Insight into Positional Isomerism of N-(Benzo[d]thiazol-2-yl)-o/m/p-Nitrobenzamide: Crystal Structure, Hirshfeld Surface Analysis and Interaction Energy


Abstract
1. Introduction
2. Materials and Methods
2.1. General Experiment
2.2. Synthetic Procedures
2.3. Single Crystal X-ray Diffraction Studies
2.4. Computational Methods
3. Results
3.1. Photophysical Properties
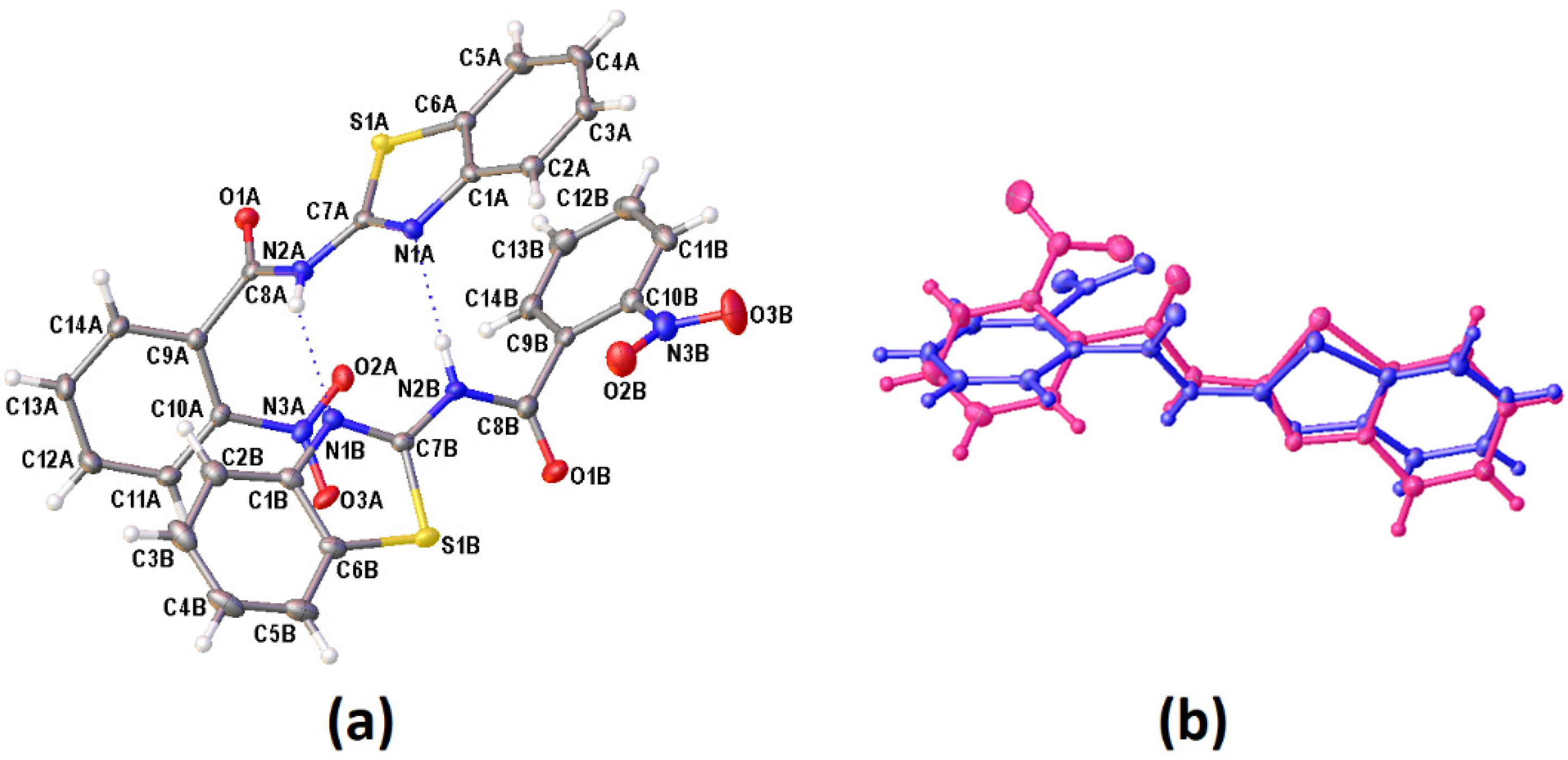
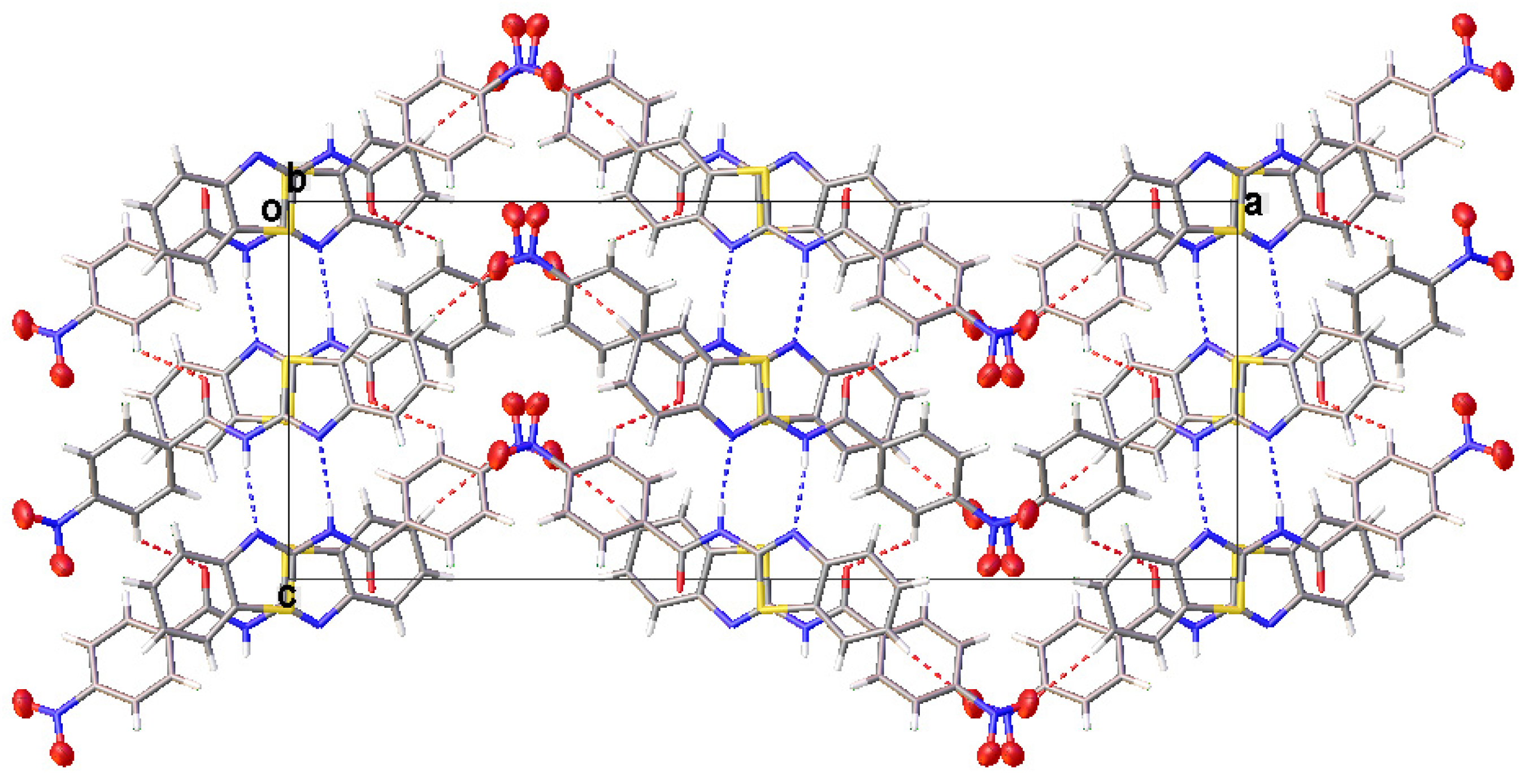
3.2. Crystal Structural Descriptions
3.3. Hirshfeld Surface Analysis
3.4. Interaction Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dadmal, T.L.; Katre, S.D.; Mandewale, M.C.; Kumbhare, R.M. Contemporary Progress in the Synthesis and Reactions of 2-Aminobenzothiazole: A review. New J. Chem. 2018, 42, 776–797. [Google Scholar] [CrossRef]
- Malik, S.; Jain, B.; Mirza, A.S.; Singh, A. Review Study on Metal Complexes Synthesized from Therapeutically Important Schiff. Pharm. Chem. J. 2016, 3, 119–124. [Google Scholar]
- Cuevas-Hernández, R.I.; Padilla-Martínez, I.I.; Martínez-Cerón, S.; Vásquez-Moctezuma, I.; Trujillo-Ferrara, J.G. Helical Arrangement of 2-(4-Hydroxy-3-Methoxyphenyl)-Benzothiazole in Crystal Formation and Biological Evaluation on HeLa Cells. Crystals 2017, 7, 171. [Google Scholar] [CrossRef]
- Shiran, J.A.; Yahyazadeh, A.; Yamin, B.M.; Mamaghani, M.; Kiyani, H. Three-component synthesis of new substituted bis[2-imino-3-(substituted)-4-phenyl-3H-thiazole] derivatives and evaluation of their antibacterial activity. Heterocycles 2015, 91, 123–133. [Google Scholar] [CrossRef]
- Sarau Devi, A.; Aswathy, V.V.; Sheena Mary, Y.; Yohannan Panicker, C.; Armaković, S.; Armaković, S.J.; Ravindran, R.; Van Alsenoy, C. Synthesis, XRD Single Crystal Structure Analysis, Vibrational Spectral Analysis, Molecular Dynamics and Molecular Docking Studies of 2-(3-Methoxy-4-Hydroxyphenyl)Benzothiazole. J. Mol. Struct. 2017, 1148, 282–292. [Google Scholar] [CrossRef]
- Swetha, T.; Akhtaruzzaman, M.; Chowdhury, T.H.; Amin, N.; Islam, A.; Noda, T.; Upadhyaya, H.M. Benzodithiazole-Based Hole-Transporting Material for Efficient Perovskite Solar Cells. Asian J. Org. Chem. 2018, 7, 2497–2503. [Google Scholar] [CrossRef]
- Gautam, P.; Maragani, R.; Misra, R. Tuning the HOMO-LUMO Gap of Donor-substituted Benzothiazoles. Tetrahedron Lett. 2014, 55, 6827–6830. [Google Scholar] [CrossRef]
- Gudasi, K.B.; Patil, S.A.; Vadavi, R.S.; Shenoy, R.V.; Patil, M.S. Synthesis and Spectral Studies of Cu(II), Ni(II), Co(II), Mn(II), Zn(II) and Cd(II) Complexes of a New Macroacyclic ligand N,N′-Bis(2-Benzothiazolyl)-2,6-Pyridinedicarboxamide. J. Serb. Chem. Soc. 2006, 71, 529–542. [Google Scholar] [CrossRef]
- Luo, C.; Chen, H.; Cao, K.; Wang, W.; Gao, J.; Jia, J. Diode Effect Caused by Sequence of Benzothiazole on Optical Properties. Tetrahedron 2017, 73, 3066–3073. [Google Scholar] [CrossRef]
- Rittmeyer, S.P.; Groß, A. Structural and Electronic Properties of Oligo- and Polythiophenes Modified by Substituents. Beilstein J. Nanotechnol. 2012, 3, 909–919. [Google Scholar] [CrossRef]
- Diana, R.; Caruso, U.; Tuzi, A.; Panunzi, B. A Highly Water-Soluble Fluorescent and Colorimetric pH Probe. Crystals 2020, 10, 83. [Google Scholar] [CrossRef]
- Zainal, W.; Hafizi, M.; Mark-Lee, W.F.; Tahir, S.M.; Ahmad, I.B.; Kassim, M.B. Experimental and DFT investigation on the influence of electron donor/acceptor on the hydrogen bonding interactions of 1-(1,3-benzothiazol-2-y1)-3-(R-benzoylthiourea). Sains Malaysiana 2018, 47, 923–929. [Google Scholar] [CrossRef]
- Minasyan, Y.V.; Degtyarenko, A.I.; Kosmacheva, K.D.; Plekhovich, S.D.; Zelentsov, S.V. Effect of Acceptor and Donor Substituents in The ortho, meta, and para Positions in the Nitrobenzene Molecule on the Reaction of Interaction with Ethylene. Proceedings 2018, 9, 5770. [Google Scholar] [CrossRef]
- Usha, H.; Ravindra, H.J. Effect of Position of Electron Acceptor Group Substitution on NLO Properties of Chalcones Derivatives: A DFT study. Int. J. Sci. Eng. Res. 2018, 9, 2–5. [Google Scholar]
- Fragoso-Medina, A.J.; Escobedo-González, R.G.; Nicolás-Vázquez, M.I.; Arroyo-Razo, G.A.; Noguez-Córdova, M.O.; Miranda-Ruvalcaba, R. A DFT Study of the Geometrical, Spectroscopical and Reactivity Properties of Diindolylmethane-Phenylboronic Acid Hybrids. Molecules 2017, 22, 1744. [Google Scholar] [CrossRef] [PubMed]
- Ćaleta, I.; Cinčić, D.; Karminski-Zamola, G.; Kaitner, B. The Synthesis and Structure of Two Novel N-(Benzothiazol-2-yl)Benzamides. J. Chem. Crystallogr. 2008, 38, 775–780. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GAUSSIAN09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17.5; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Liu, D.; Yao, R.; Dong, R.; Jia, F.; Fu, M. A Strategy to Increase Phosphorescent Efficiency without Perturbing Emission Color for Benzothiazole-Containing Iridium Phosphors. Dye. Pigment 2017, 145, 528–537. [Google Scholar] [CrossRef]
- Alias, M.F.; Jaafer, F.S.; Sadiq, A.S. Synthesis Mixed Ligands Complexes of Some Light Metals by Microwave Assisted Using 2-Acetamide Benzothiazole and 1,10-Phenanthrolin. Int J. Pharm 2015, 5, 451–458. [Google Scholar]
- Mark-Lee, W.F.; Md Nasir, M.F.; Kassim, M.B. Structural and optical properties investigation on H-bonded 1D helical self-assembly of 1,1-dibenzyl-3-(2-bromobenzoyl)thiourea molecules for nonlinear optical application. Sains Malaysiana 2018, 47, 741–747. [Google Scholar] [CrossRef]
- Roacho, R.I.; Metta-magaña, A.J.; Peña-cabrera, E.; Pannell, K.H. Synthesis, Structural Characterization, and Spectroscopic Properties of the ortho, meta, and para isomers of 8-(HOCH2-C6H4)-BODIPY and 8-(MeOC6H4)-BODIPY. J. Phys. Org. Chem. 2013, 26, 345–351. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Mark-Lee, W.F.; Chong, Y.Y.; Kassim, M.B. Supramolecular Structures of Rhenium(I) Complexes Mediated by Ligand Planarity via The Interplay of Substituents. Acta Cryst. C 2018, 74, 997–1006. [Google Scholar] [CrossRef]
- Janiak, C. A Critical Account on π-π Stacking in Metal Complexes with Aromatic Nitrogen-Containing Ligands. J. Chem. Soc. Dalton Trans. 2000, 21, 3885–3896. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld Surface Calculations, Non-Covalent Interaction (NCI) Plots and the Calculation of Interaction Energies in the Analysis of Molecular Packing. Acta Cryst. E 2019, 75, 308–318. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy Frameworks: Insights into Interaction Anisotropy and the Mechanical Properties of Molecular Crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Absorption | Emission | ||
|---|---|---|---|---|
| λmax (nm) | εa,b | λmax (nm) | Quantum Yieldc (Φ) | |
| I | 271 | 2.16 | 380 | 0.56 |
| II | 303 | 1.48 | 367 | 0.21 |
| III | 307 | 0.95 | 342 | 0.07 |
| Compound | I | II | III | |
|---|---|---|---|---|
| Formula | C14H9N3O3S | |||
| Formula weight (g/mol) | 299.30 | |||
| Wavelength | 1.54178 Å | 1.54178 Å | 1.54178 Å | |
| Temperature (K) | 100 | 100 | 298 | |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | |
| Space group | P21/n | C2/c | Pbcn | |
| Unit cell dimensions | a (Å) | 7.60873(3) | 29.35796(17) | 29.0714(5) |
| b (Å) | 23.76332(8) | 7.18924(3) | 7.8433(2) | |
| c (Å) | 14.33447(6) | 12.63074(6) | 11.6150(2) | |
| β (°) | 92.2481(4) | 97.5932(5) | - | |
| Volume (Å3) | 2589.802(17) | 2642.49(2) | 2648.40(9) | |
| Z | 8 | |||
| Density (calculated) (g cm−3) | 1.535 | 1.505 g | 1.501 | |
| Absorption coefficient (mm−1) | 2.368 | 2.321 | 2.316 | |
| Crystal size (mm) | 0.22 × 0.16 × 0.16 | 0.13 × 0.13 × 0.08 | 0.27 × 0.09 × 0.06 | |
| F(000) | 1232 | |||
| Refinement method | Full-matrix least-squares on F2 | |||
| Data/restraints/parameters | 4620/0/380 | 2355/0/190 | 2373/0/209 | |
| Measured reflections | 60534 | 30398 | 16585 | |
| θmin/θmax | 3.603/67.059 | 3.037/67.067 | 3.040/67.047 | |
| Rint | 0.024 | 0.022 | 0.030 | |
| Goodness-of-fit on F2 | 1.053 | 1.090 | 1.027 | |
| Final R indices [I > 2σ(I)] | R1 = 0.0265 wR2 = 0.0646 | R1 = 0.0277 wR2 = 0.0720 | R1 = 0.0316 wR2 = 0.0886 | |
| R indices (all data) | R1 = 0.0266 wR2 = 0.0646 | R1 = 0.0278 wR2 = 0.0721 | R1 = 0.0357 wR2 = 0.0930 | |
| Largest diff. peak and hole (e/Ǻ3) | −0.29 and 0.27 | −0.36 and 0.18 | −0.17 and 0.28 | |
| Compound | D−H···A | D−H | H···A | D···A | D−H···A |
|---|---|---|---|---|---|
| I | N(2A)−H(2AA)···N(1B) | 0.832(13) | 2.226(11) | 2.9505(15) | 145.7(3) |
| N(2B)−H(2BB)···N(1A) | 0.833(12) | 2.138(12) | 2.9512(15) | 165.2(6) | |
| C(4A)−H(4A)···O(3A)ia | 0.954(16) | 2.541(13) | 3.2989(17) | 136.5(3) | |
| C(5A)−H(5A)···O(2B)iia | 0.958(15) | 2.479(16) | 3.3822(18) | 157.3(7) | |
| C(13A)−H(13A)···O(3A)iiia | 0.950(14) | 2.450(13) | 3.0805(17) | 123.7(5) | |
| II | N(2)−H(2A)···N(1)iib | 0.800(16) | 2.137(15) | 2.9046(15) | 160.7(2) |
| C(4)−H(4)···O(3)ivb | 0.975(12) | 2.504(12) | 3.4614(18) | 167.4(10) | |
| C(12)−H(12)···O(2)ib | 0.943(12) | 2.582(10) | 3.4288(18) | 149.7(8) | |
| C(13)−H(13)···O(1)iiib | 0.933(14) | 2.513(8) | 3.1676(16) | 127.4(5) | |
| III | N(2)−H(2A)···N(1)iic | 0.74(2) | 2.286(19) | 3.0072(17) | 165.3(3) |
| C(4)−H(4)···O(2A)*iiic | 0.996(16) | 2.534(18) | 3.495(9) | 162.1(7) | |
| C(13)−H(13)···O(1)ic | 0.956(17) | 2.536(8) | 3.172(2) | 124.1(6) |
| Compound | Cg–Cg | Symmetry Code | Cg–Cg (Å) | Slippage (Å) |
|---|---|---|---|---|
| I | Cg(1)–Cg(7) | - | 3.7900(7) | 1.306 |
| Cg(2)–Cg(7) | - | 3.6808(8) | 0.938 | |
| Cg(5)–Cg(6) | 1 − x, − y, 1 − z | 3.4893(7) | 0.538 | |
| Cg(6)–Cg(5) | 1 − x, − y, 1 − z | 3.4893(7) | 0.569 |
| Compounds | Eelec | Epolc | Edisc | Erepc | Etotal | ΣEtotald |
|---|---|---|---|---|---|---|
| IAa | −80.6 | −18.6 | −86.6 | 123.7 | −97.9 | −336.0 (IB = −279.4) |
| IIb | −97.4 | −26.6 | −50.7 | 126.1 | −88.9 | −313.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binti Abdul Latiff, A.; Chong, Y.Y.; Mark-Lee, W.F.; Kassim, M.B. Insight into Positional Isomerism of N-(Benzo[d]thiazol-2-yl)-o/m/p-Nitrobenzamide: Crystal Structure, Hirshfeld Surface Analysis and Interaction Energy. Crystals 2020, 10, 348. https://doi.org/10.3390/cryst10050348
Binti Abdul Latiff A, Chong YY, Mark-Lee WF, Kassim MB. Insight into Positional Isomerism of N-(Benzo[d]thiazol-2-yl)-o/m/p-Nitrobenzamide: Crystal Structure, Hirshfeld Surface Analysis and Interaction Energy. Crystals. 2020; 10(5):348. https://doi.org/10.3390/cryst10050348
Chicago/Turabian StyleBinti Abdul Latiff, Aqilah, Yan Yi Chong, Wun Fui Mark-Lee, and Mohammad B. Kassim. 2020. "Insight into Positional Isomerism of N-(Benzo[d]thiazol-2-yl)-o/m/p-Nitrobenzamide: Crystal Structure, Hirshfeld Surface Analysis and Interaction Energy" Crystals 10, no. 5: 348. https://doi.org/10.3390/cryst10050348
APA StyleBinti Abdul Latiff, A., Chong, Y. Y., Mark-Lee, W. F., & Kassim, M. B. (2020). Insight into Positional Isomerism of N-(Benzo[d]thiazol-2-yl)-o/m/p-Nitrobenzamide: Crystal Structure, Hirshfeld Surface Analysis and Interaction Energy. Crystals, 10(5), 348. https://doi.org/10.3390/cryst10050348