Effect of Chitosan Electrospun Fiber Mesh as Template on the Crystallization of Calcium Oxalate

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Electrospun Chitosan Fibers as Organic Template
2.2. Electrocrystallization of CaOx
2.3. Characterization of Chitosan Meshes and CaOx Crystals
3. Results
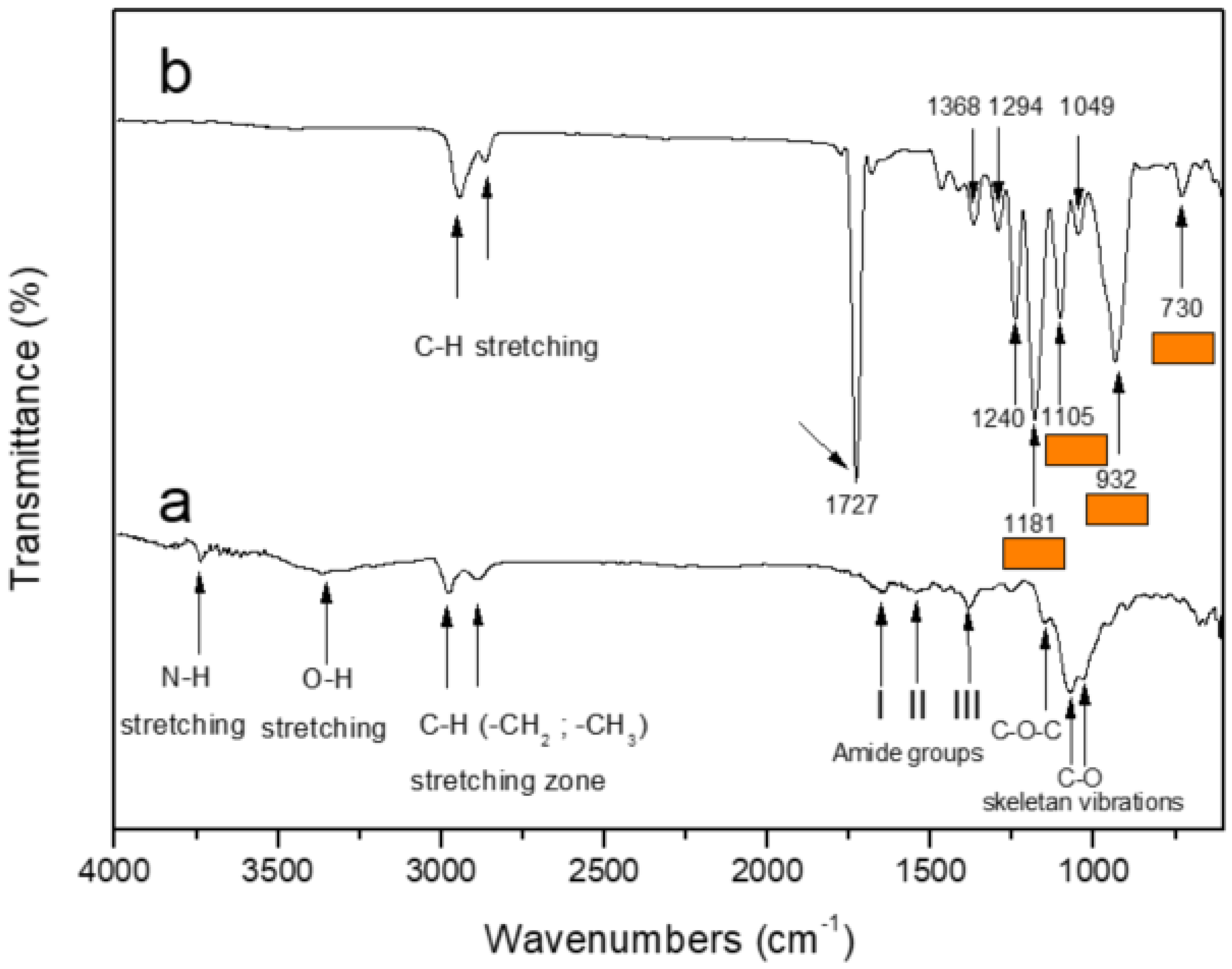
3.1. Fourier Transform Infrared Spectroscopy (FTIR)
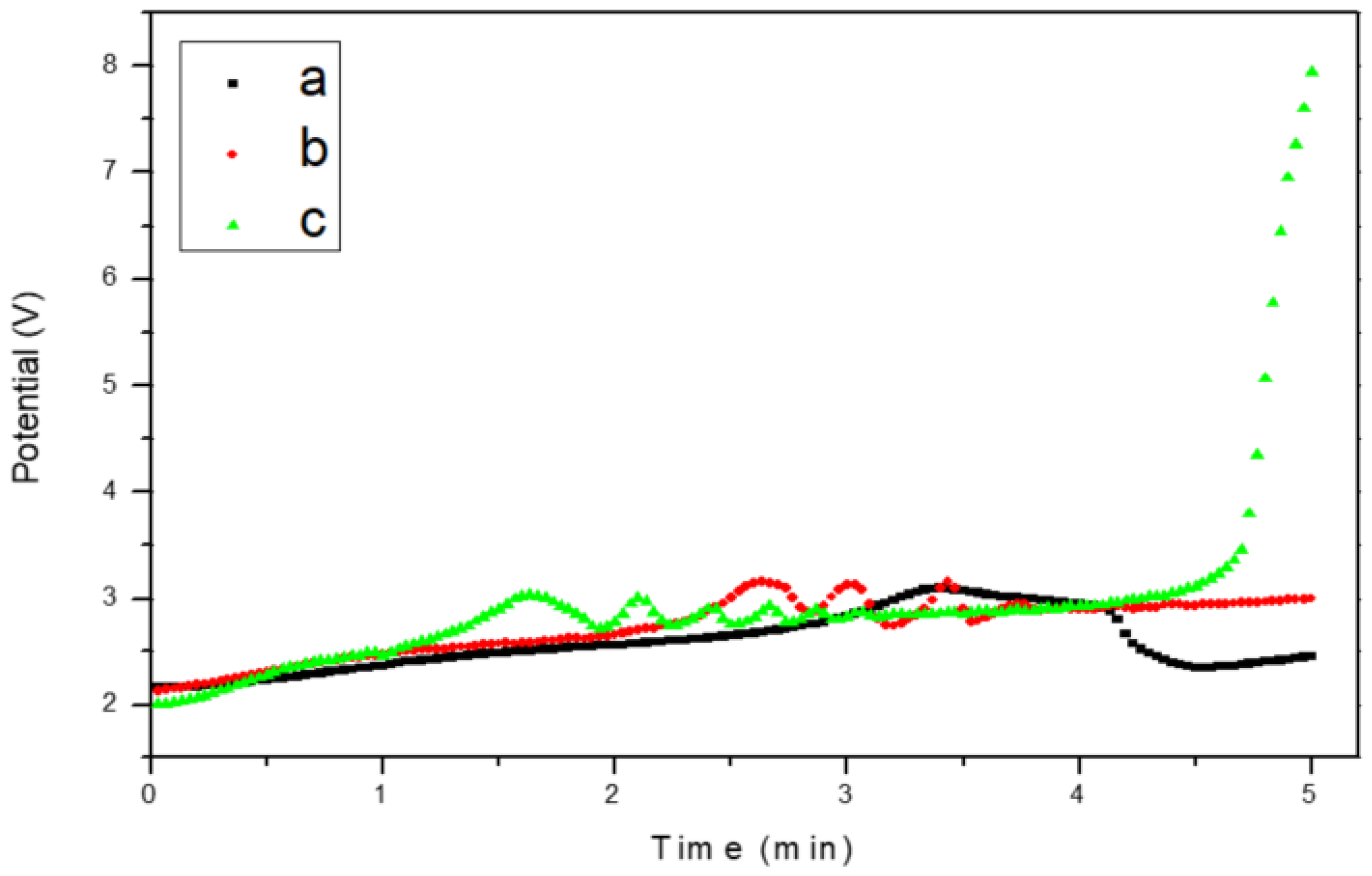
3.2. Chronopotentiometry
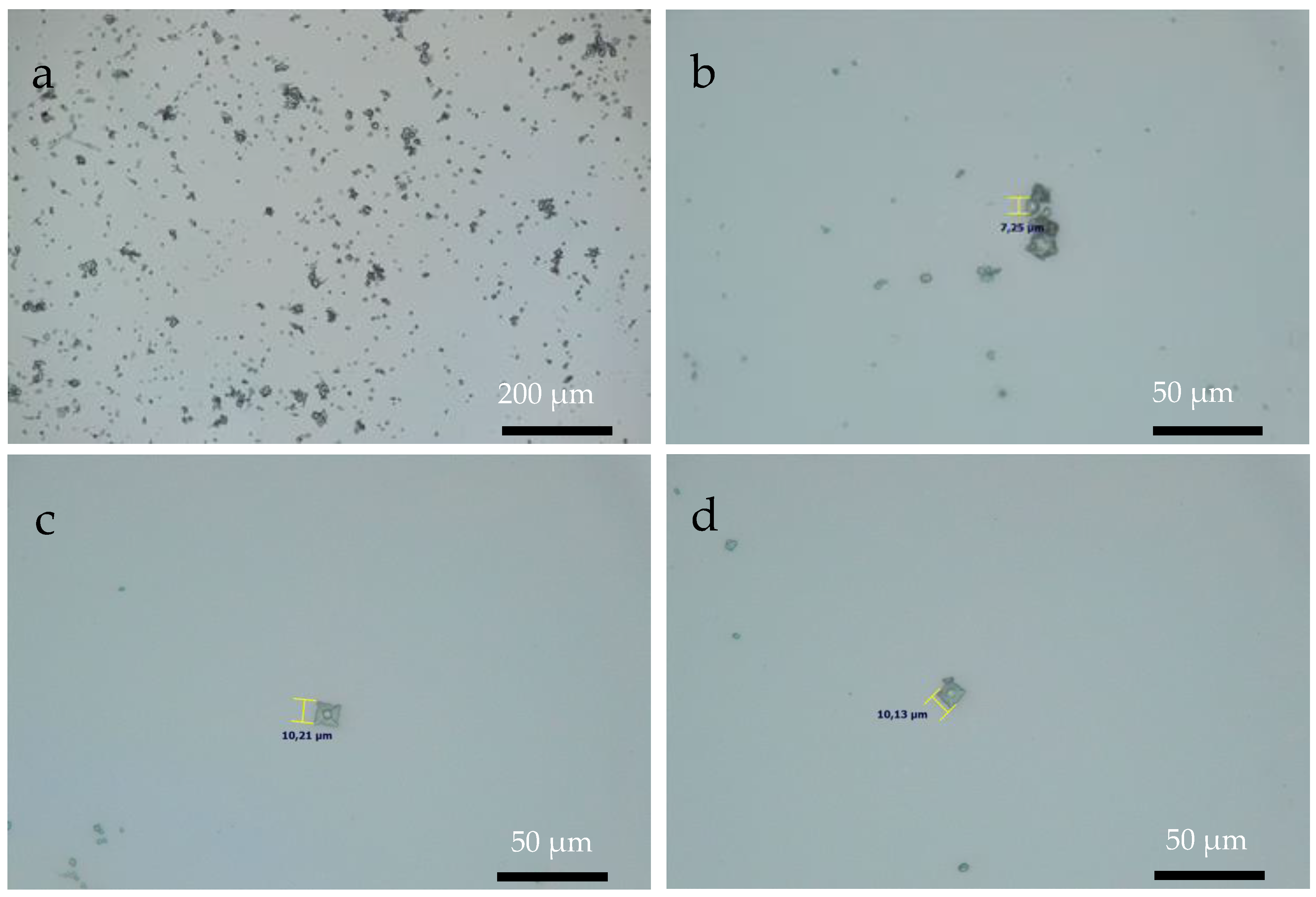
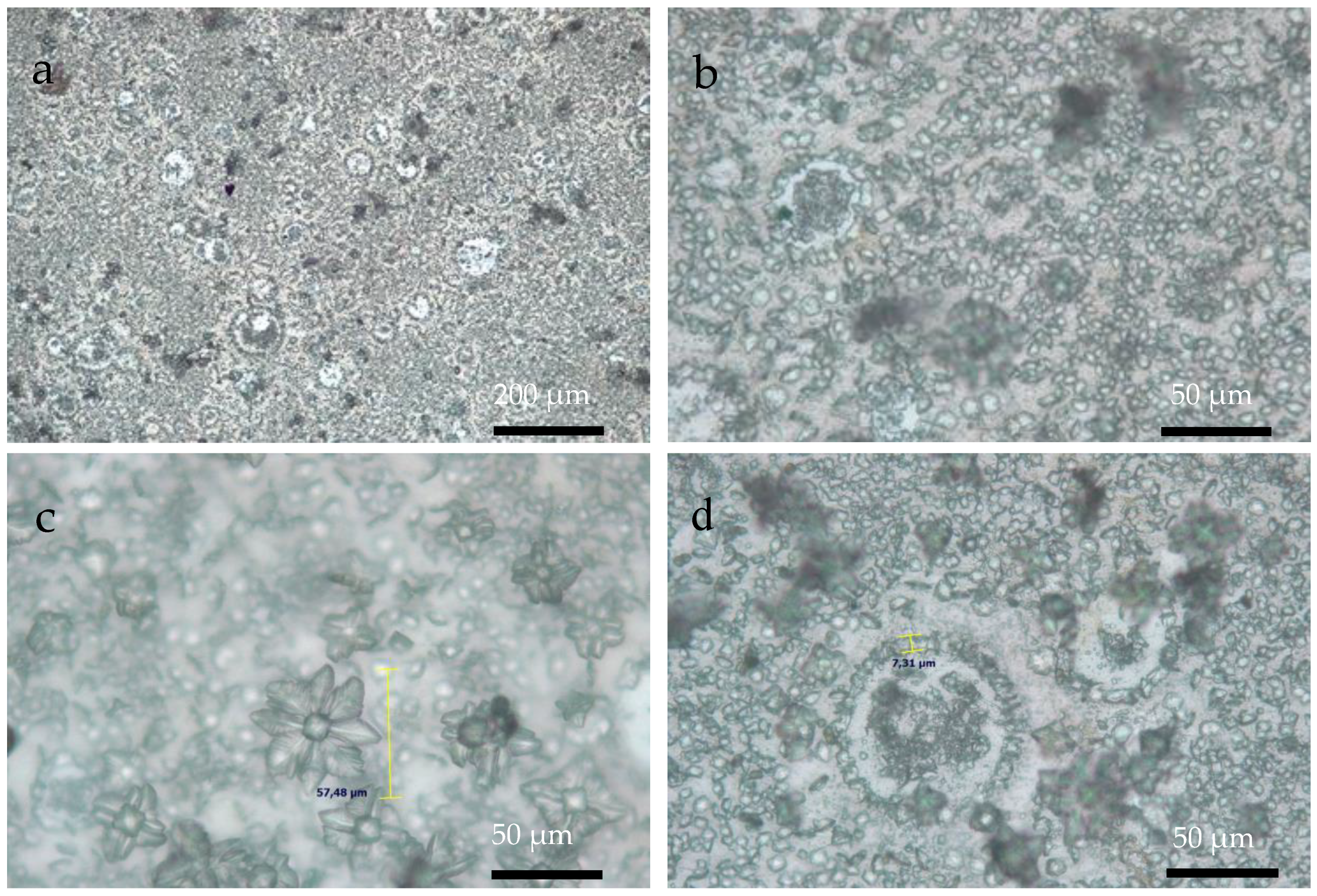
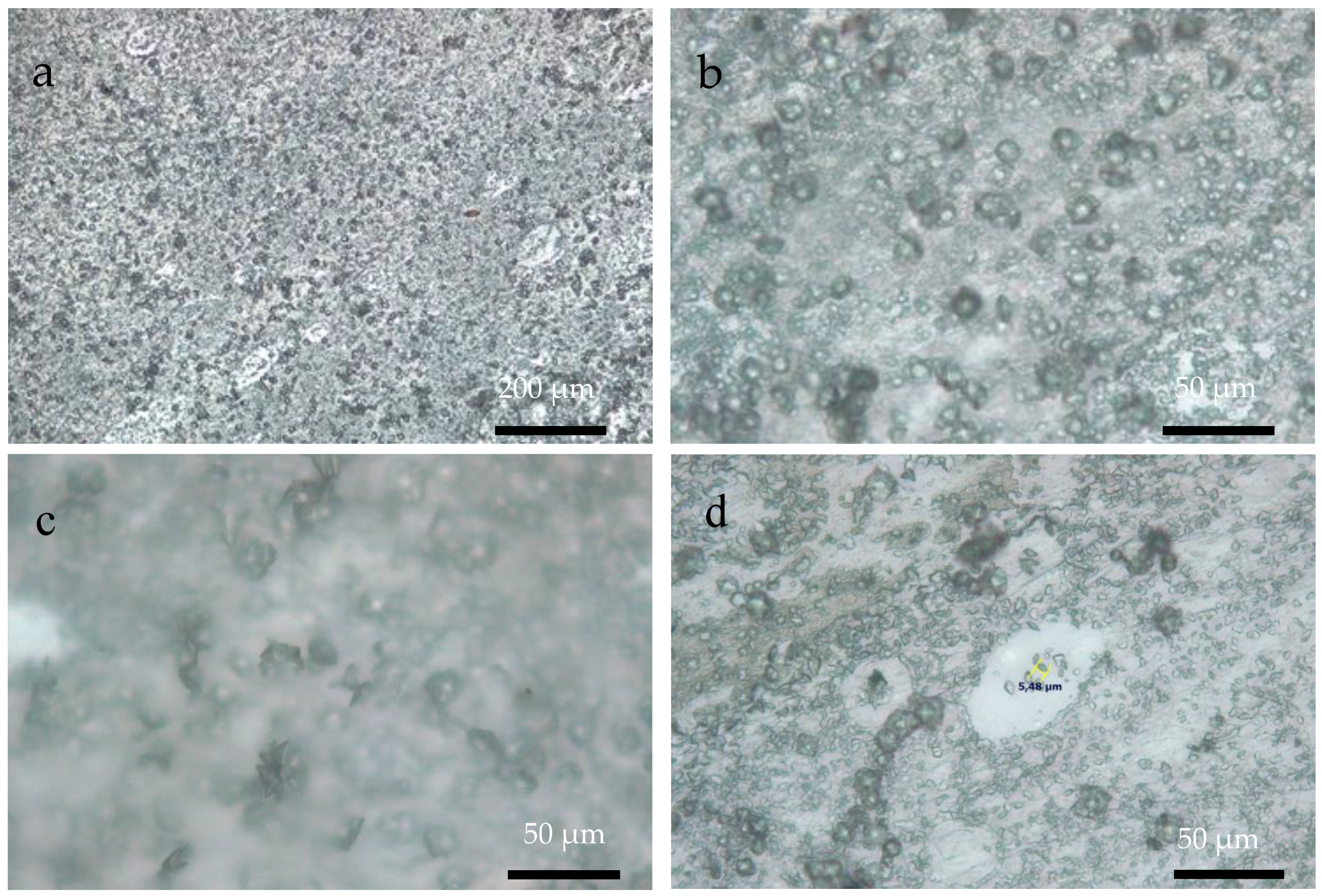
3.3. Optical Microscopy
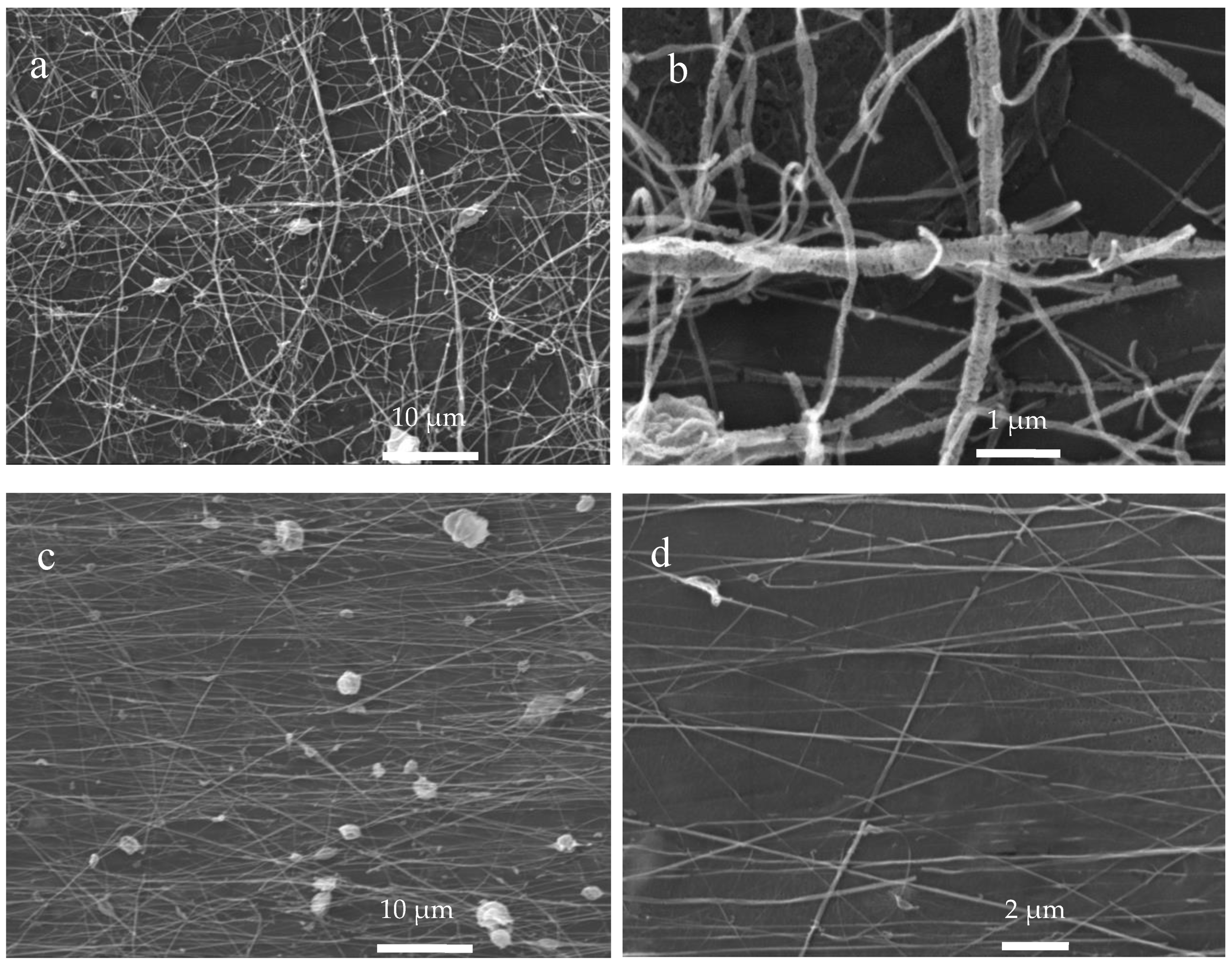
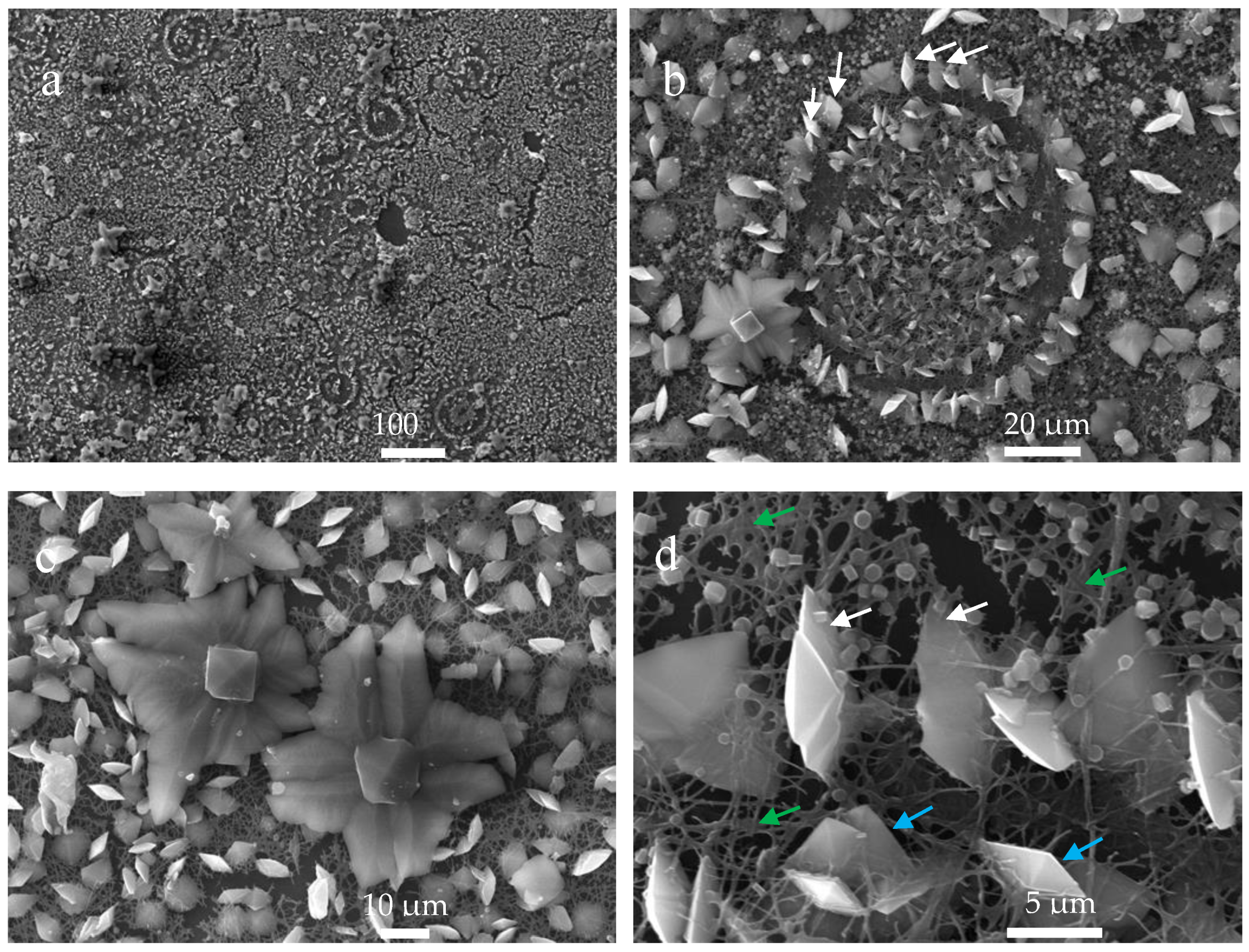
3.4. Scanning Electron Microscopy
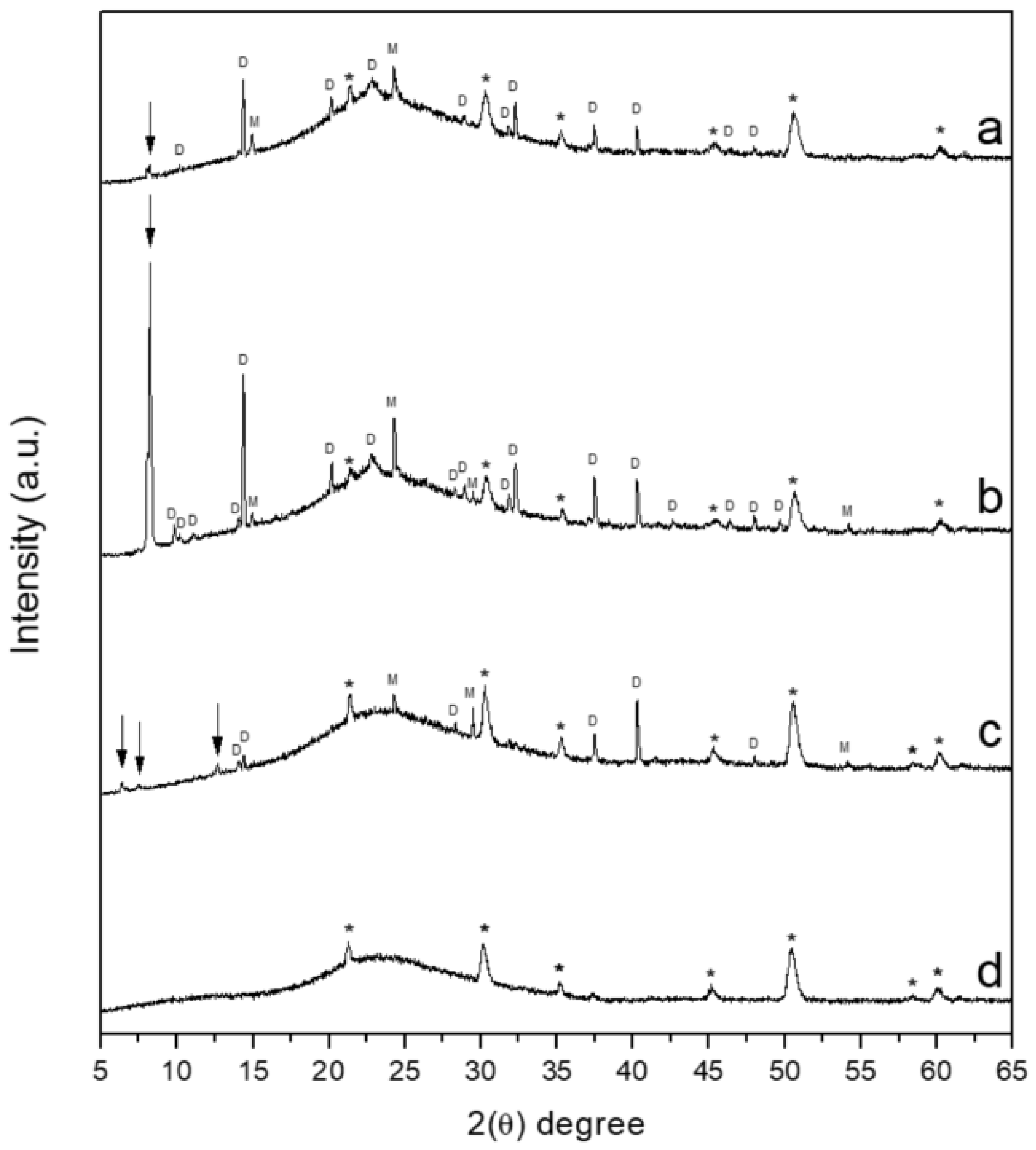
3.5. X-ray Diffraction (XRD)
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fratzl, P.; Kolednik, O.; Fischer, F.D.; Dean, M.N. The mechanics of tessellations–bioinspired strategies for fracture resistance. Chem. Soc. Rev. 2016, 45, 252–267. [Google Scholar] [PubMed]
- Demichelis, R.; Schuitemaker, A.; Garcia, N.A.; Koziara, K.B.; De La Pierre, M.; Raiteri, P.; Gale, J.D. Simulation of crystallization of biominerals. Ann. Rev. Mater. Res. 2018, 48, 327–352. [Google Scholar] [CrossRef]
- Weiner, S.; Dove, P.M. An overview of biomineralization processes and the problem of the vital effect. Rev. Mineral. Geochem. 2003, 54, 1–29. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Vásquez-Quitral, P.; Sánchez, M.; Vargas-Fernández, A.; Silva, J.F. Control of calcium oxalate morphology through electrocrystallization as an electrochemical approach for preventing pathological disease. Ionics 2015, 21, 3141–3149. [Google Scholar]
- Ruiz-Agudo, E.; Burgos-Cara, A.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Cölfen, H.; Rodriguez-Navarro, C. A non-classical view on calcium oxalate precipitation and the role of citrate. Nat. Commun. 2017, 8, 768. [Google Scholar] [CrossRef] [PubMed]
- Daudon, M.; Bazin, D.; Letavernier, E. Randall’s plaque as the origin of calcium oxalate kidney stones. Urolithiasis 2015, 43, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Ouyang, J.M.; Xu, M. Synthesis, characterization, and cytotoxicity assay of calcium oxalate dihydrate crystals in various shapes. CrystEngComm 2016, 18, 5463–5473. [Google Scholar] [CrossRef]
- Sánchez, M.; Vásquez-Quitral, P.; Butto, N.; Díaz-Soler, F.; Yazdani-Pedram, M.; Silva, J.F.; Neira-Carrillo, A. Effect of alginate from Chilean Lessonia nigrescens and MWCNTs on CaCO3 crystallization by classical and non-classical methods. Crystals 2018, 8, 69. [Google Scholar]
- Arias, J.L.; Fernández, M.S. Polysaccharides and proteoglycans in calcium carbonate-based biomineralization. Chem. Rev. 2008, 108, 4475–4482. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Yazdani-Pedram, M.; Retuert, J.; Diaz-Dosque, M.; Gallois, S.; Arias, J.L. Selective crystallization of calcium salts by poly (acrylate)-grafted chitosan. J. Colloid Interface Sci. 2005, 286, 134–141. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Fernández, M.S.; Hevia, G.P.; Arias, J.L.; Gebauer, D.; Cölfen, H. Retrosynthesis of CaCO3 via amorphous precursor particles using gastroliths of the Red Claw lobster (Cherax quadricarinatus). J. Struct. Biol. 2017, 199, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Younes, I.; Rinaudo, M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Zargar, V.; Asghari, M.; Dashti, A. A review on chitin and chitosan polymers: Structure, chemistry, solubility, derivatives, and applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Ohkawa, K.; Cha, D.; Kim, H.; Nishida, A.; Yamamoto, H. Electrospinning of Chitosan. Macromol. Rapid Commun. 2004, 25, 1600–1605. [Google Scholar] [CrossRef]
- Pham, Q.P.; Sharma, U.; Mikos, A.G. Electrospinning of polymeric nanofibers for tissue engineering applications: A review. Tissue Eng. 2006, 12, 1197–1211. [Google Scholar] [CrossRef]
- Rodríguez-Vásquez, M.; Vega-Ruiz, B.; Ramos-Zúñiga, R.; Saldaña-Koppel, D.A.; Quiñones-Olvera, L.F. Chitosan and its potential use as a scaffold for tissue engineering in regenerative medicine. BioMed Res. Int. 2015, 2015, 15. [Google Scholar] [CrossRef]
- Qasim, S.B.; Zafar, M.S.; Najeeb, S.; Khurshid, Z.; Shah, A.H.; Husain, S.; Rehman, I.U. Electrospinning of chitosan-based solutions for tissue engineering and regenerative medicine. Int. J. Mol. Sci. 2018, 19, 407. [Google Scholar] [CrossRef]
- Küther, J.; Seshadri, R.; Knoll, W.; Tremel, W. Templated growth of calcite, vaterite and aragonite crystals onself-assembled monolayers of substituted alkylthiols on gold. J. Mater. Chem. 1998, 8, 641–650. [Google Scholar] [CrossRef]
- Chun, Y.W.; Khang, D.; Haberstroh, K.M.; Webster, T.J. The role of polymer nanosurface roughness and submicron pores in improving bladder urothelial cell density and inhibiting calcium oxalate stone formation. Nanotechnology 2009, 20, 085104. [Google Scholar] [CrossRef]
- Fernandes Queiroz, M.; Melo, K.; Sabry, D.; Sassaki, G.; Rocha, H. Does the use of chitosan contribute to oxalate kidney stone formation? Mar. Drugs 2015, 13, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wang, D.; Lyu, L.; Gong, Y.; Li, Y. Fabrication and characterization of PCL/CaCO3 electrospun composite membrane for bone repair. RSC Adv. 2016, 6, 10641–10649. [Google Scholar] [CrossRef]
- Fujihara, K.; Kotaki, M.; Ramakrishna, S. Guided bone regeneration membrane made of polycaprolactone/calcium carbonate composite nano-fibers. Biomaterials 2005, 26, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Reneker, D.H.; Chun, I. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnology 1996, 7, 216–223. [Google Scholar] [CrossRef]
- Burger, C.; Hsiao, B.S.; Chu, B. Nanofibrous materials and their applications. Ann. Rev. Mater. Res. 2006, 36, 333–368. [Google Scholar] [CrossRef]
- Kishan, A.P.; Cosgriff-Hernandez, E.M. Recent advancements in electrospinning design for tissue engineering applications: A review. J. Biomed. Mater. Res. Part A 2017, 105, 2892–2905. [Google Scholar] [CrossRef]
- Xiao, Y.; Shen, M.; Shi, X. Design of functional electrospun nanofibers for cancer cell capture applications. J. Mater. Chem. B 2018, 6, 1420–1432. [Google Scholar] [CrossRef]
- Joseph, S.; Kamath, P.V. Synthesis of different polymorphic modifications of calcium oxalate by electrodeposition. J. Solid State Electrochem. 2010, 14, 1481–1486. [Google Scholar] [CrossRef]
- Dinamani, M.; Kamath, P.V.; Seshadri, R. Deposition of oriented SrSO4 coatings by electrogeneration of acid. Solid State Sci. 2003, 5, 805–810. [Google Scholar] [CrossRef]
- Kameda, T.; Kojima, K.; Miyazawa, M.; Fujiwara, S. Film Formation and Structural Characterization of Silk of the Hornet Vespa simillima xanthoptera Cameron. Z. Naturforschung C 2005, 60, 906–914. [Google Scholar] [CrossRef]
- Ketrane, R.; Saidani, B.; Gil, O.; Leleyter, L.; Baraud, F. Efficiency of five scale inhibitors on calcium carbonate precipitation from hard water: Effect of temperature and concentration. Desalination 2009, 249, 1397–1404. [Google Scholar] [CrossRef]
- Jáuregui-Zúñiga, D.; Reyes-Grajeda, J.P.; Sepúlveda-Sánchez, J.D.; Whitaker, J.R.; Moreno, A. Crystallochemical characterization of calcium oxalate crystals isolated from seed coats of Phaseolus vulgaris and leaves of Vitis vinifera. J. Plan Physiol. 2003, 160, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.; Mathew, A.P.; Girandon, L.; Fröhlich, M.; Oksman, K. Porous electrospun nanocomposite mats based on chitosan–cellulose nanocrystals for wound dressing: Effect of surface characteristics of nanocrystals. Cellulose 2015, 22, 521–534. [Google Scholar] [CrossRef]
- Mo, X.; Chen, Z.; Weber, H.J. Electrospun nanofibers of collagen-chitosan and P (LLA-CL) for tissue engineering. Front. Mater. Sci. China 2007, 1, 20–23. [Google Scholar] [CrossRef]
- Ohkawa, K.; Minato, K.I.; Kumagai, G.; Hayashi, S.; Yamamoto, H. Chitosan nanofiber. Biomacromolecules 2006, 7, 3291–3294. [Google Scholar] [CrossRef]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar] [CrossRef]
- Rajkhowa, R.; Gil, E.S.; Kluge, J.; Numata, K.; Wang, L.; Wang, X.; Kaplan, D.L. Reinforcing Silk Scaffolds with Silk Particles. Macromol. Biosci. 2010, 10, 599–611. [Google Scholar] [CrossRef]
- Homayoni, H.; Ravandi, S.A.H.; Valizadeh, M. Electrospinning of chitosan nanofibers: Processing optimization. Carbohydr. Polym. 2009, 77, 656–661. [Google Scholar] [CrossRef]
- Pillai, C.K.S.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Mirjalili, M.; Zohoori, S. Review for application of electrospinning and electrospun nanofibers technology in textile industry. J. Nanostruct. Chem. 2016, 6, 207–213. [Google Scholar] [CrossRef]
- Ostwald, W. Studien über die Umwandlung fester körper. Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Szewczyk, P.K.; Metwally, S.; Karbowniczek, J.E.; Marzec, M.M.; Stodolak-Zych, E.; Gruszczyński, A.; Bernasik, A.; Stachewicz, U. Surface-potential-controlled cell proliferation and collagen mineralization on electrospun polyvinylidene fluoride (PVDF) fiber scaffolds for bone regeneration. ACS Biomater. Sci. Eng. 2018, 5, 582–593. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butto, N.; Cotrina Vera, N.; Díaz-Soler, F.; Yazdani-Pedram, M.; Neira-Carrillo, A. Effect of Chitosan Electrospun Fiber Mesh as Template on the Crystallization of Calcium Oxalate. Crystals 2020, 10, 453. https://doi.org/10.3390/cryst10060453
Butto N, Cotrina Vera N, Díaz-Soler F, Yazdani-Pedram M, Neira-Carrillo A. Effect of Chitosan Electrospun Fiber Mesh as Template on the Crystallization of Calcium Oxalate. Crystals. 2020; 10(6):453. https://doi.org/10.3390/cryst10060453
Chicago/Turabian StyleButto, Nicole, Nicole Cotrina Vera, Felipe Díaz-Soler, Mehrdad Yazdani-Pedram, and Andrónico Neira-Carrillo. 2020. "Effect of Chitosan Electrospun Fiber Mesh as Template on the Crystallization of Calcium Oxalate" Crystals 10, no. 6: 453. https://doi.org/10.3390/cryst10060453
APA StyleButto, N., Cotrina Vera, N., Díaz-Soler, F., Yazdani-Pedram, M., & Neira-Carrillo, A. (2020). Effect of Chitosan Electrospun Fiber Mesh as Template on the Crystallization of Calcium Oxalate. Crystals, 10(6), 453. https://doi.org/10.3390/cryst10060453