Regium Bonds between Silver(I) Pyrazolates Dinuclear Complexes and Lewis Bases (N2, OH2, NCH, SH2, NH3, PH3, CO and CNH)





Abstract
1. Introduction
2. Materials and Methods
2.1. Cambridge Structural Database Search
2.2. Ab Initio Calculations
3. Results and Discussion
3.1. CSD Search
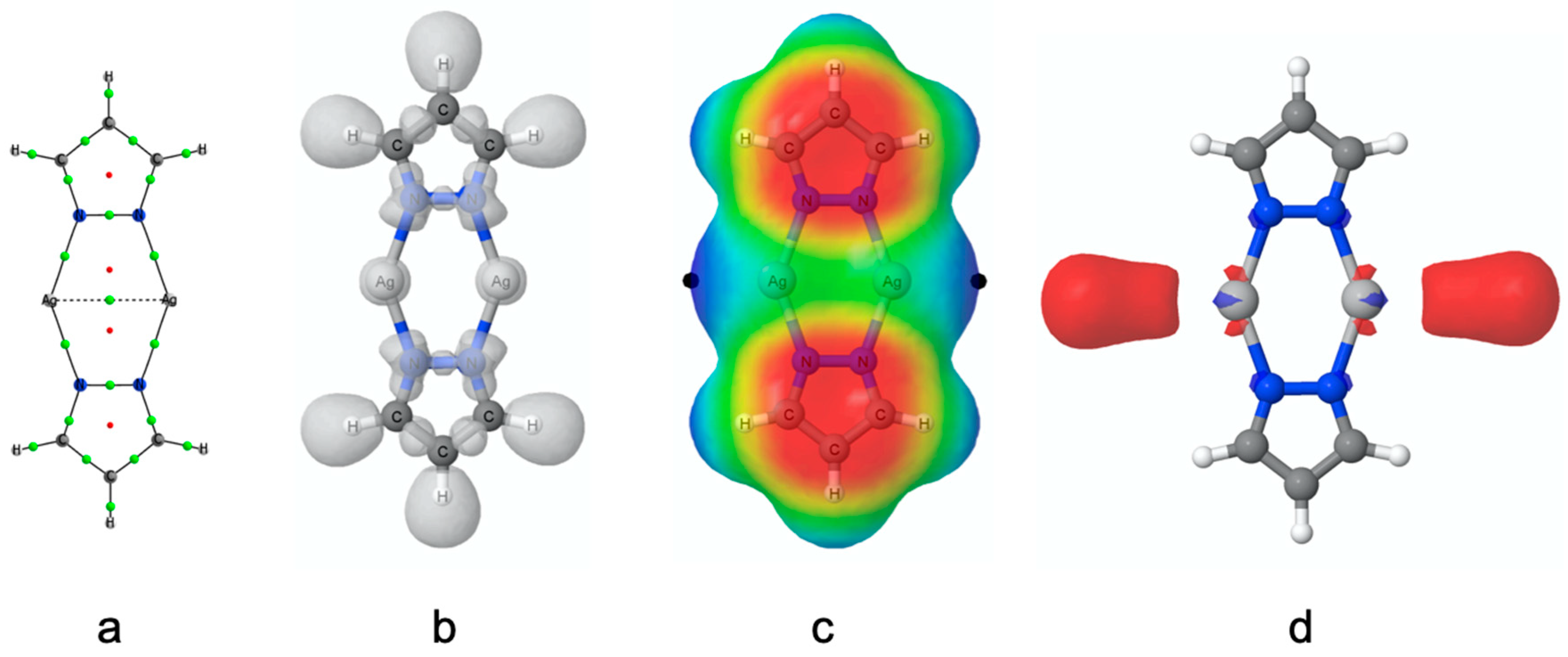
3.2. Electronic Properties of the Isolated (PzAg)2 System
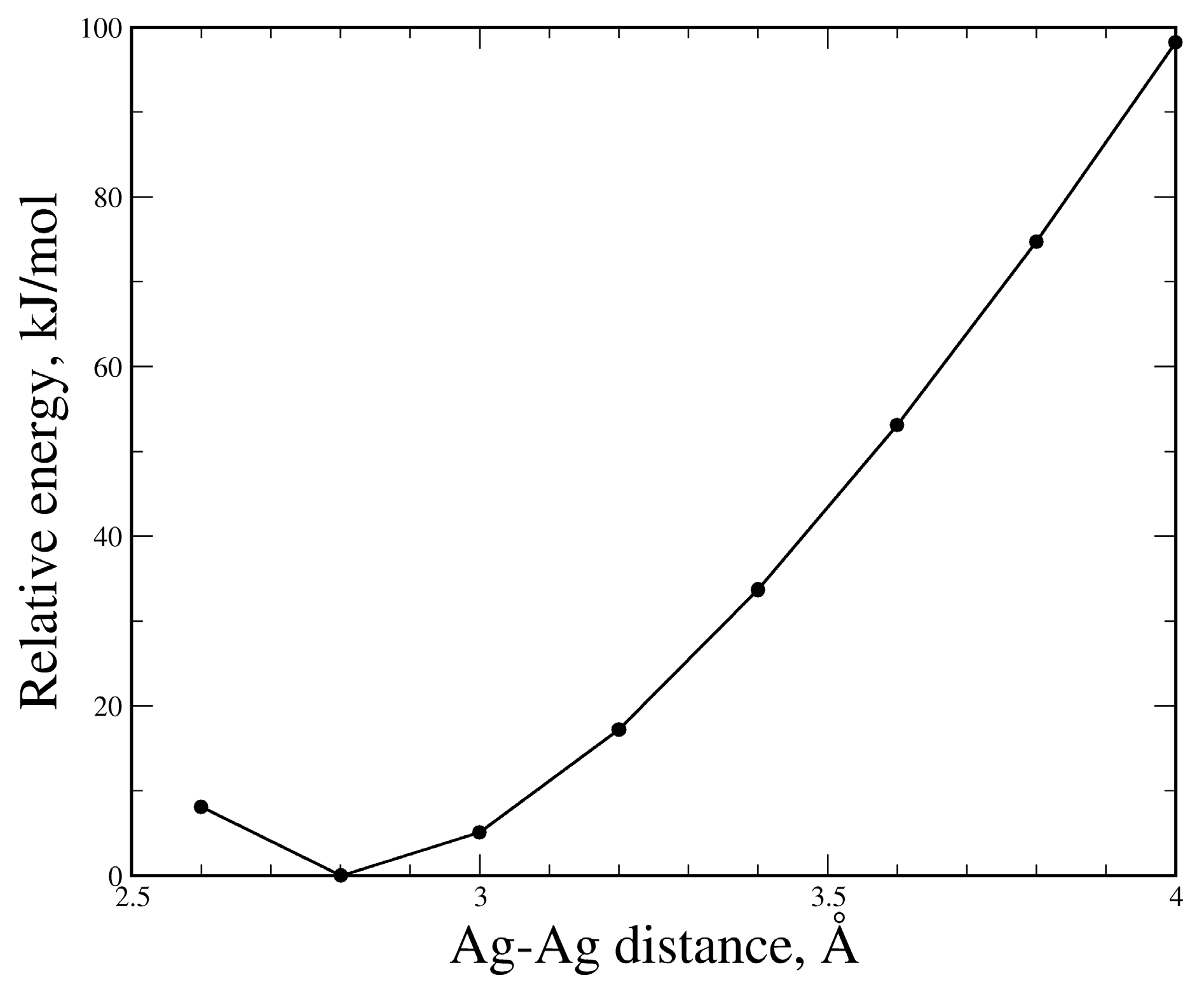
3.3. The (PzAg)2 Free Complex and the Effect of the Ag–Ag Distance on its Stability
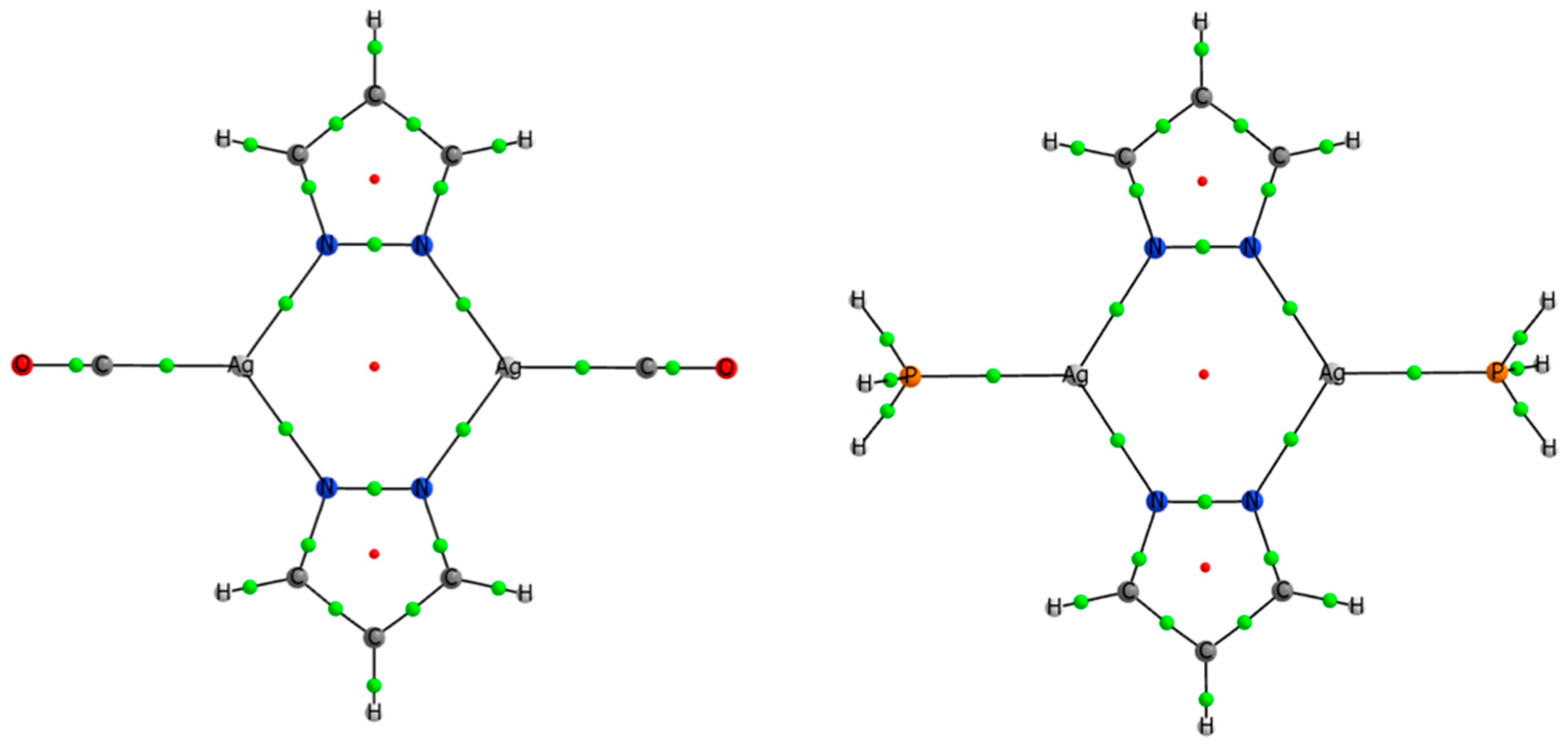
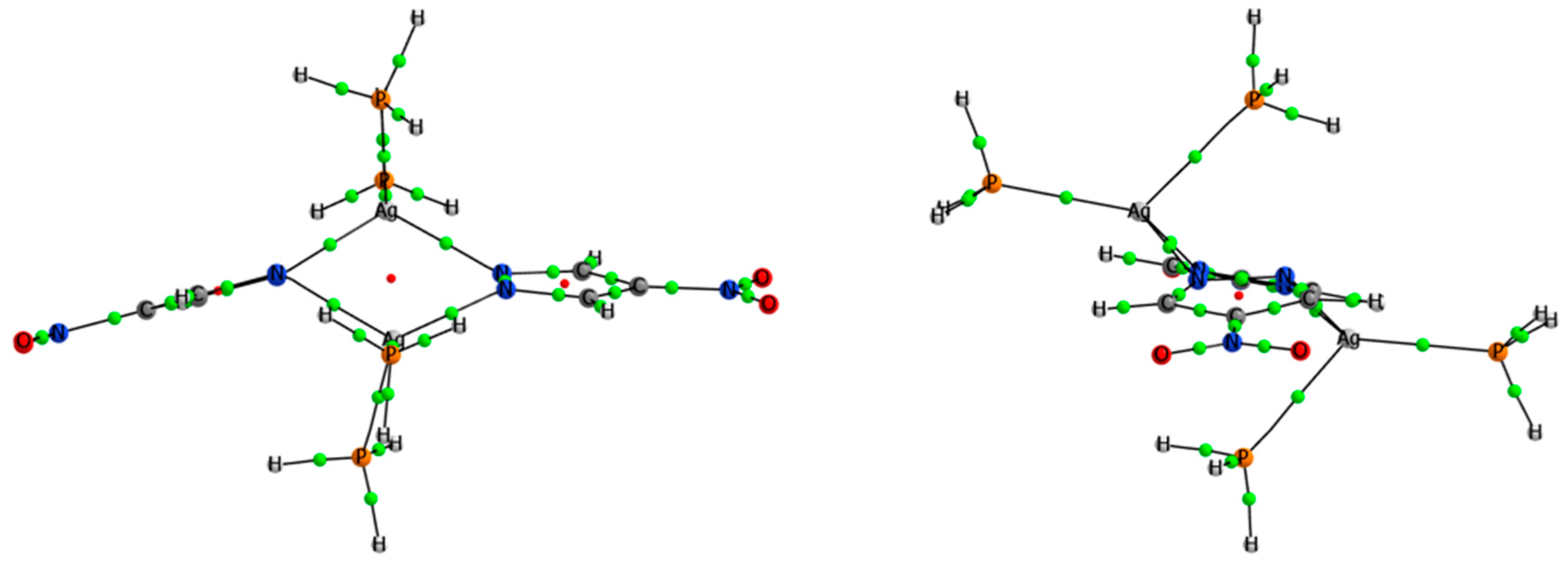
3.4. Effect of the Ligands and Substituents on the Structure and Dissociation Energy (De)
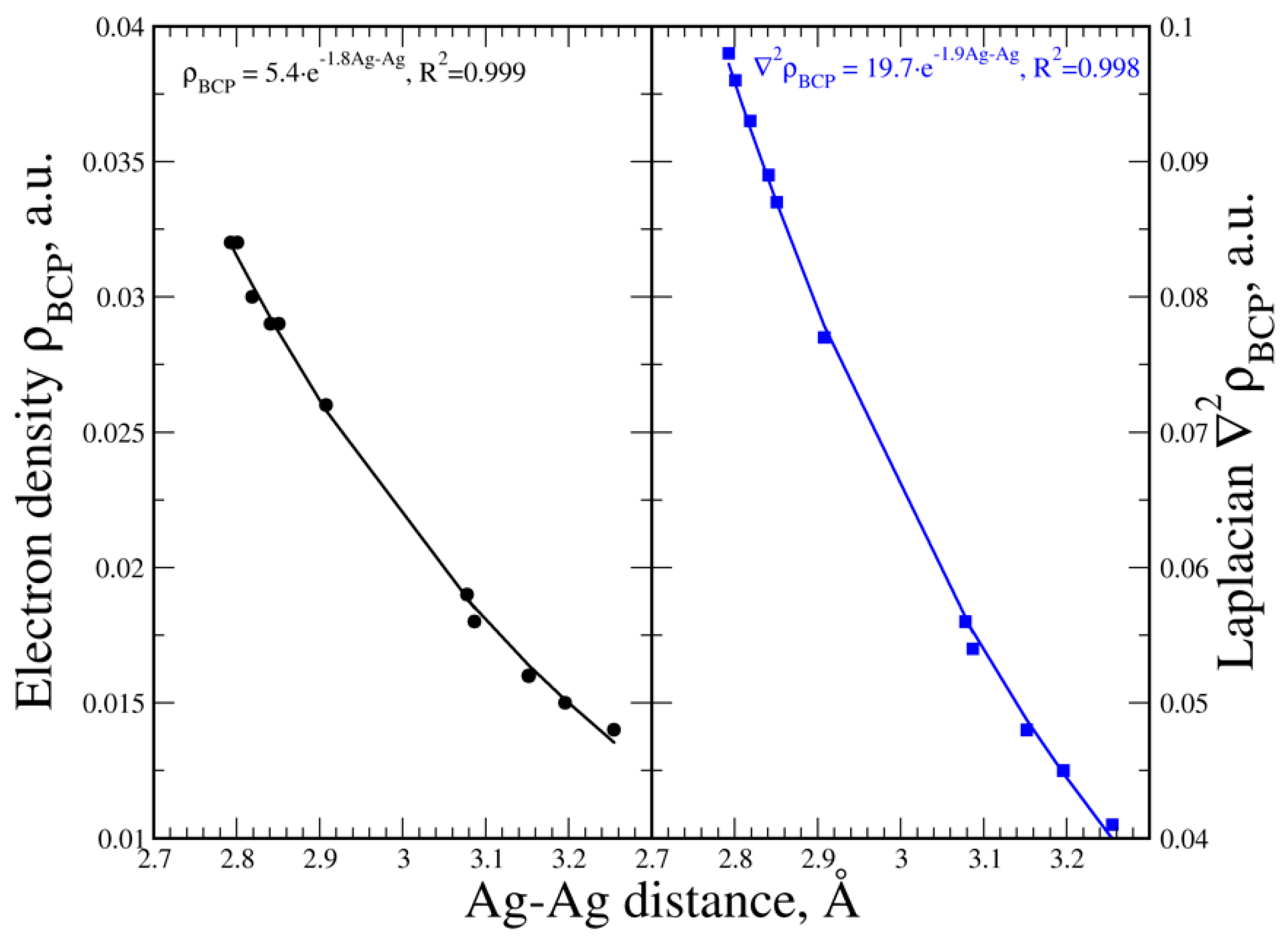
3.5. Electron Density
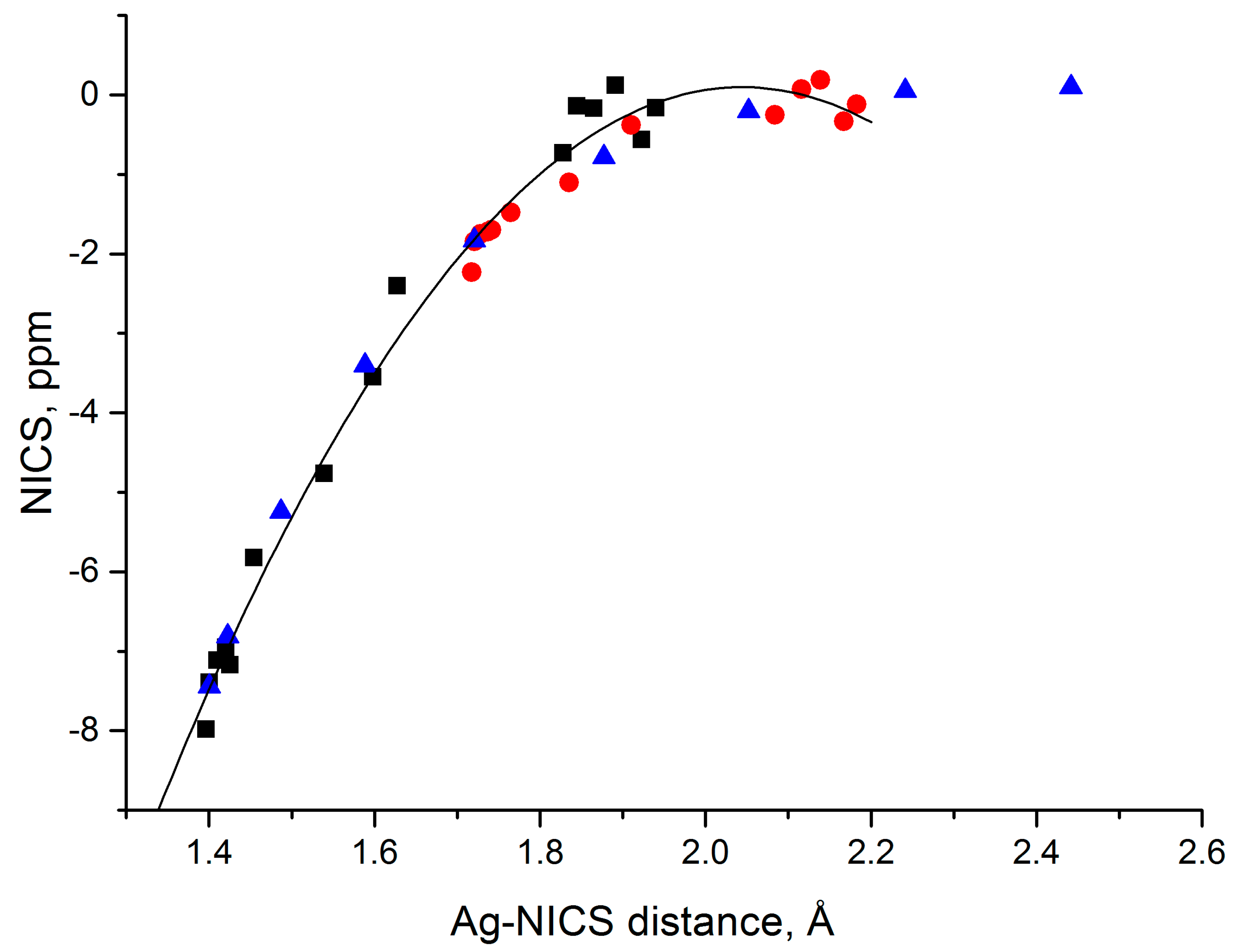
3.6. Magnetic Properties and Aromaticity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming Interactions from the Electrophilic Site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An Overview of σ-Hole Bonding, an Important and Widely-Occurring Noncovalent Interaction Practical Aspects of Computational Chemistry; Leszczynski, J., Shukla, M.K., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 149–163. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. PCCP 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other [sigma]-hole interactions: A perspective. PCCP 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Lane, P.; Clark, T.; Riley, K.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Solimannejad, M.; Ramezani, V.; Trujillo, C.; Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. Competition and Interplay between σ-Hole and π-Hole Interactions: A Computational Study of 1:1 and 1:2 Complexes of Nitryl Halides (O2NX) with Ammonia. J. Phys. Chem. A 2012, 116, 5199–5206. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds—Mechanisms protecting doublet and octet electron structures. PCCP 2017, 19, 29742–29759. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Directionality of π-holes in nitro compounds. Chem. Commun. 2015, 51, 1491–1493. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. On the ability of pnicogen atoms to engage in both σ and π-hole complexes. Heterodimers of ZF2C6H5 (Z = P, As, Sb, Bi) and NH3. J. Mol. Model. 2019, 25, 152. [Google Scholar] [CrossRef]
- Azofra, L.; Alkorta, I.; Scheiner, S. Noncovalent interactions in dimers and trimers of SO3 and CO. Theor. Chem. Acc. 2014, 133, 1–8. [Google Scholar] [CrossRef]
- Blanco, F.; Alkorta, I.; Rozas, I.; Solimannejad, M.; Elguero, J. A theoretical study of the interactions of NF3 with neutral ambidentate electron donor and acceptor molecules. PCCP 2011, 13, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Exploring the PX3:NCH and PX3:NH3 potential surfaces, with X=F, Cl, and Br. Chem. Phys. Lett. 2015, 641, 84–89. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. σ-Hole Opposite to a Lone Pair: Unconventional Pnicogen Bonding Interactions between ZF3 (Z=N, P, As, and Sb) Compounds and Several Donors. ChemPhysChem 2016, 17, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.H.; Brinck, T. Extending the σ-Hole Concept to Metals: An Electrostatic Interpretation of the Effects of Nanostructure in Gold and Platinum Catalysis. JACS 2017, 139, 11012–11015. [Google Scholar] [CrossRef]
- Shahi, A.; Arunan, E. Hydrogen bonding, halogen bonding and lithium bonding: An atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. PCCP 2014, 16, 22935–22952. [Google Scholar] [CrossRef]
- Solimannejad, M.; Rabbani, M.; Ahmadi, A.; Esrafili, M.D. Cooperative and diminutive interplay between the sodium bonding with hydrogen and dihydrogen bondings in ternary complexes of NaC3N with HMgH and HCN (HNC). Mol. Phys. 2014, 112, 2017–2022. [Google Scholar] [CrossRef]
- Yáñez, M.; Sanz, P.; Mó, O.; Alkorta, I.; Elguero, J. Beryllium Bonds, Do They Exist? J. Chem. Theor. Comput. 2009, 5, 2763–2771. [Google Scholar] [CrossRef]
- Alkorta, I.; Legon, A.C. Non-Covalent Interactions Involving Alkaline-Earth Atoms and Lewis Bases B: An ab Initio Investigation of Beryllium and Magnesium Bonds, B·MR2 (M = Be or Mg, and R = H, F or CH3). Inorganics 2019, 7, 35. [Google Scholar] [CrossRef]
- Montero-Campillo, M.M.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Chapter Three—The beryllium bond. In Advances in Inorganic Chemistry; van Eldik, R., Puchta, R., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 73, pp. 73–121. [Google Scholar]
- Frontera, A.; Bauzá, A. Regium–π bonds: An Unexplored Link between Noble Metal Nanoparticles and Aromatic Surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Regium bonds between Mn clusters (M = Cu, Ag, Au and n = 2–6) and nucleophiles NH3 and HCN. PCCP 2018, 20, 22498–22509. [Google Scholar] [CrossRef]
- Legon, A.C.; Walker, N.R. What’s in a name? ‘Coinage-metal’ non-covalent bonds and their definition. PCCP 2018, 20, 19332–19338. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules 2015, 20, 11297–11316. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Alkorta, I.; Blanco, F.; Elguero, J.; Dobado, J.A.; Ferrer, S.M.; Vidal, I. Carbon·Carbon Weak Interactions. J. Phys. Chem. A 2009, 113, 8387–8393. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond-[sigma]-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin-Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 191–263. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Minyaev, R.M.; Minkin, V.I. Theoretical study of O- > X (S, Se, Te) coordination in organic compounds. Can. J. Chem. 1998, 76, 776–788. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Sanz, P.; Yáñez, M.; Mó, O. Competition between X···H···Y Intramolecular Hydrogen Bonds and X····Y (X = O, S, and Y = Se, Te) Chalcogen−Chalcogen Interactions. J. Phys. Chem. A 2002, 106, 4661–4668. [Google Scholar] [CrossRef]
- Azofra, L.M.; Alkorta, I.; Scheiner, S. Strongly bound noncovalent (SO3)n:H2CO complexes (n = 1, 2). Phys. Chem. Chem. Phys. 2014, 16, 18974–18981. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. Prereactive Complexes of Dihalogens XY with Lewis Bases B in the Gas Phase: A Systematic Case for the Halogen Analogue B⋅⋅⋅XY of the Hydrogen Bond B⋅⋅⋅HX. Angew. Chem. Int. Ed. 1999, 38, 2686–2714. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef] [PubMed]
- Halldin Stenlid, J.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. PCCP 2018, 20, 2676–2692. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Li, R.; Jing, B.; Liu, Z.; Li, W.; Luan, F.; Cheng, J.; Gong, B.; Sun, J. Influence of Hybridization and Cooperativity on the Properties of Au-Bonding Interaction: Comparison with Hydrogen Bonds. J. Phys. Chem. A 2011, 115, 2853–2858. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Alkorta, I.; Elguero, J.; Yáñez, M.; Mó, O. Strong interactions between copper halides and unsaturated systems: New metallocycles? Or the importance of deformation. PCCP 2012, 14, 11468–11477. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Liu, S.; Cheng, J.; Li, W.; Li, Q. Synergistic and diminutive effects between triel bond and regium bond: Attractive interactions between π-hole and σ-hole. Appl. Organomet. Chem. 2019, 33, e4806. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Understanding Regium Bonds and their Competition with Hydrogen Bonds in Au2:HX Complexes. ChemPhysChem 2019, 20, 1572–1580. [Google Scholar] [CrossRef]
- Wang, R.; Yang, S.; Li, Q. Coinage-Metal Bond between [1.1.1]Propellane and M2/MCl/MCH3 (M = Cu, Ag, and Au): Cooperativity and Substituents. Molecules 2019, 24, 2601. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liu, Y.; Wang, Z.; Zhou, F.; Liu, Y.; Ding, X.; Lu, T. Regium bonds formed by MX (M = Cu, Ag, Au; X = F, Cl, Br) with phosphine-oxide/phosphinous acid: Comparisons between oxygen-shared and phosphine-shared complexes. Mol. Phys. 2019, 117, 2443–2455. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Dias, H.V.R.; Parasar, D.; Martín-Pastor, M. An experimental and computational NMR study of organometallic nine-membered rings: Trinuclear silver(I) complexes of pyrazolate ligands. Magn. Reson. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Allen, F. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Pople, J.A.; Binkley, J.S.; Seeger, R. Theoretical models incorporating electron correlation. Int. J. Quantum Chem. 1976, 10, 1–19. [Google Scholar] [CrossRef]
- Krishnan, R.; Pople, J.A. Approximate fourth-order perturbation theory of the electron correlation energy. Int. J. Quantum Chem. 1978, 14, 91–100. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Silver, D.M. Many-body perturbation theory applied to electron pair correlation energies. I. Closed-shell first-row diatomic hydrides. J. Chem. Phys. 1975, 62, 3258–3268. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D. Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadruple excitations for the correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Del Bene, J.E. Proton affinities of ammonia, water, and hydrogen fluoride and their anions: A quest for the basis-set limit using the Dunning augmented correlation-consistent basis sets. J. Phys. Chem. 1993, 97, 107–110. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Peterson, A.K.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Frisch, M.J.T.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 24 February 2020).
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Atoms in Molecules An Introduction; Prentice Hall: Harlow, UK, 2000. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Keith, T.A. AIMAll, 17.11.14 B; Version 17.11.14 B; TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. TopMod Package; Universite Pierre et Marie Curie: Paris, France, 1997. [Google Scholar]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Silvi, B.; Coionna, F. Topological analysis of the electron localization function applied to delocalized bonds. Can. J. Chem. 1996, 74, 1088–1096. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Ziegler, T. Calculation of NMR Shielding Tensors Using Gauge-Including Atomic Orbitals and Modern Density Functional Theory. J. Phys. Chem. 1995, 99, 606–611. [Google Scholar] [CrossRef]
- Lenthe, E.V.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.V.; Maerker, C.; Dransfeld, A.; Jiao, H.J.; Hommes, N.J.R.V. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. JACS 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef]
- Gershoni-Poranne, R.; Stanger, A. Magnetic criteria of aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Omary, M.A.; Rawashdeh-Omary, M.A.; Diyabalanage, H.V.K.; Dias, H.V.R. Blue Phosphors of Dinuclear and Mononuclear Copper(I) and Silver(I) Complexes of 3,5-Bis(trifluoromethyl)pyrazolate and the Related Bis(pyrazolyl)borate. Inorg. Chem. 2003, 42, 8612–8614. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(i) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Kandel, S.; Stenger-Smith, J.; Chakraborty, I.; Raptis, R.G. Syntheses and X-ray crystal structures of a family of dinuclear silver(I)pyrazolates: Assessment of their antibacterial efficacy against P. aeruginosa with a soft tissue and skin infection model. Polyhedron 2018, 154, 390–397. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A. σ-Donor and π-Acceptor Properties of Phosphorus Ligands: An Insight from the Natural Orbitals for Chemical Valence. Inorg. Chem. 2010, 49, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal Features of the Electron Density Distribution in Hydrogen-Bonding Regions: A Comprehensive Study Involving H⋅⋅⋅X (X=H, C, N, O, F, S, Cl, π) Interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Solimannejad, M.; Provasi, P.; Elguero, J. Theoretical Study of Complexes and Fluoride Cation Transfer between N2F+ and Electron Donors. J. Phys. Chem. A 2007, 111, 7154–7161. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond accepters. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viglione, R.G.; Zanasi, R. Local Aromaticity of the Six-Membered Rings in Pyracylene. A Difficult Case for the NICS Indicator of Aromaticity. J. Org. Chem. 2004, 69, 7537–7542. [Google Scholar] [CrossRef]
- Poater, J.; Bofill, J.M.; Alemany, P.; Solà, M. Role of Electron Density and Magnetic Couplings on the Nucleus-Independent Chemical Shift (NICS) Profiles of [2.2]Paracyclophane and Related Species. J. Org. Chem. 2006, 71, 1700–1702. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G. Aromatic behaviour of benzene and naphthalene upon pnicogen substitution. Tetrahedron 2015, 71, 826–839. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Rozas, I.; Alkorta, I. Influence of fluoro and cyano substituents in the aromatic and antiaromatic characteristics of cyclooctatetraene. Phys. Chem. Chem. Phys. 2015, 17, 14961–14971. [Google Scholar] [CrossRef]
- Trujillo, C.; Sánchez-Sanz, G. A Study of π–π Stacking Interactions and Aromaticity in Polycyclic Aromatic Hydrocarbon/Nucleobase Complexes. ChemPhysChem 2016, 17, 395–405. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Alkorta, I.; Trujillo, C.; Elguero, J. A theoretical NMR study of the structure of benzynes and some of their carbocyclic and heterocyclic analogs. Tetrahedron 2012, 68, 6548–6556. [Google Scholar] [CrossRef]
- Kusakiewicz-Dawid, A.; Porada, M.; Dziuk, B.; Siodlak, D. Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups. Molecules 2019, 24, 2632. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
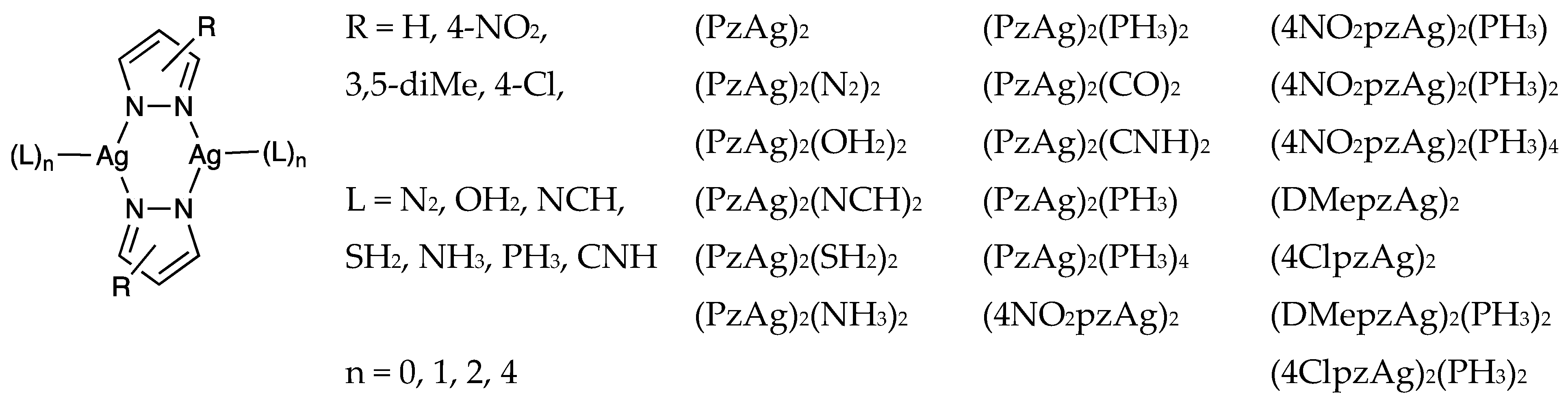{kind=link}
| Refcode | Pyrazolate | Ligand (L) | Nº of Ls | Ag–Ag | Ag–L | Conformation | τ4 index |
|---|---|---|---|---|---|---|---|
| FINWOR | 3,5-bis-CF3 | 3,5-bis-CF3-pyrazole-Ag | 2 | 3.392 | 2.250 | Nearly planar | ---- |
| PIRCUR | 3,5-bis-CF3 | PPh3 | 2 | 3.425 | 2.343 | Boat | ---- |
| PIRCUR | 3,5-bis-CF3 | PPh3 | 2 | 3.425 | 2.414 | Boat | ---- |
| FINWIL | 3,5-bis-CF3 | PPh3 | 2 | 3.479 | 2.366 | Twisted boat | ---- |
| IPIGOD | 3,5-bis-CF3 | 2,4,6-collidine | 2 | 3.499 | 2.277 | Boat + flattened chair | ---- |
| IPIGOD | 3,5-bis-CF3 | 2,4,6-collidine | 2 | 3.499 | 2.254 | Boat + lattened chair | ---- |
| IPIGOD | 3,5-bis-CF3 | 2,4,6-collidine | 2 | 3.502 | 2.269 | Boat + flattened chair | ---- |
| IPIGOD01 | 3,5-bis-CF3 | 2,4,6-collidine | 2 | 3.562 | 2.253 | Half boat | ---- |
| IPIGOD01 | 3,5-bis-CF3 | 2,4,6-collidine | 2 | 3.562 | 2.253 | Half boat | ---- |
| SOJCIG | 3,5-bis-CF3 | 3,5-bis-CF3-pyrazole-Ag | 2 | 3.632 | 3.218 | Chair | ---- |
| SOJCIG | 3,5-bis-CF3 | pyrazole-Ag | 2 | 3.632 | 2.258 | Chair | ---- |
| RATFEX | Parent | PPh3 | 3 | 3.706 | 2.370 | Flattened chair | 0.90 |
| RATFEX | Parent | PPh3 | 3 | 3.706 | 2.484 | Flattened chair | |
| RATFEX | Parent | PPh3 | 3 | 3.706 | 2.461 | Flattened chair | |
| ZIGRUF | 4-Cl | PPh3 | 3 | 3.707 | 2.374 | Flattened boat | 0.83 |
| ZIGRUF | 4-Cl | PPh3 | 3 | 3.707 | 2.475 | Flattened boat | |
| ZIGRUF | 4-Cl | PPh3 | 3 | 3.707 | 2.485 | Flattened boat | |
| ZIGSIU | 4-NO2 | PPh3 | 3 | 3.827 | 2.379 | Boat | 0.90 |
| ZIGSIU | 4-NO2 | PPh3 | 3 | 3.827 | 2.495 | Boat | |
| ZIGSIU | 4-NO2 | PPh3 | 3 | 3.827 | 2.469 | Boat | |
| RATFAT | Parent | PPh3 | 2 | 3.870 | 2.376 | Planar | ---- |
| KIRXIV | Parent | (a) | 2 (4) | 3.900 | 2.532 | Planar | 0.78 |
| KIRXIV | Parent | PPh3 | 4 | 3.900 | 2.539 | Planar | |
| ZIGSAM | 4-Cl | (b): PR3 | 4 | 4.205 | 2.460 | Twisted | 0.78 |
| ZIGSAM | 4-Cl | PR3 | 4.205 | 2.534 | Twisted | ||
| ZIGROZ | 4-Cl | PPh3 | 4 | 4.209 | 2.484 | Twisted | 0.89 |
| ZIGROZ | 4-Cl | PPh3 | 4 | 4.209 | 2.500 | Twisted | |
| ZIGSEQ | 4-NO2 | PPh3 | 4 | 4.305 | 2.502 | Twisted | 0.88 |
| ZIGSEQ | 4-NO2 | PPh3 | 4 | 4.305 | 2.487 | Twisted | |
| max | ---- | ---- | 4.305 | 3.218 | ---- | ---- | |
| min | ---- | ---- | 3.392 | 2.250 | ---- | ---- |

| Compound | Ag–Ag dist. (Å) | Pz-Pz dist. (Å) a | Ag-Z dist. (Å) b | De (kJ·mol−1) |
|---|---|---|---|---|
| (PzAg)2 | 2.801 | 3.968 | — | 0.0 |
| (PzAg)2(N2)2 | 2.851 | 3.968 | 2.837 | 22.7 |
| (PzAg)2(OH2)2 | 2.908 | 3.994 | 2.630 | 54.5 |
| (PzAg)2(NCH)2 | 3.078 | 3.933 | 2.425 | 56.3 |
| (PzAg)2(SH2)2 | 3.196 | 3.889 | 2.631 | 71.2 |
| (PzAg)2(NH3)2 | 3.255 | 3.905 | 2.358 | 91.7 |
| (PzAg)2(PH3)2 | 3.689 | 3.657 | 2.373 | 122.9 |
| (PzAg)2(CO)2 | 3.845 | 3.519 | 1.998 | 90.0 |
| (PzAg)2(CNH)2 | 3.880 | 3.534 | 2.018 | 136.6 |
| Compound | Ag–Ag dist. (Å) | De (kJ·mol−1) |
|---|---|---|
| (DMepzAg)2 | 2.793 | ---- |
| (PzAg)2 | 2.801 | ---- |
| (4ClpzAg)2 | 2.819 | ---- |
| (4NO2pzAg)2 | 2.841 | ---- |
| (DMepzAg)2(PH3)2 | 3.656 | 120.0 |
| (PzAg)2(PH3)2 | 3.689 | 122.9 |
| (4ClpzAg)2(PH3)2 | 3.730 | 135.1 |
| (4NO2pzAg)2(PH3)2 | 3.782 | 150.8 |
| Compound | Number of Ls | Ag–Ag dist. (Å) | De (kJ·mol−1) | τ4 index |
|---|---|---|---|---|
| (PzAg)2 | 0 | 2.801 | 0.0 | ---- |
| (PzAg)2(PH3) | 1 | 3.087 | 64.2 | ---- |
| (PzAg)2(PH3)2 | 2 | 3.689 | 122.9 | ---- |
| (PzAg)2(PH3)4 | 4 | 3.798 | 214.5 | 0.81 & 0.83 |
| (4NO2pzAg)2 | 0 | 2.841 | 0.0 | ---- |
| (4NO2pzAg)2(PH3) | 1 | 3.152 | 77.4 | |
| (4NO2pzAg)2(PH3)2 | 2 | 3.782 | 150.8 | ---- |
| (4NO2pzAg)2(PH3)4 | 4 | 3.870 | 251.7 | 0.80 & 0.81 |
| System | σ109Ag (ppm) | 6-Membered Ring | Pz Ring | ||
|---|---|---|---|---|---|
| NICS(0) | NICS(1) | NICS(0) | NICS(1) | ||
| (PzAg)2 | 3765.7 | −7.39 | −1.84 | −13.06 | −10.97 |
| (PzAg)2(N2)2 | 3648.3 | −7.17 | −1.70 | −12.70 | −10.95 |
| (PzAg)2(OH2)2 | 3752.8 | −5.82 | −1.48 | −12.88 | −11.22 |
| (PzAg)2(NCH)2 | 3515.5 | −4.76 | −1.10 | −12.67 | −11.03 |
| (PzAg)2(SH2)2 | 3288.3 | −3.55 | −0.65 | −12.75 | −11.04 |
| (PzAg)2(NH3)2 | 3511.9 | −2.40 | −0.38 | −12.74 | −11.12 |
| (PzAg)2(PH3)2 | 2900.9 | −0.14 | 0.12 | −12.27 | −11.11 |
| (PzAg)2(CO)2 | 2706.9 | −0.56 | −0.33 | −12.37 | −11.13 |
| (PzAg)2(CNH)2 | 2825.4 | −0.16 | −0.12 | −12.29 | −11.16 |
| (DMepzAg)2 | 3720.4 | −7.98 | −2.23 | −10.50 | −9.34 |
| (4ClpzAg)2 | 3815.9 | −7.11 | −1.75 | −12.62 | −9.98 |
| (4NO2pzAg)2 | 3835.5 | −6.95 | −1.72 | −11.98 | −9.50 |
| (DMepzAg)2(PH3)2 | 2893.1 | −0.73 | −0.25 | −9.32 | −9.29 |
| (4ClpzAg)2(PH3)2 | 2954.4 | −0.17 | 0.07 | −11.74 | −10.09 |
| (4NO2pzAg)2(PH3)2 | 3010.2 | 0.12 | 0.19 | −10.81 | −9.41 |
| (PzAg)2(PH3)4 | 2561.6 | ||||
| 2550.5 | |||||
| (4NO2pzAg)2(PH3)4 | 2619.5 | ||||
| 2627.7 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkorta, I.; Trujillo, C.; Sánchez-Sanz, G.; Elguero, J. Regium Bonds between Silver(I) Pyrazolates Dinuclear Complexes and Lewis Bases (N2, OH2, NCH, SH2, NH3, PH3, CO and CNH). Crystals 2020, 10, 137. https://doi.org/10.3390/cryst10020137
Alkorta I, Trujillo C, Sánchez-Sanz G, Elguero J. Regium Bonds between Silver(I) Pyrazolates Dinuclear Complexes and Lewis Bases (N2, OH2, NCH, SH2, NH3, PH3, CO and CNH). Crystals. 2020; 10(2):137. https://doi.org/10.3390/cryst10020137
Chicago/Turabian StyleAlkorta, Ibon, Cristina Trujillo, Goar Sánchez-Sanz, and José Elguero. 2020. "Regium Bonds between Silver(I) Pyrazolates Dinuclear Complexes and Lewis Bases (N2, OH2, NCH, SH2, NH3, PH3, CO and CNH)" Crystals 10, no. 2: 137. https://doi.org/10.3390/cryst10020137
APA StyleAlkorta, I., Trujillo, C., Sánchez-Sanz, G., & Elguero, J. (2020). Regium Bonds between Silver(I) Pyrazolates Dinuclear Complexes and Lewis Bases (N2, OH2, NCH, SH2, NH3, PH3, CO and CNH). Crystals, 10(2), 137. https://doi.org/10.3390/cryst10020137