Monitoring the Production of High Diffraction-Quality Crystals of Two Enzymes in Real Time Using In Situ Dynamic Light Scattering

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Enzyme Samples
2.2. Crystallization in the XtalController
2.3. Standard DLS Measurements
2.4. Crystal Analysis
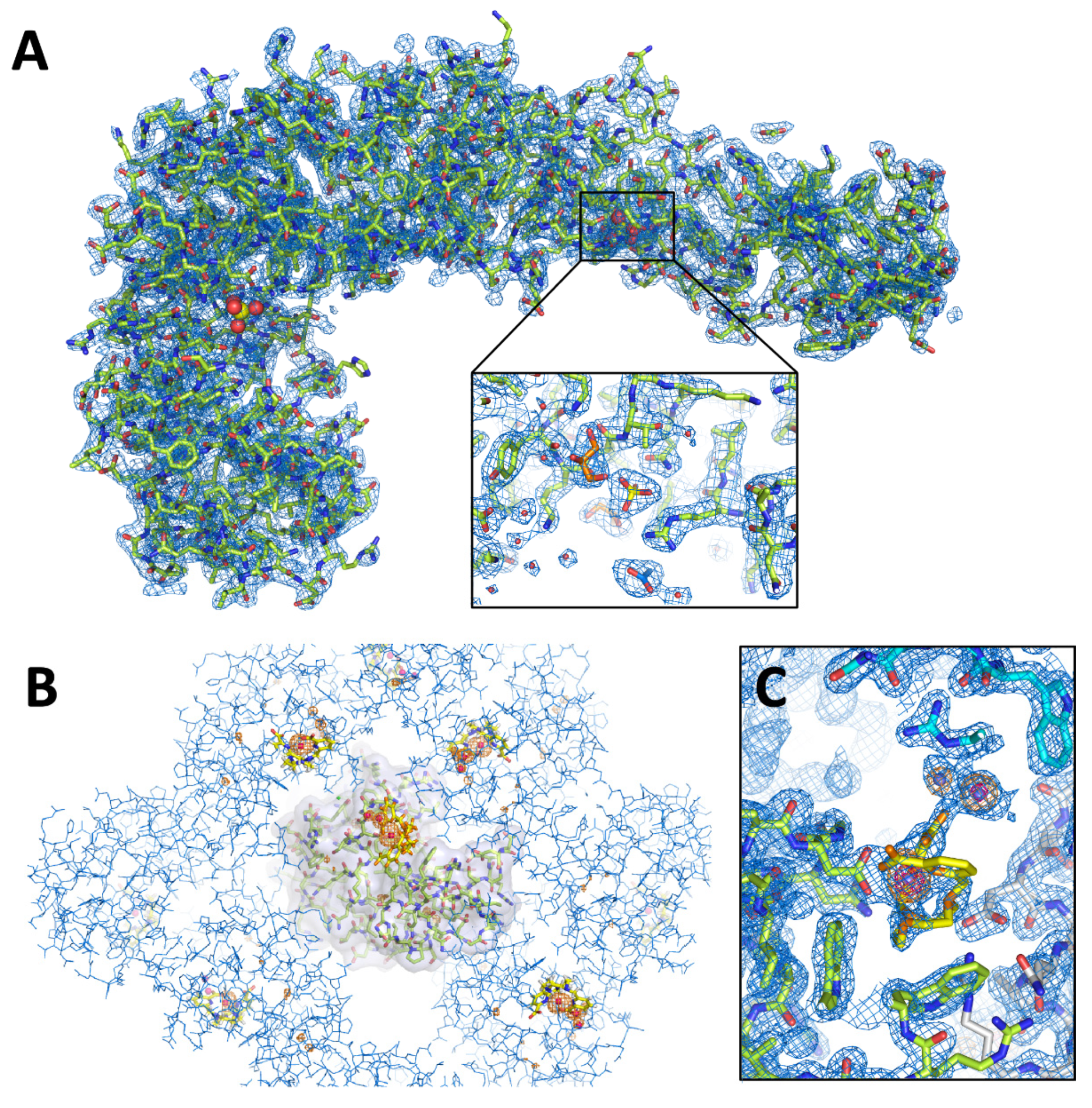
2.5. Structure Determination
3. Results and Discussion
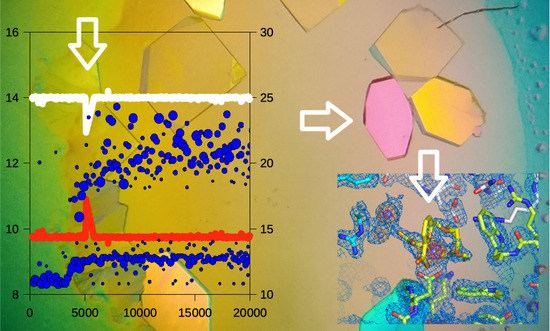
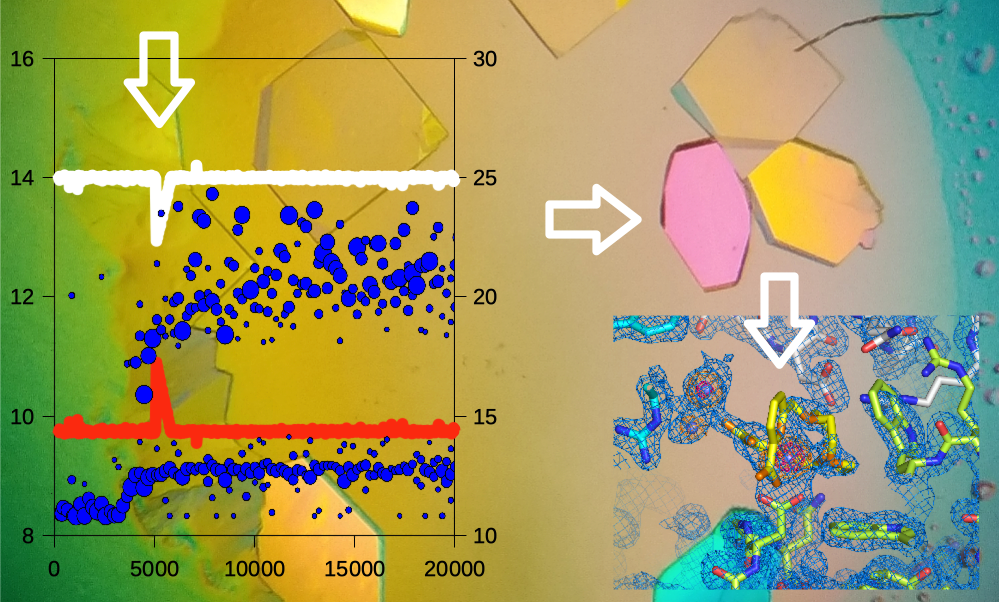
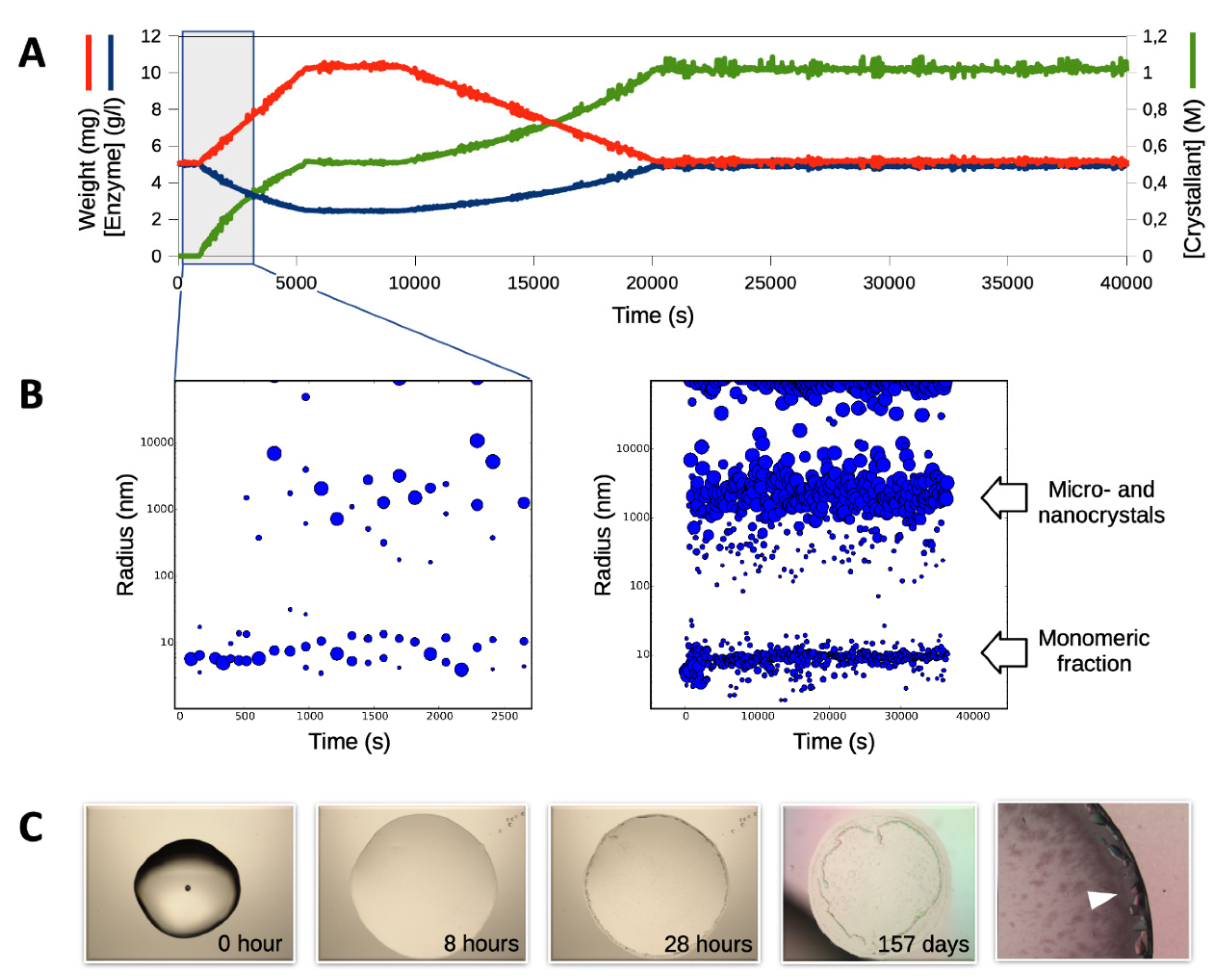
3.1. Triggering the Growth of Large Crystals of PhaCCA
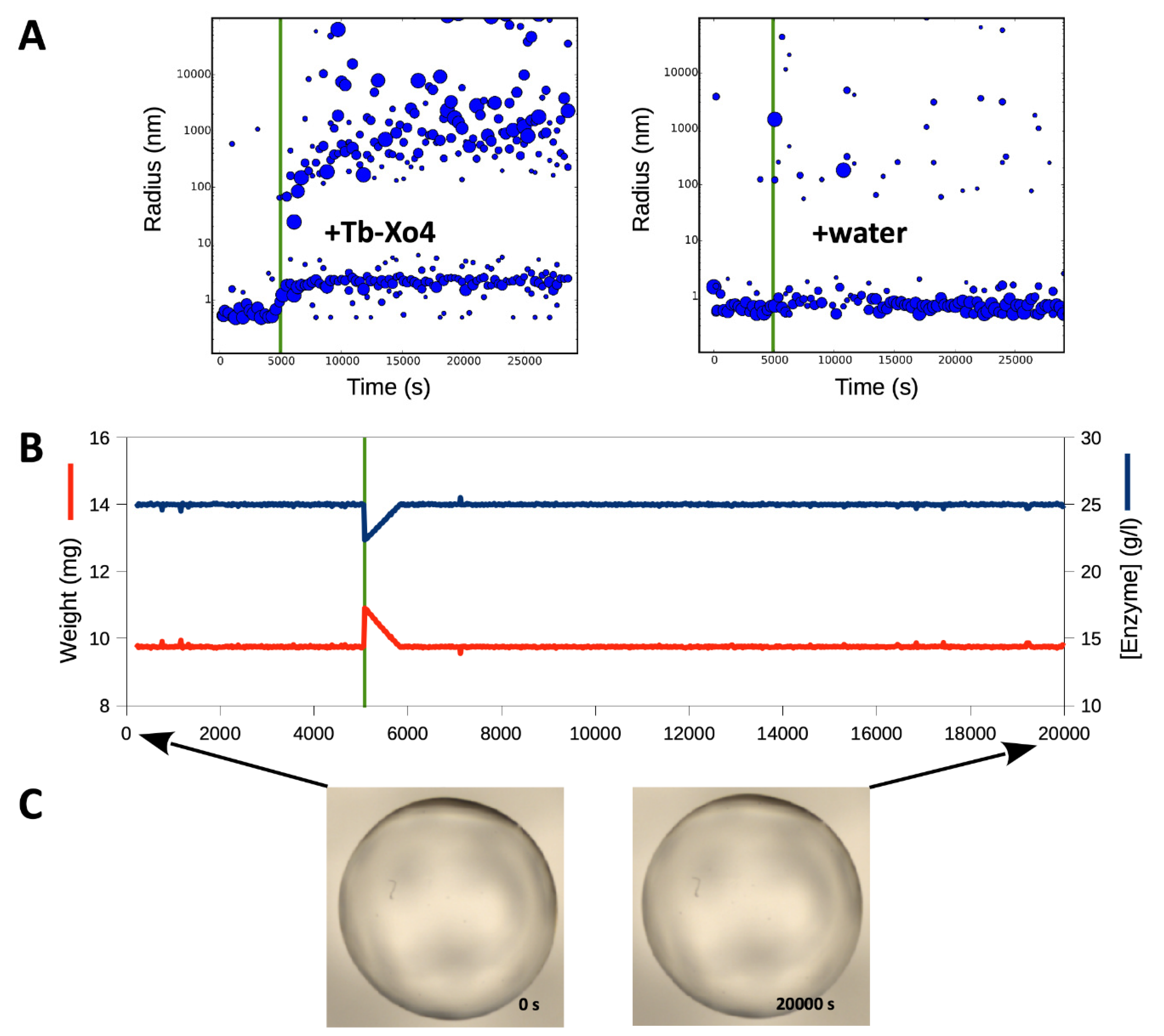
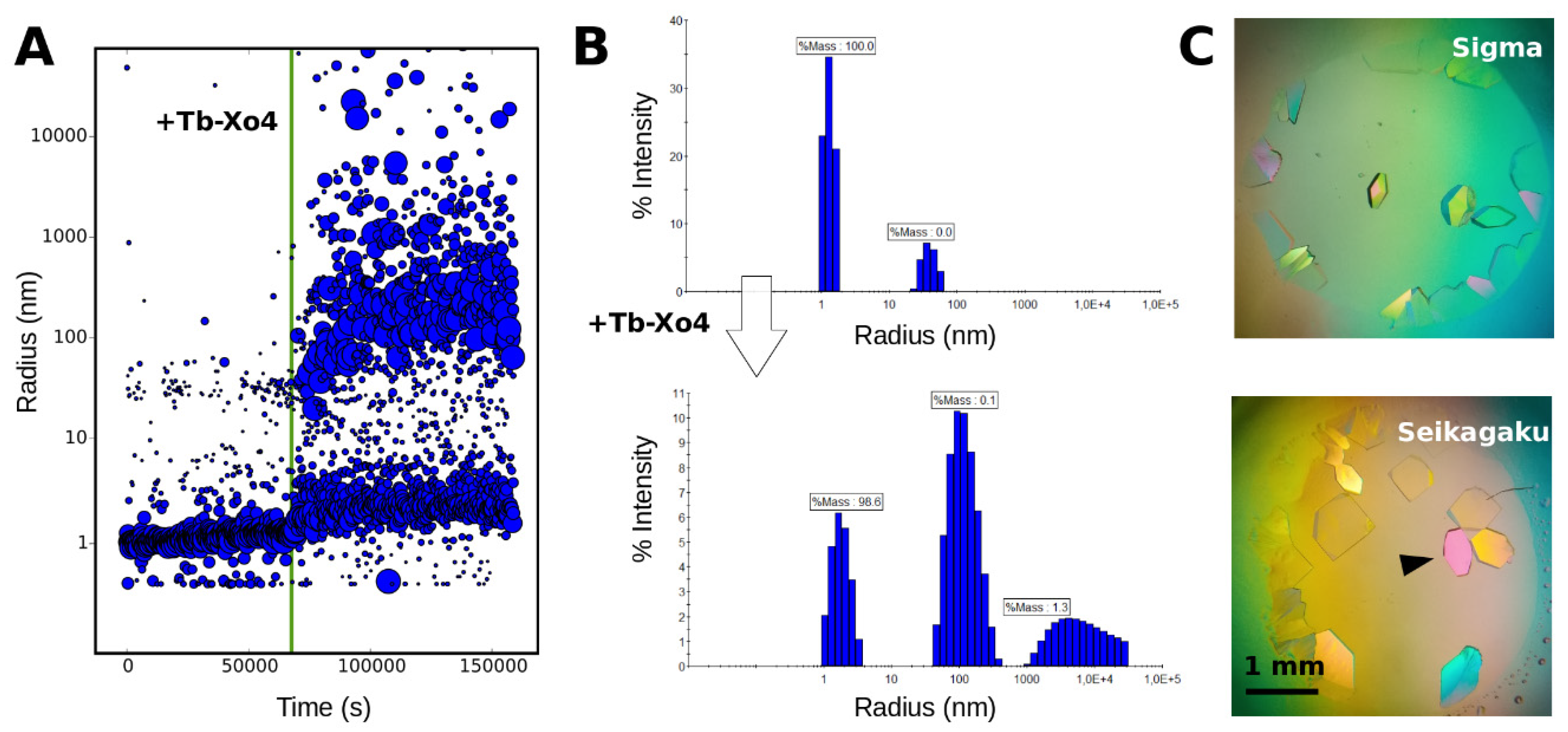
3.2. Tracking the Nucleant Effect of the Crystallophore Tb-Xo4on HEWL
3.3. Characterization of Crystals Grown in the XtalController
4. Conclusions
- -
- Explore phase diagrams of biomolecules with a direct feedback on nucleation events;
- -
- Study the stability of biomolecules in solution with respect to various parameters such as temperature, pH, ligands, etc.;
- -
- Determine the optimum conditions for introducing a cryoprotectant;
- -
- Ensure the reproducibility of crystals in the context of structural biology investigations, rational drug design, and fragment-based screening.
- -
- Produce calibrated nanocrystals on demand (difficult to monitor and control otherwise) for diffraction analyses using X-ray free electron lasers and µED, or, conversely, to promote the selective growth of large crystals for neutron diffraction.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giegé, R.; Sauter, C. Biocrystallography: Past, present, future. HFSP J. 2010, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.C.H.; Chapman, H.N. The birth of a new field. Philos. T. Roy. Soc. B 2014, 369, 20130309. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.C.; Stauch, B.; Ishchenko, A.; Cherezov, V. A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem. Sci. 2017, 42, 749–762. [Google Scholar] [CrossRef]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Three-dimensional electron crystallography of protein microcrystals. Elife 2013, 2, e01345. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Shi, D.; Leslie, A.G.W.; Gonen, T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat. Methods 2014, 11, 927–930. [Google Scholar] [CrossRef]
- Clabbers, M.T.B.; van Genderen, E.; Wan, W.; Wiegers, E.L.; Gruene, T.; Abrahams, J.P. Protein structure determination by electron diffraction using a single three-dimensional nanocrystal. Acta Cryst. D 2017, 73, 738–748. [Google Scholar] [CrossRef]
- Nederlof, I.; Li, Y.W.; van Heel, M.; Abrahams, J.P. Imaging protein three-dimensional nanocrystals with cryo-EM. Acta. Crystallogr. D. Biol Crystallogr. 2013, 69, 852–859. [Google Scholar] [CrossRef]
- Sauter, C.; Lorber, B.; McPherson, A. Crystallization–General Methods. In International Tables of Crystallography, Vol. F, Crystallography of Biological Macromolecules, 2nd ed.; Arnold, E., Himmel, D.M., Rossmann, M.G., Eds.; John Wiley and Sons: Chichester, UK, 2012; pp. 99–120. [Google Scholar]
- Garcia-Caballero, A.; Gavira, J.A.; Pineda-Molina, E.; Chayen, N.E.; Govada, L.; Khurshid, S.; Saridakis, E.; Boudjemline, A.; Swann, M.J.; Shaw Stewart, P.; et al. Optimization of Protein Crystallization: The OptiCryst Project. Cryst. Growth Des. 2011, 11, 2112–2121. [Google Scholar] [CrossRef]
- Meyer, A.; Dierks, K.; Hilterhaus, D.; Klupsch, T.; Mühlig, P.; Kleesiek, J.; Schöpflin, R.; Einspahr, H.; Hilgenfeld, R.; Betzel, C. Single-drop optimization of protein crystallization. Acta Crystallog. F 2012, 68, 994–998. [Google Scholar] [CrossRef]
- Mikol, V.; Hirsch, E.; Giegé, R. Diagnostic of precipitant for biomacromolecule crystallization by quasi-elastic light-scattering. J. Mol. Biol. 1990, 213, 187–195. [Google Scholar] [CrossRef]
- Kadima, W.; McPherson, A.; Dunn, M.F.; Jurnak, F.A. Characterization of precrystallization aggregation of canavalin by dynamic light scattering. Biophys. J. 1990, 57, 125–132. [Google Scholar] [CrossRef]
- George, A.; Wilson, W.W. Predicting protein crystallization from a dilute solution property. Acta Cryst. D Biol Cryst. 1994, 50, 361–365. [Google Scholar] [CrossRef]
- Zulauf, M.; D’Arcy, A. Light scattering of proteins as a criterion for crystallization. J. Cryst. Growth. 1992, 122, 102–106. [Google Scholar] [CrossRef]
- Dierks, K.; Meyer, A.; Einspahr, H.; Betzel, C. Dynamic Light Scattering in Protein Crystallization Droplets: Adaptations for Analysis and Optimization of Crystallization Processes. Cryst. Growth Des. 2008, 8, 1628–1634. [Google Scholar] [CrossRef]
- Schubert, R.; Meyer, A.; Baitan, D.; Dierks, K.; Perbandt, M.; Betzel, C. Real-Time Observation of Protein Dense Liquid Cluster Evolution during Nucleation in Protein Crystallization. Cryst. Growth Des. 2017, 17, 954–958. [Google Scholar] [CrossRef]
- Baitan, D.; Schubert, R.; Meyer, A.; Dierks, K.; Perbandt, M.; Betzel, C. Growing Protein Crystals with Distinct Dimensions using Automated Crystallization Coupled with in Situ Dynamic Light Scattering. JoVE. 2018, e57070. [Google Scholar] [CrossRef]
- Engilberge, S.; Riobé, F.; Di Pietro, S.; Lassalle, L.; Coquelle, N.; Arnaud, C.-A.; Pitrat, D.; Mulatier, J.-C.; Madern, D.; Breyton, C.; et al. Crystallophore: A versatile lanthanide complex for protein crystallography combining nucleating effects, phasing properties, and luminescence. Chem. Sci. 2017, 8, 5909–5917. [Google Scholar] [CrossRef]
- Engilberge, S.; Riobé, F.; Wagner, T.; Pietro, S.D.; Breyton, C.; Franzetti, B.; Shima, S.; Girard, E.; Dumont, E.; Maury, O. Unveiling the Binding Modes of the Crystallophore, a Terbium-based Nucleating and Phasing Molecular Agent for Protein Crystallography. Chemistry 2018, 24, 9739–9746. [Google Scholar] [CrossRef]
- De Wijn, R.; Hennig, O.; Ernst, F.G.M.; Lorber, B.; Betat, H.; Mörl, M.; Sauter, C. Combining crystallogenesis methods to produce diffraction-quality crystals of a psychrophilic tRNA-maturation enzyme. Acta Crystallog. F 2018, 74, 747–753. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Cryst. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- De Wijn, R.; Hennig, O.; Roche, J.; Engilberge, S.; Rollet, K.; Fernandez-Millan, P.; Brillet, K.; Betat, H.; Mörl, M.; Roussel, A.; et al. A simple and versatile microfluidic device for efficient biomacromolecule crystallization and structural analysis by serial crystallography. IUCrJ 2019, 6, 454–464. [Google Scholar] [CrossRef]
- Junius, N.; Oksanen, E.; Terrien, M.; Berzin, C.; Ferrer, J.-L.; Budayova-Spano, M. A crystallization apparatus for temperature-controlled flow-cell dialysis with real-time visualization. J. Appl. Cryst. 2016, 49, 806–813. [Google Scholar] [CrossRef]
- Engilberge, S.; Wagner, T.; Santoni, G.; Breyton, C.; Shima, S.; Franzetti, B.; Riobé, F.; Maury, O.; Girard, E. Protein crystal structure determination with the crystallophore, a nucleating and phasing agent. J. Appl. Cryst. 2019, 52, 722–731. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | PhaCCA | HEWL |
|---|---|---|
| Data collection | ||
| X-ray source | FIP/BM30A – ESRF | Rigaku Fr-X – IGBMC |
| Wavelength (Å) | 0.9799 | 1.5418 |
| Detector | ADSC Q315r | EIGER R 4M |
| Temperature (K) | 100 | 293 |
| Space group | P43212 | P43212 |
| Cell parameters (Å) | 70.53, 70.53, 291.48 | 78.81, 78.81, 38.33 |
| Mosaicity (°) | 0.31 | 0.12 |
| Solvent content | 67.4 | 40.8 |
| Resolution range (Å) | 47 – 2.28 (2.42 – 2.28) | 35 – 1.51 (1.60 – 1.51) |
| Number of reflections | 237664 (37915) | 164648 (14601) |
| Number of unique reflections | 34470 (5385) | 19535 (3017) |
| Multiplicity | 6.9 (7.0) | 8.4 (4.8) |
| Completeness (%) | 99.5 (98.5) | 99.2 (96.5) |
| Mean I/sigma(I) | 15.5 (1.5) | 29.7 (1.9) |
| Rmeas (%)1 | 8.6 (126) | 3.6 (72.6) |
| Rpim (%)2 | 3.4 (44.8) | 1.1 (31.7) |
| SigAno | – | 2.1 (0.6) |
| CC1/2 | 99.9 (76.0) | 100 (78.1) |
| Structure refinement | ||
| Rwork, Rfree | 0.213, 0.257 | 0.141, 0.175 |
| Number of non-H atoms enzyme, ligands, solvent | 2989, 36, 105 | 1001, 35, 54 |
| RMSD on bonds (Å) and angles (°) | 0.009, 0.96 | 0.004, 0.69 |
| Average ADPs (Å2) overall, enzyme, ligands, solvent | 51.0, 50.8, 62.9, 51.6 | 30.1, 28.7, 51.7, 41.9 |
| Ramachandran plot: % of residues in favored, allowed, unfavored regions | 96.4, 3.3, 0.3 | 99.2, 0.8, 0 |
| PDBid | 6TVZ | 6TVY |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Wijn, R.; Rollet, K.; Engilberge, S.; McEwen, A.G.; Hennig, O.; Betat, H.; Mörl, M.; Riobé, F.; Maury, O.; Girard, E.; et al. Monitoring the Production of High Diffraction-Quality Crystals of Two Enzymes in Real Time Using In Situ Dynamic Light Scattering. Crystals 2020, 10, 65. https://doi.org/10.3390/cryst10020065
de Wijn R, Rollet K, Engilberge S, McEwen AG, Hennig O, Betat H, Mörl M, Riobé F, Maury O, Girard E, et al. Monitoring the Production of High Diffraction-Quality Crystals of Two Enzymes in Real Time Using In Situ Dynamic Light Scattering. Crystals. 2020; 10(2):65. https://doi.org/10.3390/cryst10020065
Chicago/Turabian Stylede Wijn, Raphaël, Kévin Rollet, Sylvain Engilberge, Alastair G. McEwen, Oliver Hennig, Heike Betat, Mario Mörl, François Riobé, Olivier Maury, Eric Girard, and et al. 2020. "Monitoring the Production of High Diffraction-Quality Crystals of Two Enzymes in Real Time Using In Situ Dynamic Light Scattering" Crystals 10, no. 2: 65. https://doi.org/10.3390/cryst10020065
APA Stylede Wijn, R., Rollet, K., Engilberge, S., McEwen, A. G., Hennig, O., Betat, H., Mörl, M., Riobé, F., Maury, O., Girard, E., Bénas, P., Lorber, B., & Sauter, C. (2020). Monitoring the Production of High Diffraction-Quality Crystals of Two Enzymes in Real Time Using In Situ Dynamic Light Scattering. Crystals, 10(2), 65. https://doi.org/10.3390/cryst10020065