1. Introduction
The theory and modelling of the interaction of matter with intense X-ray pulses [
1,
2,
3] has played an important role in the recent rise of X-ray free-electron lasers (XFELs) as a powerful new X-ray source for structural biology. Under the right experimental conditions, XFEL crystallography can be radiation-damage free for all practical purposes [
4,
5,
6]. XFEL pulses are, however, violently destructive and the absence of radiation damage effects can not be taken for granted. Damage modelling still plays a key role in understanding when radiation damage can by mitigated by a suitable choice beam conditions [
7,
8,
9].
XFELs are particularly advantageous for time-resolved protein crystallography experiments [
10,
11]. XFELs are able access sub-picosecond timescales that are inaccessible at synchrotron sources. Even at longer biologically relevant timescales of microseconds or milliseconds, XFELs provide a number of practical advantages for avoiding radiation damage, permitting micron-sized crystals, room temperature measurements and for studying irreversible processes [
12]. Targets of time-resolved XFEL studies frequently contain heavy elements in the active site of the protein, such as in metalloproteins [
10]. The uptake of light during photosynthesis, for example, occurs via a series of subtle structural changes at a manganese cluster [
13]. The absorption of oxygen by myoglobin occurs at the site of an ion-containing cluster [
14]. The heavy element constituents are, however, hotspots for damage because they ionize at a faster rate than light elements. Furthermore, the molecular coordination and exchange of energy between these heavier atoms and their immediate environment adds complexity to the manifestation of damage locally, which are processes that are not well understood. Simulations have predicted reproducible local damage effects in the manganese cluster of photosystem II [
15] and near the iron-sulfur clusters in ferrodoxin [
16]. Local damage has been observed experimentally in ferrodoxin crystals [
17]. Damage has clearly not prevented time-resolved measurements of the structure of many protein crystals, but it remains an important consideration for the interpretation of the data. Sub-Angström changes in bond distances can be interpreted as chemical changes in a dynamical process, but these are also the length scales at which the residual effects of damage may be present. Theoretical modelling of these damage hotspots plays a crucial, ongoing role in ensuring the accurate interpretation of time-resolved data.
Computational modelling of XFEL damage processes faces the formidable challenges of modelling ionization, thermal motion, plasma formation and Coulomb-driven ion dynamics on samples that are effectively bulk materials. Monte Carlo molecular dynamics (MC/MD) simulations model most of these processes by explicitly treating the electrons ejected from ionization events as classical point particles [
18]. In particular they have been used to study the local damage effects of heavier elements in proteins, such as photosystem [
15] and ferrodoxin [
16] by tracking the ion motion due to Coulomb repulsion. A drawback of MC/MD simulations for XFEL damage is their large computational expense, which puts a severe limitation on the simulated sample volume and the number of experimental conditions that can be simulated in a reasonable time. Compared to conventional MD simulations for structural dynamics, XFEL MC/MD simulations track fast moving electrons that require very short time steps (
s) compared to the total simulation time (
s). Physically, this arises because of the different dynamical timescales between the fast moving electrons and slow moving ions.
Rate-equation methods (REM) are an alternative to MC/MD that predict the populations of bound electrons, trapped electrons and escaped electrons [
19,
20], but they do not track the ion positions. They are good for predicting global damage effects and the variations of ionization levels in the different elemental species. REMs are computationally fast and inexpensive and do not have a limit on sample size. REMs are commonly used in studies that span a large range of experimental parameters [
8,
21]. They do not track the motion of individual ions and do not, as a consequence, provide a detailed picture of the dynamics.
Here, we present a new hybrid model of XFEL damage by combining the MD and REM approaches. Both MC/MD and plasma/MD simulations require detailed knowledge of the ionization rates for photoionization, radiative decay, Auger decay and electron-electron impact ionization. We use a detailed atomic model [
22] to predict the ionization rates used in the REM part of our model, consistent with practices in state-of-art XFEL MC/MD simulations. We treat the ion motion using molecular dynamics and the ejected and trapped electrons using a continuum (plasma) model. The parameters of the trapped electron gas, such as density and temperature, are predicted by an REM. We then use these predicted electron gas parameters to modify the ion-ion interactions with Debye screening and plasma drag forces. The ion dynamics are then predicted with MD using effective potentials that incorporate the screening effects of the plasma electrons. Modelling only the ion trajectories explicitly allows larger sample volumes and shorter simulation times compared with existing XFEL MD simulations that track electrons as classical particles. In turn, this enables more extensive and thorough simulation studies of damage hot spots at the heavy atom clusters that are relevant for time-resolved crystallography.
We show that the gain in computational speed provided by plasma/MD model can be leveraged to estimate the radiation damage induced disulfide (S-S) bond elongation on the example of a lysozyme nano-crystal. This effect has recently been demonstrated in an X-ray pump/X-ray probe serial femtosecond crystallography (SFX) experiment [
23]. It was shown in that work that the observed bond length depends on the fluence of the incident pump pulse as well as the time delay between the pump and probe pulses. We investigate how an observed S-S bond length depends on the incident XFEL pulse duration under a constant incident pulse fluence. Such simulations can be used to fine-tune the pulse parameters prior to an experiment with radiation-sensitive samples. Our simulation can be carried out in a few hours on a desktop computer for molecules with
non-hydrogen atoms. An equivalent calculation with classical electron trajectories would require high performance computing resources.
2. Theory: Ionization Simulation
The rate equation model utilizes precalculated rates for Auger decay, fluorescence and scattering cross-section [
22]. Atomic rates and cross-sections are calculated using our computer code “Atomic code for damage calculation” (AC4DC). Below, we give a brief description of the underlying theory.
The wave function of an atom is approximated by a single Slater determinant [
24], which is an antisymmetrized product of single-particle atomic orbitals. Each atomic orbital satisfies an average-over-configuration restricted Hartree-Fock equation [
22] and is characterized by a principal quantum number,
n, and orbital angular momentum quantum number,
l. These equations are of the form
where
is the number of electrons in orbital
. The maximum number of electrons in the orbital
is
. Matrix elements for transitions are averaged over initial and summed over final values of magnetic quantum numbers and spin projections. Excited states, such as final state continuum orbitals in photo-absorption and Auger decay, are calculated using the
approximation [
22] which modifies Equations (2) and (3). This approximation accounts for a reduction in the number of bound electrons in the final state and yields accurate transition energies. The energies of the required excited orbitals, as well as their quantum numbers, can be easily found using well-known selection rules for photo-ionization, fluorescence and Auger decay.
Once a set of bound and excited states is generated for a given electron configuration
, all atomic transition rates that involve this configuration as an initial state are calculated. A full set of rates and transition energies, required by REM, is obtained by repeating the above calculation for all possible electron configurations. For heavier elements, such as iron or magnesium, a high degree of ionization in valence shells can result in the K-shell ionization potential dropping below the incident photon energy. In the absence of a resonance the matrix element for a multi-photon absorption of X-rays with energy
keV is small. For typical typical values of incident X-ray intensities we ignore multi-photon absorption and consider these electrons to be locked in their orbitals. This assumption considerably reduces the number of possible electron configurations for which Equation (
1) needs to be solved.
3. Theory: Plasma Simulation
We use a rate-equations model (REM) that is closely related to existing models [
19,
20]. We have used REM code in previous studies of the relationship between damage and diffraction [
21,
25]. Since the models are given in detail in previous publications, we briefly review here the essential components of an REM model and highlight the most relevant parts for the hybrid model.
A REM calculates the populations
of several species
i of electrons or ions. In XFEL damage simulations, it is common to include every electron configuration for each ionization state of each element, as well as the populations of trapped unbound electrons and escaped photoelectrons. The rate equations are a series of couple equations for the populations of each electron/ion species tracked:
where
is the total transition rate of all processes involving electron(ion) species
j that increases
and
is the total rate of all processes that decrease the
.
The rates of all photoionization, Auger decay, radiative processes and secondary ionization can be included by with the appropriate cross-sections that can be found in the literature (see Ref. in [
19]) or calculated from atomic code [
22,
24], as described above. Other inputs to the rate equations model are: (i) the beam parameters, such as size and intensity; (ii) elemental composition of the sample and (iii) the particle size. The particle size is typically used in the estimation of the energy required to escape the particle. We will also use it to define the density of trapped unbound electrons, which will be required for the MD simulations.
The trapped electron gas is typically assumed to be a Maxwell-Boltzmann distribution. The total energy of the gas is tracked as a function time and defines the temperature of the gas. The electron gas temperature can then be used to solve for the temperature of the ions as a function of time [
26]. It is assumed that electrons added to the gas are instantly thermalized, which may lead to an overestimation of the secondary ionization caused by the gas [
23].
The two key parameters that we extract from the REM are the density and temperature of the trapped electron gas. These parameters are need to define the extent of screening force between the ions.
4. Theory: Molecular Dynamics Using Plasma Parameters
The trajectories of the ions are subject to forces
, that are calculated from the effective two-body potentials,
, using
These potentials can include Coulomb repulsion forces, short-range collision potentials, bonding potentials or any other relevant pairwise potentials.
Here, we focus primarily on modifying the Coulomb term with an electrostatic screening factor to account for the effect of the trapped electron gas. We also include a short-range collision potential to avoid unphysically small ion separation distances. In most XFEL damage simulations, bonding interactions are ignored because their effect is assumed to be diminished when the sample becomes highly ionized. Moreover, the form and magnitude of bonding effects in a simple two-body form are not known in detail when the sample is a highly ionized complex molecule. In the results presented here, we do not include bonding effects.
The screened electrostatic interaction (in atomic units) between the ions labelled
i and
j is of the conventional Debye form,
where
is the charge of ion
i at time
t provided by the quantum-mechanical calculation detailed in
Section 2. The parameter
is the distance between the two atoms. The parameter
is the Debye length, which is calculated from the temperature and density of the trapped electron gas present at time
t during the interaction, with its time dependence determined by the REM calculation. The effect of the Debye parameter is to attenuate pairwise Coulomb interactions that arise over distances greater than
. It is utilized as an input parameter in the molecular dynamics simulation that follows as a separate step in the calculation.
The accuracy of the plasma/MD model lies in the validity of using Debye screened two-body potentials to capture the effect of the trapped electrons on the ion motion [
27]. The Debye term is the analytic solution of the Poisson equation for an initially uniform electron density in the presence of an ion. The validity of this form in a dense sample is quantified by the plasma parameter
The use of pairwise screened potentials is valid in the weak-coupling regime where
. We calculate the plasma parameter in our simulations as a check on the validity of Equation (
6) in each calculation.
An ionic collision potential may be employed to exclude an unphysically close approach of two ions. In our model this potential takes the form
The parameters are the effective radii of the interacting ionic species. The ionic collision potential may be adjusted through the parameters and to modify the strength of the collisional interaction.
The trajectories of the ions are calculated by solving coupled classical equations of motion using standard Runge-Kutta integration. To focus on a region of interest in the protein crystal and to reduce computational labour, a spherical simulation target region may be defined centred on an atom of interest. All atoms within this spherical region are free to move, while all other atoms in the unit cell are fixed. The electrostatic interactions that act on the target atoms are calculated by summing periodic replications of the unit cell. At the start of the simulation, the Debye screening length is assumed to be infinitely large, but rapidly reduces as the sample ionizes. When the Debye screening become significant, reduces to atomic length scales and the number of atoms applying a significant force to a specified target atom reduces. As a consequence, far fewer Coulomb interactions need to be calculated, greatly reduceing the time required to complete the simulation.
5. Results
Disulfide bonds are found in a large number of biological molecules and are among the simplest radiation sensitive complexes. The dynamics of sulfur-sulfur bonds under an XFEL pulse illumination has been investigated recently [
23] in an X-ray pump/X-ray probe serial femtosecond crystallography (SFX) experiment. Here we apply our model to evaluate the dependence of observed S-S bond lengths in nano-crystalline sample of lysozyme illuminated with three XFEL pulses that have the same total fluence but different pulse widths. We simulated a hydrated lysozyme (PDB code: 4ET8) unit cell with dimensions of
Å which contains 9491 non-hydrogen atoms that include 80 sulfur atoms, 64 of which form the S-S bonds of interest. We considered incident XFEL pulses with a total fluence of
J/cm
, a photon energy of 8 keV and widths (FWHM) of 5, 15 and 30 femtoseconds (fs). All three pulses were assumed to have a Gaussian temporal profile. Each simulation spanned a total time equal to 4 pulse widths with a peak intensity that was reached in the middle of the time interval. Re-scaling the time in each simulation by the inverse of the corresponding XFEL pulse width yields identical temporal pulse profiles. This allows us to compare the sulfur atom separations at different stages of the incident pulse with pulses of different widths; these results are depicted in
Figure 1.
The green, blue and red dashed lines represent the average separation between sulfur atoms that initially formed S-S bonds, for incident pulses of 5, 15 and 30 fs, respectively. The shaded areas correspond to sulfur-sulfur distances one standard deviation above and below the average value. The variation in bond distances for different sulfur pairs originates from differences in the surrounding biomolecular structure, with higher density regions retarding the motions of the sulfur atoms. The observed S-S bond distance is a weighted average of the bond distances throughout the pulse. The weights are determined by the time-dependent scattering factors and the instantaneous pulse intensity. For simplicity, we approximate the average bond distance by the value of sulfur-sulfur separation taken at −0.25 pulse widths from the peak. A bias towards the earlier stages of the pulse is justified by the reduction in the number of bound electrons capable of coherently scattering X-rays [
25] and a damage gating effect caused by the breakdown of crystalline periodicity when atomic motion becomes significant [
7]. A detailed quantitative analysis of these effects will be the topic of future studies.
The approximate pulse-averaged sulfur-sulfur separations are Å, Å and Å for 5, 15 and 30 fs pulses respectively. The undamaged average bond length at the start of the pulse is Å. For 30 fs, the S-S bond elongation induced by the radiation damage is of the order of 25% for the 30 fs pulse, but only 3% for a 15 fs pulse and ∼0.1% for a 5 fs pulse. The average charges of sulfur ions at the same time point (−0.25 pulse width) are +2.23, +3.12 and +3.74 for 5, 15 and 30 fs pulses, respectively. During shorter pulses the Coulomb repulsion forces tend to be weaker and the atoms have less time to move during the exposure. This inverse dependence of total ionic charge on the pulse length is largely caused by the Auger decay half-life, which is 5–10 fs in light elements. As these second row elements are the major source of secondary electrons, a short pulse leads less secondary electron impact ionization. The simulations demonstrate that for XFEL pulses under 15 fs one can expect to observe only a marginal S-S bond disintegration for a given value of incident fluence.
6. Discussion
The hybrid plasma/MD model presented above makes possible ab initio simulations of XFEL imaging of protein nano-crystals that contain in excess of
atoms in a unit cell. Computational efficiency is achieved by adopting a plasma description of the free electron density and the consequent attenuation of electrostatic interactions between ions in the MD simulation due to the Debye screening mechanism. The combined computation time of all three simulations presented above was under 10 h on a desktop computer with Intel Core i7-9800X 4.4 GHz CPU (14/16 threads utilized) and 64 Gb of DDR4 memory. The conventional MC/MD approach to XFEL pulse-sample interaction treats free electrons as a point-like classical particles, with each atom (except hydrogen atoms) emitting at least 2–3 electrons during the pulse of a moderate fluence. This increases the number of particles that must be treated explicitly inside a unit cell. Free electrons have velocities that are
times higher than ions. The accurate description of their motions requires, therefore, a proportionally smaller time step in free-electron and ion MD simulation approaches, compared to ion-only simulation. When the plasma parameter
, a free electron gas is polarized in the vicinity of positive ions in such a way that a time-averaged net charge inside a sphere with an ion at its centre exponentially approaches zero as the radius of the sphere is increased. The Debye screening effect naturally introduces a cutoff distance for the interactions between the ions, beyond which the screened Coulomb repulsion forces between ions are negligible and can be ignored. A similar approach, an Ewald summation [
28], is routinely used in conventional MD simulations, where a calculation of a force acting on the atom is carried out in a real space for all atoms within a sphere of a cutoff radius and in a Fourier space for atoms located beyond the sphere.
Two key assumptions that underpin our model are the weak coupling regime for the free electron gas and the independence of the time-dependent ionic charge on the positions of the atoms in a unit cell. The first assumption of weak coupling enables the averaging of the electron gas distribution in the vicinity of an ion which yields a Debye shielding factor for the inter-ionic potential [
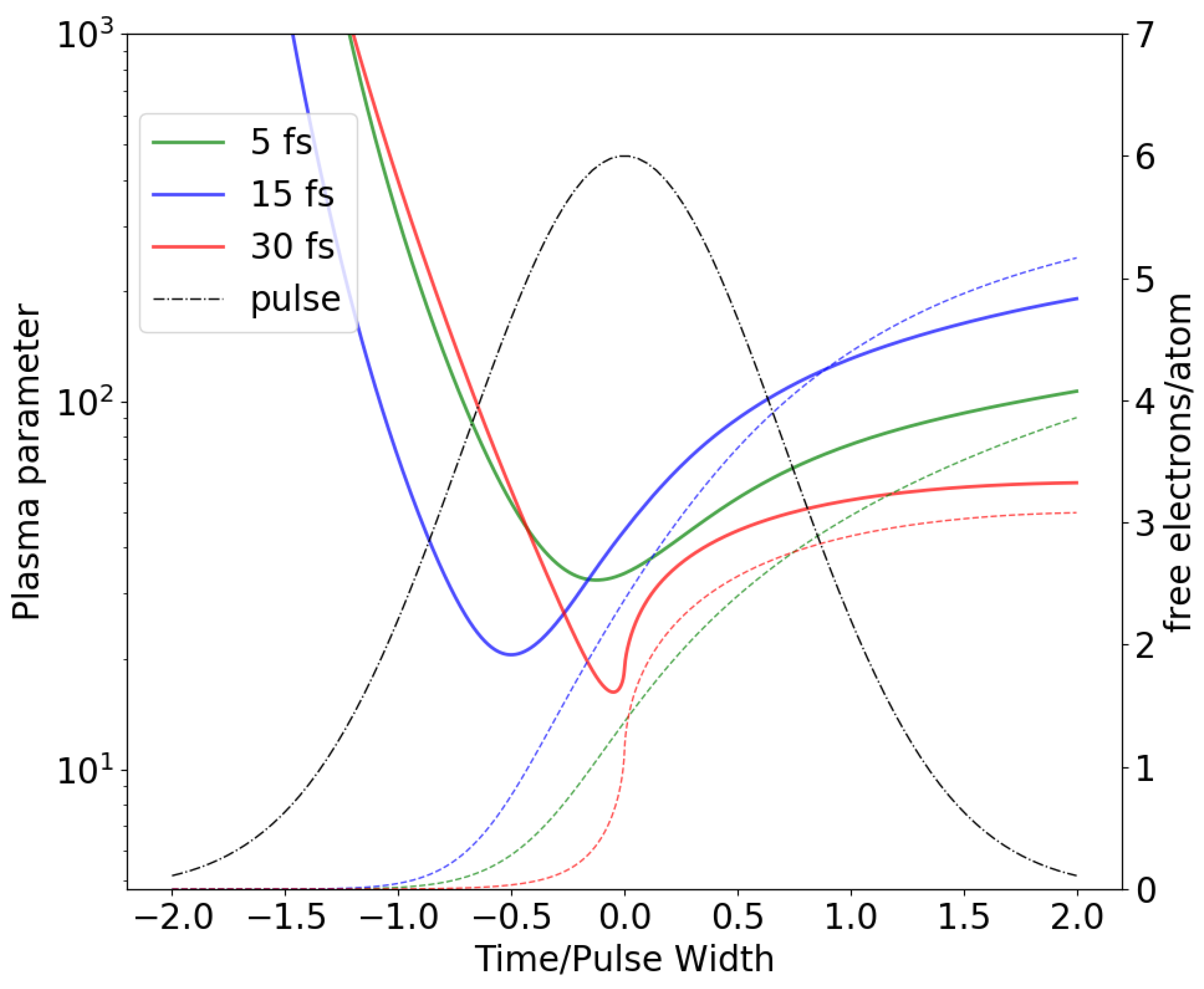
27]. It is evident that at the early stages of the pulse, when a number of free electrons and the degree of ionization are both small, such averaging is not valid. However, for this early stage of an incident XFEL pulse Coulomb forces are unable to cause any significant motion of atoms. Shortly after the start of the pulse, when a number of free electrons in a unit cell becomes comparable to the number of atoms, a continuum description of free electron gas becomes valid. In all presented simulations this time point occurs at least a half pulse width before the pulse peak. The plasma parameter as a function of time, re-scaled by the pulse width, is depicted in
Figure 2. The smallest values of the plasma parameter throughout the pulse was
(5 fs),
(15 fs) and
(30 fs) which corresponds to the weak coupling regime (
) for the secondary electron plasma. The second assumption, an independence of a time-dependent ionic charges on atomic position, is a direct consequence of an independent atom approximation to ionization dynamics in both the plasma/MD model as well as conventional MC/MD. It is straightforward to show that the expected value of a force acting between any two atoms is determined by the expectation values of their charges under an independent atom approximation. Even though atomic charges are independent under this assumption, they are correlated since they are created by the same incident pulse. This leads to an increase in the variance of observable quantities such as S-S bond distances depicted in
Figure 1 compared to those predicted by the plasma/MD model. The S-S bond distance variances predicted by the plasma/MD model should, as a consequence, be considered to be lower bounds.
The proposed plasma/MD model was used to investigate S-S bond elongation in a Lysozyme crystal in an X-ray pump—X-ray probe experiment [
23] as a function of time delay between two pulse. Comparison with a MC/MD simulation, carried out using XMDYN [
18], was found to be in a good agreement with plasma/MD model, although some minor discrepancies were observed. The discrepancies originate from the difference in samples used in both simulations. In XMDYN simulation only a small part of a Lysozyme unit cell (
Å with a total of 300 atoms) with periodic boundary conditions was used, while plasma/MD model was applied to an entire Lysozyme unit cell. Both models were in good agreement with experimental observations up to 50 fs delay between pump and probe pulses.



{kind=link}
{kind=link}

