 |
| Compounds | TS[1]_RA_B_AB_nCi_2_M8_MP1_T_LGL[2–3]_c-s_MID | SD_B_AB_nCi_2_M5_NS3_P_KA_v-h_MID | S_B_AB_nCi_2_M11_NS4_T_KA_psa-r_MID | VC_B_AB_nCi_2_M15_NS7_P_KA_v-m_MID | P2_B_AB_nCi_2_M1_SS0_A_LGL[1,2]_c-h_MID |
|---|---|---|---|---|---|
| 1 | 0.0044 | 14.7938 | 2.7992 | 1.8229 | 0.0012 |
| 2 | 0.0069 | 13.8815 | 2.2173 | 2.0817 | 0.0024 |
| 3 | 0.0045 | 9.9907 | 4.9058 | 3.8930 | 0.0024 |
| 4 | 0.0033 | 10.9633 | 3.7194 | 3.3114 | 0.0001 |
| 5 | 0.0044 | 9.2263 | 5.3614 | 3.7377 | 0.0011 |
| 6 | 0.0052 | 10.5953 | 3.7704 | 3.7133 | 0.0029 |
| 7 | 0.0046 | 10.2373 | 3.7369 | 3.4611 | 0.0035 |
| 8 | 0.0058 | 9.7606 | 4.3982 | 2.5146 | 0.0015 |
| 9 | 0.0054 | 11.3406 | 5.8630 | 3.7567 | 0.0008 |
| 10 | 0.0068 | 11.1807 | 5.1249 | 2.8569 | 0.0020 |
| 11 | 0.0055 | 10.4532 | 3.7345 | 3.5327 | 0.0025 |
| 12 | 0.0029 | 11.6010 | 5.1244 | 3.3131 | 0.0002 |
| 13 | 0.0040 | 9.5737 | 4.0032 | 3.6647 | 0.0011 |
| 14 | 0.0036 | 11.1387 | 4.9855 | 2.6120 | 0.0030 |
| 15 | 0.0041 | 10.6774 | 4.1333 | 2.3554 | 0.0037 |
| 16 | 0.0072 | 10.1870 | 3.8763 | 3.6022 | 0.0015 |
| 17 | 0.0053 | 12.2729 | 5.5835 | 3.7104 | 0.0010 |
| 18 | 0.0077 | 11.9375 | 4.8680 | 3.5326 | 0.0026 |
| 19 | 0.0080 | 12.3688 | 3.6870 | 3.3128 | 0.0020 |
| 20 | 0.0073 | 10.7380 | 4.4719 | 3.1839 | 0.0021 |
| 21 | 0.0034 | 11.0070 | 5.1107 | 2.6703 | 0.0004 |
| 22 | 0.0027 | 11.1369 | 3.6025 | 3.3132 | 0.0004 |
| 23 | 0.0035 | 10.9987 | 4.4855 | 3.8152 | 0.0002 |
| 24 | 0.0026 | 8.8067 | 5.7488 | 3.9288 | 0.0001 |
| Compounds | TS[1]_RA_B_AB_nCi_2_M8_MP1_T_LGL[2,3]_c-s_MID | Q1_Tr_AB_nCi_3_M26(M5)_NS3_P_KA_a-e-v_MID | AC[1]_VC_B_AB_nCi_2_M1_MP3_T_LGL[5,6]_p-h_MID | AC[1]_RA_B_AB_nCi_2_M1_MP6_T_KA_h-s_MID | AC[1]_VC_F_AB_nCi_2_M3_NS2_A_KA_psa_MID |
|---|---|---|---|---|---|
| 1 | 0.0044 | −60.3582 | 2.4964 | 0.0622 | 1.4648 |
| 2 | 0.0069 | −67.2432 | 0.0000 | 0.0850 | 0.6886 |
| 3 | 0.0045 | −22.9006 | 1.0652 | 0.0313 | 0.9884 |
| 4 | 0.0033 | −42.3669 | 1.3793 | 0.0309 | 1.4035 |
| 5 | 0.0044 | −23.9372 | 1.2764 | 0.0234 | 1.2500 |
| 6 | 0.0052 | −30.8211 | 1.6685 | 0.0214 | 1.2913 |
| 7 | 0.0046 | −31.5012 | 1.8934 | 0.0121 | 1.4092 |
| 8 | 0.0058 | −26.9075 | 2.8690 | 0.0616 | 0.9605 |
| 9 | 0.0054 | −56.5838 | 2.0773 | 0.0265 | 1.2378 |
| 10 | 0.0068 | −36.1725 | 2.7731 | 0.0280 | 1.1236 |
| 11 | 0.0055 | −28.9726 | 2.2279 | 0.0153 | 1.5304 |
| 12 | 0.0029 | −39.7408 | 1.0434 | 0.1213 | 1.4034 |
| 13 | 0.0040 | −31.2046 | 1.3216 | 0.0072 | 1.2847 |
| 14 | 0.0036 | −30.0663 | 1.0286 | 0.1658 | 1.4694 |
| 15 | 0.0041 | −31.0154 | 1.6328 | 0.0069 | 1.4986 |
| 16 | 0.0072 | −29.2698 | 1.2807 | 0.0083 | 1.2959 |
| 17 | 0.0053 | −64.2540 | 2.2565 | 0.0194 | 1.2696 |
| 18 | 0.0077 | −52.5670 | 1.3425 | 0.0338 | 0.9095 |
| 19 | 0.0080 | −74.3838 | 1.5881 | 0.0094 | 1.5915 |
| 20 | 0.0073 | −47.7595 | 1.8794 | 0.2085 | 1.6470 |
| 21 | 0.0034 | −42.2837 | 1.4434 | 0.1388 | 0.9999 |
| 22 | 0.0027 | −43.1504 | 0.8824 | 0.0375 | 0.9359 |
| 23 | 0.0035 | −29.8853 | 2.3293 | 0.0834 | 0.9010 |
| 24 | 0.0026 | −23.1060 | 1.0684 | 0.0130 | 1.1991 |
| Compounds | MW | #RB | #HBA | #HBD | MR (mol/cm3) | TPSA | Log P |
|---|---|---|---|---|---|---|---|
| 1 | 312.49 | 6.0 | 1.0 | 1.0 | 102.79 | 20.23 | 5.92 |
| 2 | 314.46 | 6.0 | 2.0 | 2.0 | 96.93 | 40.46 | 4.31 |
| 8 | 330.50 | 7.0 | 2.0 | 1.0 | 104.83 | 29.46 | 4.61 |
| 10 | 316.48 | 9.0 | 2.0 | 2.0 | 101.96 | 40.46 | 4.70 |
| 18 | 310.43 | 4.0 | 2.0 | 1.0 | 97.09 | 29.46 | 5.21 |
| 19 | 328.49 | 6.0 | 2.0 | 2.0 | 104.40 | 40.46 | 5.54 |
| Cannabinoids | PBP | Iso-TyrRS | DNA Gyr |
|---|---|---|---|
| 2 | −8.5 | −4.6 | −3.9 |
| 3 | −3.9 | −2.5 | −3.4 |
| 10 | −6.7 | −4.7 | −3.9 |
| 14 | −5.7 | −3.6 | −2.9 |
| 19 | −6.2 | −2.8 | −4.1 |
| 23 | −4.9 | −3.2 | −4.0 |
| Norfloxacin | −5.1 | −7.2 | −8.6 |
| Oxacillin | −8.8 | −6.2 | −4.8 |
| Mupirocin | −4.9 | −7.8 | −2.7 |
| Compound | MW | #RB | #HBA | #HBD | MR (mol/cm3) | TPSA (Å2) | Log P | Scoring Values PBP (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| ClP1 | 486.22 | 7 | 5 | 4 | 133.70 | 110.1 | 4.89 | −7.9 |
| ClP2 | 486.58 | 6.0 | 5 | 4 | 128.34 | 127.2 | 4.20 | −7.4 |
| ClP3 | 474.71 | 7.0 | 5 | 4 | 128.46 | 110.1 | 3.08 | −7.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes, E.; Mora, J.; Márquez, E. Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals 2020, 10, 692. https://doi.org/10.3390/cryst10080692
Cortes E, Mora J, Márquez E. Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals. 2020; 10(8):692. https://doi.org/10.3390/cryst10080692
Chicago/Turabian StyleCortes, Eliceo, José Mora, and Edgar Márquez. 2020. "Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study" Crystals 10, no. 8: 692. https://doi.org/10.3390/cryst10080692
APA StyleCortes, E., Mora, J., & Márquez, E. (2020). Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals, 10(8), 692. https://doi.org/10.3390/cryst10080692