Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their Classification


Abstract
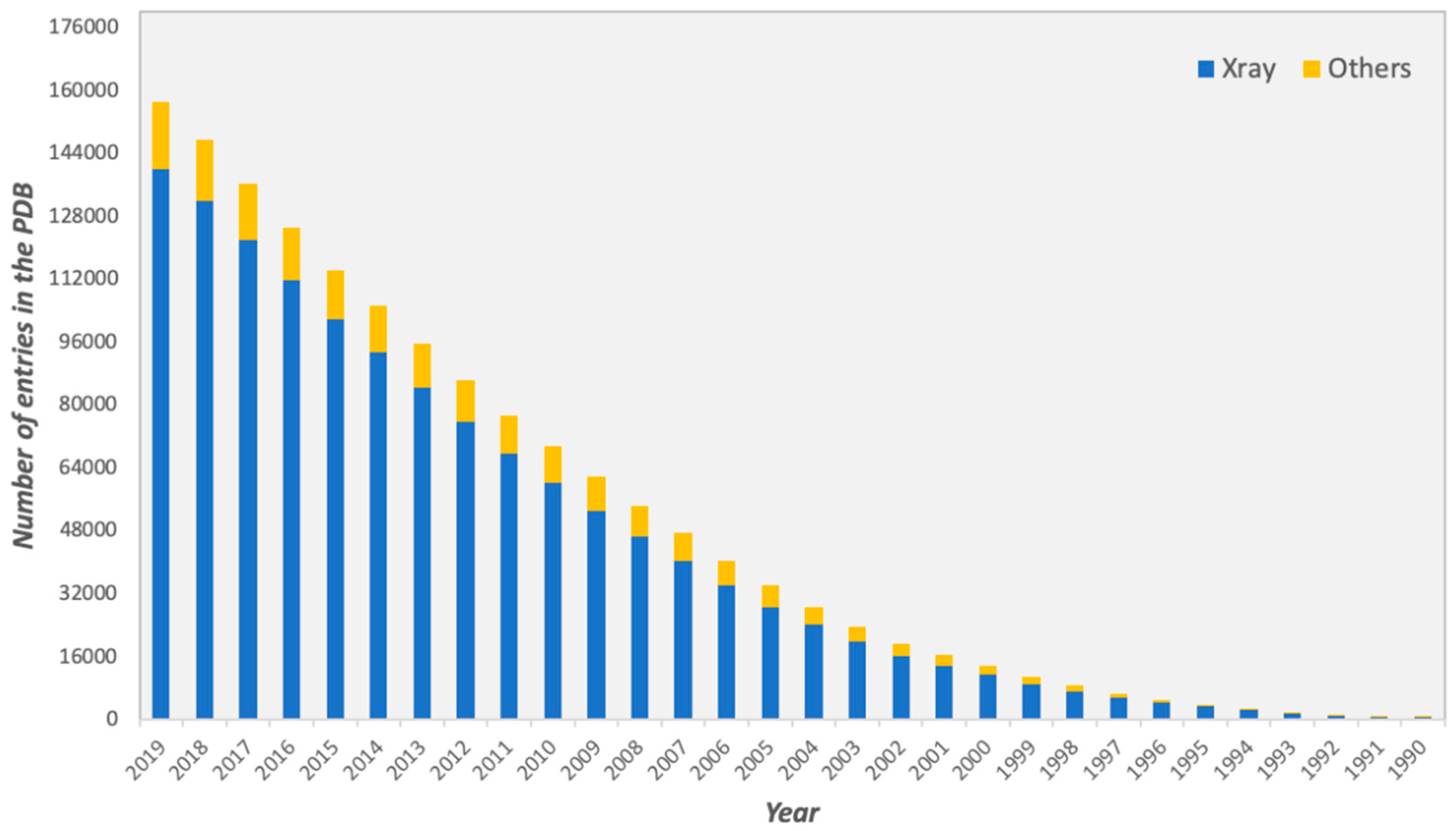
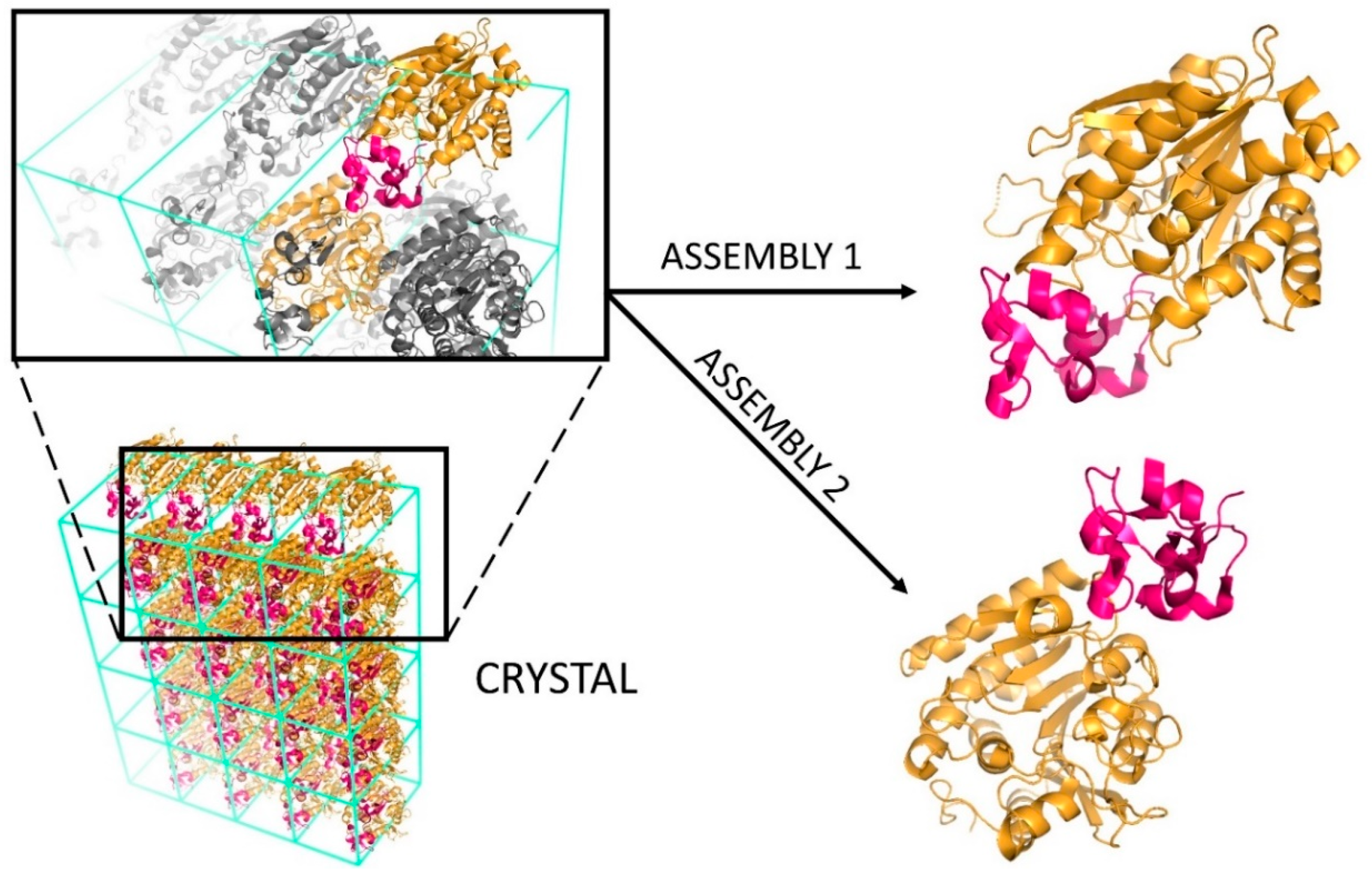
1. Introduction
2. Computational Methodologies for Classification of Biological Interfaces
2.1. Energy Based Classification Approaches
2.1.1. ClusPro-DC (2017)
2.1.2. PISA (2005, 2007)
2.2. Empirical, Knowledge-Based
2.2.1. Liu et al. (2014)
2.2.2. EPPIC (2012, 2014, 2018)
2.2.3. PreBI and COMP (2006, 2008)
2.2.4. CFPScore (2006)
2.2.5. Bahadur et al. (2004)
2.2.6. Elcock and McCammon (2001)
2.2.7. PITA (2000, 2003)
2.3. Machine Learning-Based
2.3.1. PIACO (2019)
2.3.2. PRODIGY-CRYSTAL (2018, 2019)
2.3.3. RPAIAnalyst (2018)
2.3.4. NOXclass (2016)
2.3.5. IChemPIC (2015)
2.3.6. Luo et al. (2014)
2.3.7. IPAC (2011)
2.3.8. DiMoVo (2008)
2.3.9. Valdar and Thornton (2001)
3. Datasets
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stites, W.E. Protein-Protein Interactions: Interface Structure, Binding Thermodynamics, and Mutational Analysis. Chem. Rev. 1997, 97, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.; Edwards, A. In Situ Proteolysis to Generate Crystals for Structure Determination: An Update. PLoS ONE 2009, 4, e5094. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Imperiali, B. Protein oligomerization: How and why. Bioorg. Med. Chem. 2005, 13, 5013–5020. [Google Scholar] [CrossRef] [PubMed]
- Capitani, G.; Duarte, J.M.; Baskaran, K.; Bliven, S.; Somody, J.C. Understanding the fabric of protein crystals: Computational classification of biological interfaces and crystal contacts. Bioinformatics 2015, 32, 481–489. [Google Scholar] [CrossRef]
- Nooren, I.M.A.; Thornton, J.M. Diversity of protein–protein interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef]
- Marsh, J.A.; Teichmann, S.A. Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem. 2015, 84, 551–575. [Google Scholar] [CrossRef]
- Arolas, J.L.; Popowicz, G.M.; Lorenzo, J.; Sommerhoff, C.P.; Huber, R.; Aviles, F.X.; Holak, T.A. The Three-Dimensional Structures of Tick Carboxypeptidase Inhibitor in Complex with A/B Carboxypeptidases Reveal a Novel Double-headed Binding Mode. J. Mol. Biol. 2005, 350, 489–498. [Google Scholar] [CrossRef]
- Taudt, A.; Arnold, A.; Pleiss, J. Simulation of protein association: Kinetic pathways towards crystal contacts. Phys. Rev. E 2015, 91, 033311. [Google Scholar] [CrossRef]
- Xu, Q.; Dunbrack, R.L. Principles and characteristics of biological assemblies in experimentally determined protein structures. Curr. Opin. Struct. Biol. 2019, 55, 34–49. [Google Scholar] [CrossRef]
- Xu, Q.; Dunbrack, R.L. The protein common interface database (ProtCID)—A comprehensive database of interactions of homologous proteins in multiple crystal forms. Nucleic Acids Res. 2010, 39, D761–D770. [Google Scholar] [CrossRef]
- Dey, S.; Ritchie, D.W.; Levy, E.D. PDB-wide identification of biological assemblies from conserved quaternary structure geometry. Nat. Methods 2018, 15, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Yueh, C.; Hall, D.R.; Xia, B.; Padhorny, D.; Kozakov, D.; Vajda, S. ClusPro-DC: Dimer Classification by the Cluspro Server for Protein–Protein Docking. J. Mol. Biol. 2017, 429, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. Dissecting subunit interfaces in homodimeric proteins. Proteins Struct. Funct. Bioinform. 2003, 53, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, R.P.; Chakrabarti, P.; Rodier, F.; Janin, J. A Dissection of Specific and Non-specific Protein-Protein Interfaces. J. Mol. Biol. 2004, 336, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Srebniak, A.; Schärer, M.A.; Capitani, G. Protein interface classification by evolutionary analysis. BMC Bioinform. 2012, 13, 334. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Detection of Protein Assemblies in Crystals. In Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2005; pp. 163–174. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Ponstingl, H.; Kabir, T.; Thornton, J.M. Automatic inference of protein quaternary structure from crystals. J. Appl. Crystallogr. 2003, 36, 1116–1122. [Google Scholar] [CrossRef]
- Krissinel, E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015, 43, W314–W319. [Google Scholar] [CrossRef]
- Liu, Q.; Li, Z.H.; Li, J.Y. Use B-factor related features for accurate classification between protein binding interfaces and crystal packing contacts. BMC Bioinform. 2014, 15, 3. [Google Scholar] [CrossRef]
- Zhu, H.B.; Domingues, F.S.; Sommer, I.; Lengauer, T. NOXclass: Prediction of protein-protein interaction types. BMC Bioinform. 2006, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Ponstingl, H.; Henrick, K.; Thornton, J.M. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins Struct. Funct. Bioinform. 2000, 41, 47–57. [Google Scholar] [CrossRef]
- Schärer, M.A.; Grütter, M.G.; Capitani, G. CRK: An evolutionary approach for distinguishing biologically relevant interfaces from crystal contacts. Proteins Struct. Funct. Bioinform. 2010, 78, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P.; Janin, J. Dissecting protein-protein recognition sites. Proteins Struct. Funct. Bioinform. 2002, 47, 334–343. [Google Scholar] [CrossRef]
- Levy, E.D. A Simple Definition of Structural Regions in Proteins and Its Use in Analyzing Interface Evolution. J. Mol. Biol. 2010, 403, 660–670. [Google Scholar] [CrossRef]
- Baskaran, K.; Duarte, J.M.; Biyani, N.; Bliven, S.; Capitani, G. A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct. Biol. 2014, 14, 22. [Google Scholar] [CrossRef]
- Bliven, S.; Lafita, A.; Parker, A.; Capitani, G.; Duarte, J.M. Automated evaluation of quaternary structures from protein crystals. PLoS Comput. Biol. 2018, 14, e1006104. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Kinoshita, K.; Ito, N.; Nakamura, H. PreBI: Prediction of biological interfaces of proteins in crystals. Nucleic Acids Res. 2006, 34, W20–W24. [Google Scholar] [CrossRef][Green Version]
- Tsuchiya, Y.; Nakamura, H.; Kinoshita, K. Discrimination between biological interfaces and crystal-packing contacts. Adv. Appl. Bioinform. Chem. 2008, 1, 99–113. [Google Scholar] [CrossRef]
- Liu, S.Y.; Li, Q.L.; Lai, L.H. A combinatorial score to distinguish biological and nonbiological protein–protein interfaces. Proteins Struct. Funct. Bioinform. 2006, 64, 68–78. [Google Scholar] [CrossRef]
- Elcock, A.H.; McCammon, J.A. Identification of protein oligomerization states by analysis of interface conservation. Proc. Natl. Acad. Sci. USA 2001, 98, 2990–2994. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, Y.; Tomii, K. Accurate Classification of Biological and non-Biological Interfaces in Protein Crystal Structures using Subtle Covariation Signals. Sci. Rep. 2019, 9, 12603. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Buchan, D.W.A.; Cozzetto, D.; Pontil, M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 2011, 28, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK 2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Elez, K.; Bonvin, A.M.J.J.; Vangone, A. Distinguishing crystallographic from biological interfaces in protein complexes: Role of intermolecular contacts and energetics for classification. BMC Bioinform. 2018, 19, 438. [Google Scholar] [CrossRef]
- Jiménez-García, B.; Elez, K.; Koukos, P.I.; Bonvin, A.M.; Vangone, A. PRODIGY-crystal: A web-tool for classification of biological interfaces in protein complexes. Bioinformatics 2019, 35, 4821–4823. [Google Scholar] [CrossRef]
- Hu, J.; Liu, H.F.; Sun, J.; Wang, J.; Liu, R. Integrating co-evolutionary signals and other properties of residue pairs to distinguish biological interfaces from crystal contacts. Protein Sci. 2018, 27, 1723–1735. [Google Scholar] [CrossRef]
- Da Silva, F.; Desaphy, J.; Bret, G.; Rognan, D. IChemPIC: A Random Forest Classifier of Biological and Crystallographic Protein–Protein Interfaces. J. Chem. Inf. Model. 2015, 55, 2005–2014. [Google Scholar] [CrossRef]
- Luo, J.S.; Guo, Y.Z.; Fu, Y.Y.; Wang, Y.; Li, W.L.; Li, M.L. Effective discrimination between biologically relevant contacts and crystal packing contacts using new determinants. Proteins Struct. Funct. Bioinform. 2014, 82, 3090–3100. [Google Scholar] [CrossRef]
- Mitra, P.; Pal, D. Combining Bayes Classification and Point Group Symmetry under Boolean Framework for Enhanced Protein Quaternary Structure Inference. Structure 2011, 19, 304–312. [Google Scholar] [CrossRef]
- Bernauer, J.; Bahadur, R.P.; Rodier, F.; Janin, J.; Poupon, A. DiMoVo: A Voronoi tessellation-based method for discriminating crystallographic and biological protein–protein interactions. Bioinformatics 2008, 24, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Valdar, W.S.J.; Thornton, J.M. Conservation helps to identify biologically relevant crystal contacts. J. Mol. Biol. 2001, 313, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.D. PiQSi: Protein Quaternary Structure Investigation. Structure 2007, 15, 1364–1367. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Methodology | Training Dataset | Webserver or Source Code | Ref. |
|---|---|---|---|---|
| Methods with a functioning webserver or source code | ||||
| ClusPro-DC | Number of near-native docking poses in the top 1000 lowest energy structures | Bahadur et al. [14,15] | https://cluspro.bu.edu/dimer_predict | [12] |
| EPPIC | Number of core residues, core-rim and core-surface entropy ratios | DC [16], Many [27] 1 | http://www.eppic-web.org | [16,27,28] |
| IPAC | Interface area, normalized surface complementarity, accessible surface area variation, normalized interface packing, interface packing gradient, hydrophobicity of interface and surface, patch ratio and normalized solvation energy -> NB | Mitra and Pal [41] | http://pallab.serc.iisc.ernet.in/IPAC | [41] |
| PIACO | Covariation signal, number of core residues, amino acid compositions of the interface and of core residues, amino acid pair frequency, local density index, residue propensity score and gap volume index -> RF | DC [16] | https://github.com/yfukasawa/piaco | [33] |
| PISA | Binding energy and entropy of dissociation | Ponstingl et al. [19] | https://www.ebi.ac.uk/msd-srv/prot_int http://www.ccp4.ac.uk/pisa | [17,18,20] |
| PITA | Interface area and atom-pair frequencies | Ponstingl et al. [19,23] | https://www.ebi.ac.uk/thornton-srv/databases/pita | [19,23] |
| PRODIGY-CRYSTAL | Number of residue contacts grouped by their character, number of residue contacts per amino acid and link density -> RF | Many [27] | https://haddock.science.uu.nl/services/PRODIGY-CRYSTAL | [36,37] |
| RPAIAnalyst | Co-evolutionary and conservation scores, residue pair frequency, Voronoi cell volume, secondary structure, core-rim, B-factor and hot spots -> RF | DC [16] | http://liulab.hzau.edu.cn/RPAIAnalyst | [38] |
| Methods without a functioning webserver or source code | ||||
| Bahadur et al. | Non-polar interface area, fraction of buried atoms and residue propensity scores | Bahadur et al. [14,15] | [15] | |
| CFPScore | Potential mean force and shape complementarity scores, interface size and packing density | Ponstingl et al. [23] | [31] | |
| DiMoVo | Interface area, number, Voronoi volumes and frequencies of interface residues, frequency of pairs of residues in contact and distances between their geometric centers -> SVM | Bahadur et al. [14,15] | http://fifi.ibbmc.u-psud.fr | [42] |
| Elcock and McCammon | Ratio between mean interface and surface entropies | [32] | ||
| IChemPIC | Number of interaction pseudoatoms, percentage of interaction pseudoatoms for each of the four interaction types and their corresponding distributions of buriedness -> RF | Da Silva et al. [39] | http://bioinfo-pharma.u-strasbg.fr/IChemPIC | [39] |
| Liu et al. | Average B-factor score | DC [16] | [21] | |
| Luo et al. | Core-surface and core-interface scores, residue propensity, core area, non-polar area and fully buried atoms fractions, gap volume and local density indices, number of hot spots, interface area ratio, amino acid and secondary structure compositions and propensities of interface residues, core residues and hot spots -> RF | DC [16] | http://cic.scu.edu.cn/bioinformatics/bio-cry.zip | [40] |
| NOXclass | Interface area, ratio of interface area to protein surface area and amino acid composition of the interface -> SVM | Zhu et al. [22] | http://noxclass.bioinf.mpiinf.mpg.de | [22] |
| PreBI and COMP | Interface area and shape, hydrophobicity andelectrostatic potential | Tsuchiya et al. [29] | http://pre-s.protein.osaka-u.ac.jp/prebi | [29,30] |
| Valdar and Thornton | Fraction of buried surface residues and contact conservation score -> MLP | Ponstingl et al. [23] | [43] | |
| Dataset | Content | Ref. |
|---|---|---|
| Bahadur et al. | 122 homodimers and 188 monomers | [14,15] |
| DC | 83 biological and 82 crystallographic interfaces | [16] |
| De Silva et al. | 200 biological and 200 crystallographic interfaces | [39] |
| Many | 2831 biological and 2913 crystallographic interfaces | [27] |
| Mitra and Pal | 268 dimers and 396 monomers | [41] |
| Ponstingl et al. (2000) | 76 homodimers and 96 monomers | [23] |
| Ponstingl et al. (2003) | 163 oligomers and 55 monomers | [19] |
| Zhu et al. | 137 biological (75 obligate + 62 non-obligate) and 106 crystallographic interfaces | [22] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elez, K.; Bonvin, A.M.J.J.; Vangone, A. Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their Classification. Crystals 2020, 10, 114. https://doi.org/10.3390/cryst10020114
Elez K, Bonvin AMJJ, Vangone A. Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their Classification. Crystals. 2020; 10(2):114. https://doi.org/10.3390/cryst10020114
Chicago/Turabian StyleElez, Katarina, Alexandre M. J. J. Bonvin, and Anna Vangone. 2020. "Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their Classification" Crystals 10, no. 2: 114. https://doi.org/10.3390/cryst10020114
APA StyleElez, K., Bonvin, A. M. J. J., & Vangone, A. (2020). Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their Classification. Crystals, 10(2), 114. https://doi.org/10.3390/cryst10020114