Abstract
The wide-range variation of the strength of halogen bonds (XB) not only facilitates a variety of applications of this interaction, but it also allows examining the relation (and interconversion) between supramolecular and covalent bonding. Herein, the Br…Cl halogen bonding in a series of complexes of bromosubstituted electrophiles (R-Br) with chloride anions were examined via X-ray crystallographic and computational methods. Six co-crystals showing such bonding were prepared by evaporation of solutions of R-Br and tetra-n-propylammonium chloride or using Cl− anions released in the nucleophilic reaction of 1,4-diazabicyclo[2.2.2]octane with dichloromethane in the presence of R-Br. The co-crystal comprised networks formed by 3:3 or 2:2 halogen bonding between R-Br and Cl−, with the XB lengths varying from 3.0 Å to 3.25 Å. Analysis of the crystallographic database revealed examples of associations with substantially longer and shorter Br…Cl separations. DFT computations of an extended series of R–Br…Cl− complexes confirmed that the judicious choice of brominated electrophile allows varying halogen Br…Cl bond strength and length gradually from the values common for the weak intermolecular complexes to that approaching a fully developed covalent bond. This continuity of halogen bond strength in the experimental (solid-state) and calculated associations indicates a fundamental link between the covalent and supramolecular bonding.
1. Introduction
Halogen bonding, R-X⋅⋅⋅D, is a directional attraction of electron-rich species D to electrophilic halogen atoms, X (covalently bonded to a group or atom R) [1,2]. Many experimental and computational studies demonstrated that its strength can be varied considerably by changing the nature of XB donors and acceptors [1,2,3,4,5,6,7]. The directionality of halogen bonding and the possibility of modulation of its strength open the way to a variety of applications of this intermolecular interaction in areas ranging from crystal engineering of solid-state materials and molecular recognition, to catalysis and rational drug design [1,2,8]. Besides practical applications, the XB variability provides an opportunity to explore the nature of supramolecular bonding and the boundary between such bonding and the covalent bond [6].
Most frequently, halogen bonding (XB) is explained using an electrostatic model which relates this interaction primarily to the attraction of electron-rich nucleophiles to the areas of positive potentials (σ-hole) on the surfaces of the covalently bonded halogen atoms [9]. These areas are formed along the extension of the covalent R–X bonds, and the positive potentials are enhanced by the presence of electron withdrawing substituents in R, as well as by high polarizability of the large bromine and iodine atoms. This model allows explaining directionality and variation of strength of many XB complexes, especially when the polarization of the reactants is taken into account [10].
Many studies pointed out that besides electrostatic attraction and dispersion, weakly-covalent (charge-transfer) interactions also contribute substantially to the formation of XB complexes [11,12,13,14,15,16,17]. These suggestions were supported by the computational analyses of the components of interaction energies [11,12,13], and by experimental evidence such as unexpected trends in the XB strengths [14], UV–Vis [15] and photoelectron spectra [16,17], structural features [18], and charge-density analysis [19] of the complexes, as well as by a drastic increase in the electron-transfer rates induced by halogen bonding [20,21,22]. These features are especially pronounced in strongly-bonded complexes. In fact, the XB lengths in some of the complexes are 30–35% shorter than the van der Waals separations of the corresponding atoms [6,23]. Accordingly, they are within 10–15% of the corresponding covalent bonds. In other words, the bond lengths of strong halogen bonds are closer to that of a conventional covalent bond than to weak intermolecular interactions. Such observations raise a fundamental question about the existence and location of a boundary between supramolecular and covalent bonds involving halogen atoms.
We have recently demonstrated a gradual change in Br···N bond lengths and strengths in a series of XB complexes of brominated electrophiles R-Br with 1,4-diazabicyclo[2.2.2]octane from the values typical for weak intermolecular associates to that characteristic of a covalent bond [6]. In the current work, we present the results of the X-ray structural and computational characterization of XB complexes formed between R-Br electrophiles and Cl− anions. The latter are strong nucleophiles which form complexes with various halogenated electrophiles (and several X-ray structures of such associations are available in the literature, vide infra). Importantly, they allow extending the analysis of bonding in complexes of neutral species to the associations between neutral or cationic XB donor and anionic XB acceptor.
2. Materials and Methods
2.1. Materials
Commercially available tetrabromomethane, tribromofluoromethane, hexabromoacetone, 1,4-diazabicyclo[2.2.2]octane (DABCO) and tetra-n-propylammonium chloride ((Pr4N)Cl) (all from Sigma-Aldrich) were used without additional purification. Tribromoacetamide was prepared by reaction of hexabromoacetone with NH4OH, and tribromoacetonitrile was synthesized by dehydration of CBr3CONH2 with P2O5 [24]. Tribromonitromethane was synthesized by bromination of nitromethane as described earlier [25].
2.2. Crystallization and X-ray Structural Analysis
Single crystals of XB associates suitable for X-ray measurements were obtained by evaporating solutions containing mixtures of an R-Br molecule and (Pr4N)Cl in acetone or by diffusion of hexane into dichloromethane solutions containing mixtures of R-Br and DABCO. The latter utilized the Menshutkin reaction between DABCO and CH2Cl2 resulting in formation of Cl− anions which crystallize with R-Br electrophiles and DABCO-CH2Cl+ cations (similar reaction was reported earlier by McMurtie and co-workers [26]). For example, 22 mg (0.10 mmol) of (Pr4N)Cl was dissolved together with 33 mg (0.10 mmol) of CBr4 in acetone. Slow evaporation of the solution produced colorless (Pr4N)Cl⋅CBr4 crystals. Crystals of (Pr4N)Cl·CBr3COCBr3 were obtained in a similar way from an acetone solution containing 54 mg (0.010 mmol) of hexabromoacetone and 22 mg (0.10 mmol) of (Pr4N)Cl. Co-crystals of (Pr4N)Cl·2CBr3CN and (Pr4N)Cl·CBr3F were obtained by evaporation of acetone solutions containing (Pr4N)Cl and an excess of the liquid electrophiles. (Note: X-ray analysis of (Pr4N)Cl·CBr3COCBr3 and (Pr4N)Cl·2CBr3CN co-crystals revealed that some Cl− anions are replaced by Br− anions, which were produced by the substitution reaction of Cl− with the R-Br electrophiles.) Co-crystals of (DABCO-CH2Cl)Cl·CBr3NO2 were obtained by dissolution of 30 mg (0.010 mmol) of CBr3NO2 and 12 mg (0.011 mmol) of DABCO in 5 mL of dry dichloromethane in a Schlenk tube. The resulting solution was carefully layered with 2 mL of a 1:1 dichloromethane/hexane mixture, and then with hexane (20 mL), and kept at −35 °C. Slow diffusion of hexane into the dichloromethane resulted in the formation of colorless crystals suitable for single crystal X-ray measurements. Co-crystals of (DABCO-CH2Cl)Cl·CBr3CONH2 were obtained in a similar way using 30 mg (0.010 mmol) of tribromoacetamide instead of tribromonitromethane.
The single crystal structures (except (DABCO-CH2Cl)Cl·CBr3NO2) were examined on a Bruker Quest diffractometer with a fixed chi angle, a sealed tube fine–focus X-ray tube, single crystal curved graphite incident beam monochromator, a Photon100 area detector and an Oxford Cryosystems low-temperature device (all X-ray instrumentation was from Bruker AXS Inc., Madison, WI, USA). Data were collected at 150 K. Examination and data collection were performed with Mo Kα radiation (λ = 0.71073 Å). A single crystal of (DABCO-CH2Cl)Cl·CBr3NO2 was analyzed with a Bruker Kappa APEX CCD area detector diffractometer with (microsource) Cu Kα radiation (λ = 1.54178 Å) at 100 K. Reflections were indexed and processed, and the files were scaled and corrected for absorption using APEX3 [27]. The space groups were assigned using XPREP within the SHELXTL suite of programs [28], the structures were solved by direct methods and refined by full matrix least squares against F2 with all reflections using Shelxl2018 [29,30] using the graphical interface Shelxle [31]. If not specified otherwise, H atoms attached to carbon and nitrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C-H bond distances of 1.00, 0.99 and 0.98 Å for aliphatic C-H, CH2 and CH3 moieties and N-H distances of 0.88 Å, respectively. Methyl H atoms were allowed to rotate but not to tip to best fit the experimental electron density. Uiso(H) values were set to a multiple of Ueq(C) with 1.5 for CH3, and 1.2 for C-H and N-H units, respectively. X-ray structural data were presented using the Mercury 2020.2.0 program [32].
(Pr4N)Cl·CBr3F (1). Chemical formula C12H28N·CBr3F·Cl, M = 492.54 g/mol. Monoclinic, space group Pn (no. 7), a = 8.3500 (5) Å, b = 8.8217 (5) Å, c = 13.0282 (7) Å, β = 97.1844 (19)°, V = 952.14 (9) Å3, Z = 2, T = 150 K, μ(MoKα) = 6.49 mm−1, Dcalc = 1.718 g/cm3, 22227 reflections measured, 6401 unique (Rint = 0.047). The final R1 was 0.030 (I > 2σ(I)) and wR2 was 0.065 (all data).
(Pr4N)Cl·CBr4 (2). Chemical formula C12H28N·CBr4·Cl, M = 553.45 g/mol. Monoclinic, space group P21/n (no. 14), a = 8.3237 (4), Å, b = 18.7831 (9) Å, c = 12.8399 (6) Å, β = 100.5800 (16)°, V = 1973.33 (16) Å3, Z = 4, T = 150 K, μ(MoKα) = 8.28 mm−1, Dcalc = 1.863 g/cm3, 42420 reflections measured, 7519 unique (Rint = 0.061). The final R1 was 0.034 (I > 2σ(I)) and wR2 was 0.083 (all data).
(Pr4N)Cl·CBr3COCBr3 (3). Chemical formula C12H28N·C3Br6O·0.101(Br)·0.899(Cl), M = 757.73 g/mol. Orthorhombic, space group Pbca (no. 61), a = 15.6671 (10) Å, b = 15.0131 (8) Å, c = 19.9888 (13) Å, V = 4701.6 (5) Å3, Z = 8, T = 150 K, μ(MoKα) = 10.52 mm−1, Dcalc = 2.141 g/cm3, 89148 reflections measured, 8925 unique (Rint = 0.068). The final R1 was 0.037 (I > 2σ(I)) and wR2 was 0.090 (all data).
(Pr4N)Cl·2CBr3CN (4). Chemical formula C12H28N·2(C2Br3N)·0.245(Br)·0.755(Cl), M = 788.22 g/mol. Monoclinic, space group C2/c (no. 15), a = 8.2066 (6) Å, b = 14.0978 (11) Å, c = 22.966 (3) Å, β = 94.942 (4)°, V = 2647.2 (4) Å3, Z = 4, T = 150 K, μ(MoKα) = 9.55 mm−1, Dcalc = 1.978 g/cm3, 24848 reflections measured, 4896 unique (Rint = 0.073). The final R1 was 0.050 (I > 2σ(I)) and wR2 was 0.163 (all data).
(DABCO·CH2Cl)Cl·CBr3NO2 (5). Chemical formula CBr3NO2·C8H15ClN·Cl, M = 493.86 g/mol. Monoclinic, space group P21/c (no. 14), a = 8.2066 (6) Å, b = 12.2997 (5) Å, c = 15.0315 (6) Å, β = 90.9993 (14)°, V = 1517.42 (10) Å3, Z = 4, T = 100 K, μ(CuKα) = 13.09 mm−1, Dcalc = 2.162 g/cm3, 2643 reflections measured, 2643 unique. The final R1 was 0.048 (I > 2σ(I)) and wR2 was 0.134 (all data).
(DABCO-CH2Cl)Cl·CBr3CONH2 (6). Chemical formula C7H14ClN2·C2H2Br3NO·Cl, M = 492.88 g/mol. Monoclinic, space group P21/c (no. 14), a = 6.8643 (3) Å, b = 16.6939 (8) Å, c = 13.1824 (7) Å, β = 90.029 (2)°, V = 1510.60 (13) Å3, Z = 4, T = 100 K, μ(MoKα) = 8.36 mm−1, Dcalc = 2.167 g/cm3, 36132 reflections measured, 5739 unique (Rint = 0.088). The final R1 was 0.050 (I > 2σ(I)) and wR2 was 0.106 (all data).
Complete crystallographic data, in CIF format, have been deposited with the Cambridge Crystallographic Data Centre. CCDC 2020744–2020749 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
2.3. Computations
Quantum-mechanical calculations were carried out using the Gaussian 09 suite of programs [33]. Geometries of electrophiles and halogen-bonded R–Br…Cl complexes were optimized without constraints in the gas phase and in dichloromethane via DFT calculations with the M06-2X functional and the 6-311+G(d,p) basis set [34]. Previous theoretical analyses indicated that such commutations provide excellent geometries and energies at a reasonable computational cost [35,36,37]. Importantly, our earlier studies demonstrated that characteristics of the XB complexes between R-Br electrophiles with halide or pseudohalide anions obtained via such computations were in good agreement with experimental data [15,21,38,39]. Since previous works showed that the solid-state environment is best modelled using calculations with a moderately polar solvents as the medium [15], the geometry optimizations in the current work were done via computations in the gas phase and in dichloromethane (the latter were carried out using a Polarizable Continuum Model [40]). The absence of imaginary vibrational frequencies confirmed that the optimized structures represent true minima. Values of halogen-bond energies ΔE were determined as: ΔE = Ecomp − (ER-Br + ECl) + BSSE, where Ecomp, ER-Br and ECl are sums of the electronic and ZPE of the optimized complex, R-Br and chloride, respectively, and BSSE is a basis set superposition error [41]. Since formation of complexes involve deformation of R-Br, these values can be expressed as ΔE = ΔEstrain + ΔEint, where ΔEint is an interaction energy between deformed reactants [42]. The ΔEint values follow the same trends as those of ΔE (see Figure S3 in the Supporting Information). Energies and atomic coordinates of the calculated complexes are listed in the Supporting Information.
3. Results and Discussion
3.1. Crystallization and X-ray Structural Characterization of the Solid-State Associates of R-Br with Cl−
Evaporation of a solution containing a mixture of tribromofluoromethane and Pr4NCl (1:1 molar ratio) in acetone resulted in formation of colorless block-like crystals containing both the organic molecule and the chloride salt. X-ray structural analysis of these monoclinic co-crystals 1 (Pn space group) revealed the presence of quasi-2D networks formed by brominated electrophiles and chloride anions. Each CBr3F molecule in these networks is coordinated (through its three bromine substituents) with three Cl− anions. In turn, each chloride is bonded to three CBr3F molecules. Such 3:3 CBr3F/Cl− bonding led to formation of zigzag networks consisting of twelve-membered rings. Since C-Br…Cl− fragments are close to linear, these rings resemble cyclohexane in a chair conformation (Figure 1a).
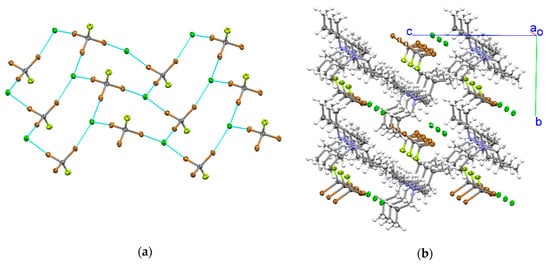
Figure 1.
(a) 2D network formed by tribromofluoromethane and Cl− anions in (Pr4N)Cl·CBr3F co-crystals 1. (Note: blue lines show halogen bonds.) (b) Crystal lattice in the co-crystals.
The CBr3F molecules and Cl− anions propagated along a axes. The Pr4N+ counter-ions are located in the channels of the XB network (Figure 1b).
The intermolecular Br…Cl distances in the networks formed by CBr3F and chloride anions were 3.0820 (10) Å, 3.1266 (9) Å and 3.1728 (10) Å. Such separations are 10–15% smaller than the sum of the van der Waals radii of bromine and chlorine of 3.66 Å [43]. The corresponding C-Br…Cl− angles were 171.80 (11)°, 168.05(11)/° and 178.51 (11)°.
Analogous co-crystallizations of Pr4NCl with tetrabromomethane produced a salt co-crystal 2 which comprised hybrid networks formed by a 3:3 halogen-bonded association between CBr4 and Cl− anions. They consisted of twelve-membered rings similar to that in the co-crystals with CBr3F (Figure 2).
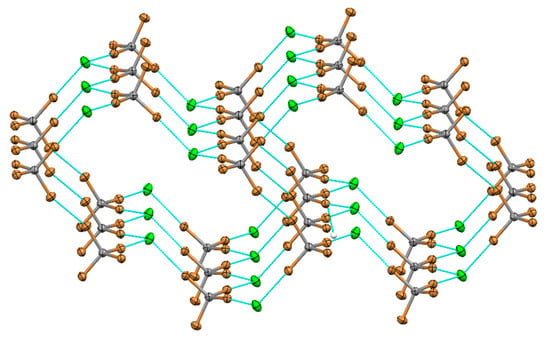
Figure 2.
Halogen-bonded (XB) network formed by tetrabromomethane and Cl− anions in (Pr4N)Cl·CBr4 co-crystals 2.
The three crystallographically non-equivalent Br…Cl halogen bond lengths of 3.0650 (7) Å, 3.1769 (7) Å and 3.2431 (7) Å in these co-crystals were in the same range as those in the crystals with CBr3F. The fourth bromine atom in CBr4 formed a symmetric contact referred to as Type I halogen–halogen contact [1] with a symmetry equivalent CBr4 bromine atom from another layer (Figure 2). The Br⋅⋅⋅Br distance is 3.3711 (7) Å and both C-Br⋅⋅⋅Br angles are 151.62 (7)°. The Pr4N+ counter-ions in these crystals filled the channels formed by stacking of the XB layers propagating along the b axis. In should be mentioned in this respect that previous works showed the existence of two types of close halogen–halogen contacts with a clear geometric and chemical distinctions. All R-Br⋅⋅⋅Cl− interactions described herein follow description of Type II contacts, which occur between a nucleophile and the positively-charged areas on the surface of XB donor. These XB contacts are characterized by the R-Br⋅⋅⋅C− angles of approximately 180°. In comparison, Type I contacts represent interactions between identical areas on the surfaces of two covalently-bonded halogens in the two R-X molecules, with the equal R-X⋅⋅⋅X angles (usually in the 150°–160° range). Resnati, Metrangolo et al. pointed out [1] that (symmetrical) Type I contacts are related to close-packing requirements and they are not halogen bonding (which occur between electrophilic and nucleophilic sites according to the IUPAC definition).
Crystallization from a solution containing a mixture of Pr4NCl and hexabromoacetone resulted in formation of co-crystals 3 showing quazi-2D layers formed by 3:3 halogen bonding between the brominated electrophile and the anion (Figure 3). In contrast to the layers described above, they consisted of two types of cells. The smaller cells were formed by pairs of CBr3COCBr3 and pairs of Cl−. The larger cells comprised four XB donors linked by four chloride anions (Figure 3).
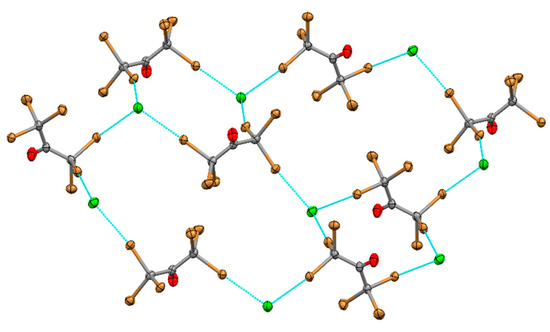
Figure 3.
XB network formed by hexabromoacetone and Cl− anions in (Pr4N)Cl·CBr3COCBr3 co-crystals.
The XB Br…Cl lengths measured in these crystals are 3.0306 (8) Å, 3.0618 (8) Å and 3.2056 (8) Å. Besides Br…Cl bonds, these crystals also show short Br…Br contacts of 3.5683 (8) Å between neighboring CBr3COCBr3 molecules.
Evaporation of a solution containing an excess of tribromoacetonitrile and Pr4NCl produced monoclinic crystals 4 (P21/c space group). They contained XB donors and acceptors in a 2:1 ratio forming layers parallel to the ab plane. These layers consist of columns of Cl− anions and double columns of CBr3CN molecules, and they are separated by the Pr4N+ counter-ions. Within the layers, each tribromoacetonitrile is bonded with three Cl− anions, with Br…Cl bond lengths of 3.0980 (4) Å, 3.1320 (4) Å and 3.1398 (4) Å (Figure 4a). In turn, each chloride coordinates six bromine atoms (Figure 4b).
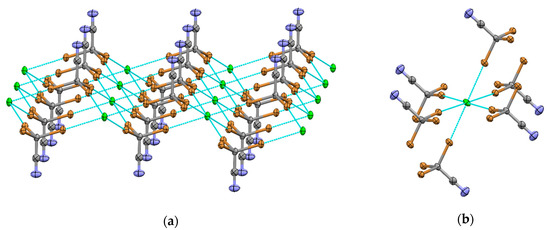
Figure 4.
(a) XB networks formed by tribromoacetonitrile and chloride anions in (Pr4N)Cl·CBr3CN co-crystals 4. (b) Fragment of the X-ray structure of co-crystals 4 showing a (hexacoordinated) Cl− anion bonded with six CBr3CN molecules.
Diffusion of hexane into a dichloromethane solution containing 1,4-diazabicyclo[2.2.2]octane (DABCO) and either tribromoacetamide or tribromonitromethane resulted in formation of co-crystals 5 and 6 containing R-Br, Cl− anions and the DABCO-CH2Cl+ counter-ions. Such crystallization utilized the Menshutkin reaction between DABCO and CH2Cl2 (producing Cl−) in the presence of the XB donors [26]. Co-crystals 5 of CBr3NO2 with Cl− anions and DABCO-CH2Cl+ counter-ions obtained in this way showed 2D layers (Figure 5).
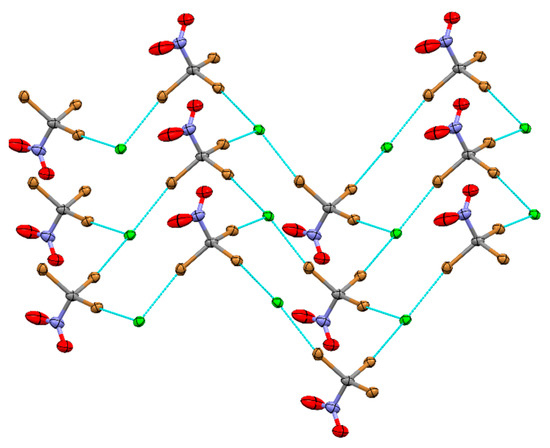
Figure 5.
XB networks in (DABCO-CH2Cl)Cl ·CBr3NO2 co-crystals 5.
The layers consisted of chair-like cells formed by 3:3 bonding of Cl− and CBr3NO2 (similar to that obtained with CBR3F or CBr4 with Pr4NCl), with Br…Cl− bond lengths of 3.1420 (17) Å, 3.1728 (16) Å and 3.1715 (16) Å. Stacking of the XB layers created channels that are filled with DABCO-CH2Cl+ counter-ions (which form also hydrogen bonds with the chloride anions, Figure S1 in the Supporting Information).
The analogous reaction between DABCO and CH2Cl2 in the presence of tribromoacetamide led to formation of (DABCO-CH2Cl)Cl·CBr3CONH2 crystals 6. They showed multiple types of halogen and hydrogen bonding (Figure 6). Most relevant for the current work was 2:2 bonding between bromine substituents of CBr3CONH2 and Cl− anions in which each chloride is bonded with two electrophiles and two substituents of the latter that are bonded to two Cl−. The lengths of these bonds were 3.0457 (12) Å and 3.2770 (12) Å. The CBr3CONH2 molecules are also hydrogen-bonded (via the NH2 group) with another Cl− anion (Figure 6). Besides, co-crystals showed a halogen Cl…Cl− bond of 3.2479 (16) Å between chlorine substituent of DABCO-CH2Cl+ and Cl− anions, as well as two hydrogen bonds between the cations and chlorides.
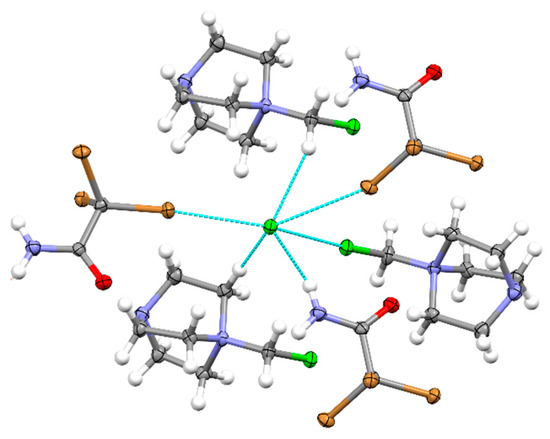
Figure 6.
Intermolecular interactions by chloride anions with tribromoacetamide and DABCO-CH2Cl+ in (DABCO-CH2Cl)Cl ·CBr3CONH2 co-crystals 6. (Note: blue line show contacts shorter than the sum of van der Waals separations, counter-ions are omitted for clarity.).
The Br…Cl− bond length measured in the solid-state XB associations prepared in the current work together with several representative examples of such bonds available in the crystallographic literature [44] are summarized in Table 1. The Br…Cl distances measured in the XB associations described above are within the range from 3.00 Å to 3.25Å. As such, they are 10% to 20% shorter than the van der Waals separations. The crystallographic database contains examples of XB associations showing substantially longer Br…Cl bonds. For example, the complexes of chloride anions with bromopyridinium or dibromopyrrolidine cations show Br…Cl distances in the 3.30 Å–3.50 Å range (which is close to the sum of the van der Waals radii of bromine and chloride of 3.66 Å [43]). On the other hand, the Br…Cl distances in complexes of Cl− anions with N-bromosuccinimide or N-bromophthalimidesome of 2.70 Å–2.80 Å are closer to the covalent Br–Cl bond (2.14 Å [45]) than to the Br…Cl− van der Waals separation.

Table 1.
Halogen Br…Cl bond length in the solid-state associations.
It should be noted, however, that the Br…Cl distances vary substantially in the solid-state associations of Cl− anion with the same R-Br electrophile (Table 1). For example, the co-crystals of CBr4 with chloride show Br…Cl distances from 3.07 Å to 3.24 Å, and those of CBr3NO2 show distances from 2.99 Å to 3.27 Å. Apparently, such bonding is characterized by a shallow energy minimum, and its length can vary considerable in response to crystal forces. Furthermore, each XB donor in these solid-state networks is bonded with several Cl− anions and each anion is bonded to several electrophiles. Such multiple bondings can noticeably affect the XB length in the solid state [46]. Thus, to verify variations in the XB length derived from the X-ray crystallographic data, we carried out computational analysis of 1:1 XB complexes.
3.2. Quantum-Mechanical Computations of Halogen-Bonded R–Br…Cl− Complexes
Optimization of R-Br…Cl− pairs (carried out in vacuum and in dichloromethane as a medium; see the Experimental section) produced energy minima showing Br…Cl halogen bonds. While some of the R-Br electrophiles could also form hydrogen-bonded complexes with Cl− anions, this work is focused on the minima showing single halogen bonding between reactants (with the X–Br–Cl angles in the range between 170° and 180°, which is common for halogen bonding). The calculated interaction energies, ΔE, and the Br…Cl− separations, dBr···Cl, in the XB complexes calculated in dichloromethane are listed in Table 2. The energies of the complexes and individual components, X–Br–Cl angles and X–Br bond length, as well as the corresponding characteristics of the complexes obtained from the calculations in vacuum and atomic coordinates of all complexes are presented in the Supporting Information. For comparison, Table 2 also presents characteristics of the BrCl molecule, which could be formally considered as a result of interaction between a Br+ electrophile and a Cl− nucleophile.
Table 2.
Characteristics of the calculated R–Br·Cl− complexes.
Comparison with the experimental data in Table 1 demonstrates that the Br…Cl distances in the complexes calculated in dichloromethane are mostly within the range of the Br…Cl separations observed in the solid-state associations. The correlation between calculated and average experimental bond length in these complexes is characterized by an R2 of 0.94 (see Figure S2 in the Supporting Information). The Br…Cl distances in the calculated gas-phase complexes are, on average, approximately 0.25 Å shorter than that in dichloromethane. As such, they are also substantially shorter than that measured for the corresponding solid-state associations (Table 1). Further, ΔE values for the XB complexes of Cl− anions with the same R-Br molecule are more negative for the pairs calculated in vacuum than for the corresponding pairs calculated in dichloromethane. These results are consistent with the data reported earlier showing a higher bonding strength of the XB complexes involving an anionic XB acceptor when calculated in the gas-phase [15,22]. Further, while most of the complexes calculated in dichloromethane and all complexes calculated in the gas phase are characterized by the negative ΔE values (i.e., they are thermodynamically stable (Table S1 in the Supporting Information), the complexes with the weakest R-Br electrophiles calculated in dichloromethane are characterized by the positive ΔE values (i.e., they are only kinetically stable).
Most notably, the data in Table 2 and Table S1 in the Supporting Information show a gradual change in the XB Br…Cl length from the values which are close to the sum of the van der Waals radii of these atoms (R~1) to those which are 40% shorter than these separations. The latter are within 10% of the covalent Br–Cl bond (2.14 Å). The decrease in the Br…Cl distances is accompanied by a progressive increase in the interaction strength (Table 2 and Table S1 in the Supporting Information). Besides, in the stronger complexes, the shortening of the Br…Cl distance is accompanied by an elongation of the X–Br bond length (see the dX–Brcom/dX–Brsep ratios in Table 2 and in Table S1 in the Supporting Information). Importantly, the appropriate choice of R-Br electrophiles in the series of the complexes in Table 1 and Table 2 (and Table S1 in the Supporting Information) allowed eliminating any large changes between subsequent entries. As such, the energies and interatomic distances in these series cover the whole range between weak van der Waals complexes and the covalently-bonded BrCl molecule leaving no substantial gaps between subsequent entries (Figure 7).
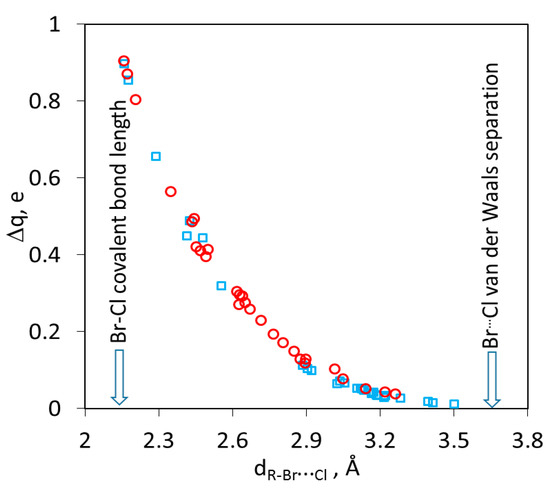
Figure 7.
Relation between interatomic Br…Cl distances and amount of charge transfer in the R–Br∙Cl− complexes calculated in vacuum (○) or dichloromethane (□).
As the distance between Br and Cl decreased, substantial parts of the negative charge which was originally residing on the chloride anions were transferred onto the R-Br electrophile (the relation between the intermolecular distances and amount of charge transfer Δq can be described by the equation dBr…Cl= −0.303ln(Δq) + 2.2121 with R2 of 0.98; see Figure S4 in the Supporting Information). Accordingly, the highest occupied molecular orbitals (HOMO) of the weak complexes are almost completely localized on the chloride anions, and their lowest unoccupied molecular orbital is on the R-Br electrophiles. The HOMOs and LUMOs of the strong complexes are delocalized over both counterparts (Figure 8). As such, while the HOMO→LUMO electronic transition in the weak complexes represents essentially a charge transfer from the donor to acceptor, the analogous transition in the strong complexes does not involve a substantial charge redistribution between the reactants. Finally, due to substantial elongation of the N–Br bond in the complexes between N-bromopyrazinium and chloride and similar complexes in the bottom of Table 2, these associations can be described as an association between a group R and a BrCl molecule (Figure 8, right). This underlines the fact that as the Br…Cl distance decreases, the intermolecular bond is gradually transformed into a covalent bond and the intramolecular R–Br bond is converted into an intermolecular one. It is important to stress the continuous character of these changes, which underscore the direct link between the limiting types (intra- and intermolecular) of bonds.
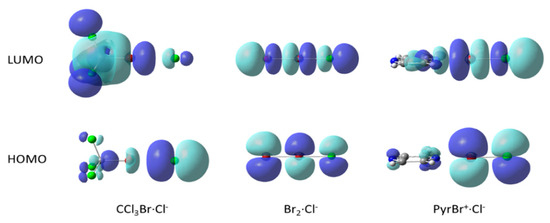
Figure 8.
Variations in the Highest Occupied Molecular Orbital (HOMO) and Lowes Unoccupied Molecular Orbital (LUMO) shapes in the associations between R-Br electrophiles and Cl− anions.
4. Conclusions
Experimental and computational studies on a series of complexes formed by R-Br electrophiles and chloride anions showed a wide variation of the structural and thermodynamic features of these associations. At the one extreme, the intermolecular Br···Cl distances were within 5% of the sum of the van der Waals radii of bromine and chloride, and the interaction energies were close to zero. Halogen bonding within these complexes was accompanied by a very small charge transfer and such intermolecular interaction practically did not affect intramolecular bond length in the R-Br electrophiles. At the other extreme, the series comprised complexes in which the Br···Cl distances were within 10% of a covalent Br–Cl bond. Charge delocalization between R-Br and Cl− in these associations was accompanied by the elongation of the intramolecular X–Br bond. As such, these complexes could be considered as an association of a neutral or anionic residue R with a BrCl molecule. The series under study contained complexes with characteristics covering the whole range of values between the limiting cases, with no substantial gaps between successive entries. It showed that the characteristics of the Br···Cl bond can change gradually from the values typical for intermolecular associates to that of a covalent Br–Cl bond. Together with the similar series of complexes between R-Br and DABCO or between diiodine and heteroaromatic N-oxides [6,7], and other systems [48,49] this indicates a generality of the continuum between the limiting types of bonding.
It should be noted that the involvement of the weakly-covalent interactions in the formation of XB complexes represents a major topic of the ongoing discussions about the nature and properties of halogen bonding (and many other supramolecular interaction, e.g., hydrogen bonding [50,51] or multicenter π-bonding between ion-radicals [52,53]). Indeed, the electrostatic (σ-hole) model explains linear geometry and (if polarization is taken into account) energy variations in a large number of XB complexes [9,10]. Yet, many computational studies indicated that covalent (charge transfer) interactions represent an important part of the bonding in many such associates [11,12,13]. Experimental studies of the XB complexes also revealed many features which can be explained by involvement of charge transfer. For example, photoelectron spectra of XB complexes and charge-density analysis directly demonstrated charge transfer from XB acceptor (nucleophile) to XB donor (electrophile) indicating covalency of halogen bonding [16,19]. Furthermore, appearance of a strong absorption bands in the UV–Vis spectra of complexes and structural characteristics of many such associates suggest that the molecular-orbital interactions between XB species play a vital role in their formation [15,18]. While such features are usually most pronounced in the strong XB complexes, the data presented herein verify continuity of the bonding from the weak supramolecular associations to a covalent bond. This continuum implies an intrinsic link between the limiting types of bonding and, therefore, it suggests an onset of covalency in intermolecular interactions. The progressive increase in its input leads to the transformation of a weak supramolecular interaction into a covalent bond.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/10/12/1075/s1, Figure S1: X-ray structure of (DABCO-CH2Cl)Cl·CBr3NO2. Figure S2: Correlation between calculated and experimental Br…Cl distances. Figure S3. Correlation between calculated ΔE and ΔEINT. Figure S4. Correlation between calculated values of charge transfer and intermolecular distances. Table S1: Characteristics of the R–Br·Cl− complexes calculated in the gas phase. Table S2: Calculated energies of the R-Br− electrophiles and the R–Br·Cl− complexes. Table S3. Atomic coordinates of the calculated R–Br·Cl− complexes.
Author Contributions
Conceptualization and methodology, S.V.R.; crystal preparation, S.V.R.; X-ray crystallographic analysis, M.Z.; quantum mechanical computations. C.L; data analysis, S.V.R.; writing—original draft preparation, S.V.R.; writing—review and editing, C.L., M.Z. and S.V.R.; visualization, S.V.R.; supervision, S.V.R.; funding acquisition, S.V.R. and M.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Division of Chemistry of the National Science Foundation, grant numbers CHE-1607746 and CHE-2003603. X-ray structural measurements were supported by the National Science Foundation through the Major Research Instrumentation Program under Grant No. CHE 1625543 (funding for the single crystal X-ray diffractometer).
Acknowledgments
We thank Charlotte Stern (Northwestern University) for X-ray structure determination of (DABCO-CH2Cl)Cl·CBr3NO2 co-crystals.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.-W.; Jiang, Y.-J.; Guo, M.; Hu, G.-X.; Zhang, B.; Liu, H.-C.; Yu, Q.-S. Ab Initio Study of the Complexes of Halogen-Containing Molecules RX (X=Cl, Br, and I) and NH3: Towards Understanding the Nature of Halogen Bonding and the Electron-Accepting Propensities of Covalently Bonded Halogen Atoms. Chem. Eur. J. 2005, 11, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of Halogen Bonding in Solution: Substituent, Structural, and Solvent Effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, C.; Hines, R.; Zeller, M.; Rosokha, S.V. Continuum of covalent to intermolecular bonding in the halogen-bonded complexes of 1,4-diazabicyclo[2.2.2]octane with bromine-containing electrophiles. Chem. Commun. 2018, 54, 8060–8063. [Google Scholar] [CrossRef] [PubMed]
- Borley, W.; Watson, B.; Nizhnik, Y.P.; Zeller, M.; Rosokha, S.V. Complexes of Diiodine with Heteroaromatic N-Oxides: Effects of Halogen-Bond Acceptors in Halogen Bonding. J. Phys. Chem. A 2019, 123, 7113–7123. [Google Scholar] [CrossRef] [PubMed]
- Pennington, W.T.; Resnati, G.; Taylor, M.S. Halogen bonding: From self-assembly to materials and biomolecules. CrystEngComm 2013, 15, 3057. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On The Nature of the Halogen Bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef] [PubMed]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Kraka, E.; Cremer, D. The intrinsic strength of the halogen bond: Electrostatic and covalent contributions described by coupled cluster theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. Unexpected trends in halogen-bond based noncovalent adducts. Chem. Commun. 2012, 48, 7708. [Google Scholar] [CrossRef] [PubMed]
- Rosokha, S.V.; Stern, C.L.; Ritzert, J.T. Experimental and Computational Probes of the Nature of Halogen Bonding: Complexes of Bromine-Containing Molecules with Bromide Anions. Chem. Eur. J. 2013, 19, 8774–8788. [Google Scholar] [CrossRef]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for Halogen Bond Covalency in Acyclic and Interlocked Halogen-Bonding Receptor Anion Recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef]
- Kellett, C.W.; Kennepohl, P.; Berlinguette, C.P. π covalency in the halogen bond. Nat. Commun. 2020, 11, 3310. [Google Scholar] [CrossRef]
- Grounds, O.; Zeller, M.; Rosokha, S.V. Structural preferences in strong anion-π and halogen-bonded complexes: π- and σ-holes vs frontier orbitals interaction. New J. Chem. 2018, 42, 10572–10583. [Google Scholar] [CrossRef]
- Eraković, M.; Cinčić, D.; Molčanov, K.; Stilinović, V. A Crystallographic charge density study of the partial covalent nature of strong N⋅⋅⋅Br halogen bonds. Angew. Chem. Int. Ed. 2019, 58, 15702–15706. [Google Scholar] [CrossRef]
- Rosokha, S.V. Electron-transfer reactions of halogenated electrophiles: A different look into the nature of halogen bonding. Faraday Discuss. 2017, 203, 315–332. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Traversa, A. From charge transfer to electron transfer in halogen-bonded complexes of electrophilic bromocarbons with halide anions. Phys. Chem. Chem. Phys. 2015, 17, 4989–4999. [Google Scholar] [CrossRef] [PubMed]
- Rosokha, S.V.; Vinakos, M.K. Halogen bond-assisted electron transfer reactions of aliphatic bromosubstituted electrophiles. Phys. Chem. Chem. Phys. 2014, 16, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Ruttreddy, R.; Jurcek, O.; Bhowmik, S.; Makela, T.; Rissanen, K. Very strong –N-X+∙∙∙-O-N+ halogen bonds. Chem. Commun. 2016, 52, 2338–2341. [Google Scholar] [CrossRef] [PubMed]
- Suero, M.I.; McNeil, L.E.; Marquez, F. 15N isotopic effects on the Raman spectra of tribromoacetonitrile. J. Raman Spectrosc. 1987, 18, 273–275. [Google Scholar] [CrossRef]
- Heasley, V.L.; Titterington, D.R.; Rold, T.L.; Heasley, G.E. Bromination of nitroalkanes with alkyl hypobromites. J. Org. Chem. 1976, 41, 1285–1287. [Google Scholar] [CrossRef]
- Pfrunder, M.C.; Micallef, A.S.; Rintoul, L.; Arnold, D.P.; Davy, K.J.P.; McMurtrie, J.C. Exploitation of the Menshutkin Reaction for the Controlled Assembly of Halogen Bonded Architectures Incorporating 1,2-Diiodotetrafluorobenzene and 1,3,5-Triiodotrifluorobenzene. Cryst. Growth Des. 2012, 12, 714–724. [Google Scholar] [CrossRef]
- Bruker. Apex3 v2019.1-0, SAINT V8.40A; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- SHELXTL Suite of Programs; Version 6.14; Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2000.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Hübschle, C.; Sheldrick, G.; Dittrich, B. ShelXle: A Qt Graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theor. Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xu, Z.; Zhu, W. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Inf. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Stern, C.L.; Swartz, A.; Stewart, R. Halogen bonding of electrophilic bromocarbons with pseudohalide anions. Phys. Chem. Chem. Phys. 2014, 16, 12968–12979. [Google Scholar] [CrossRef]
- Watson, B.; Grounds, O.; Borley, W.; Rosokha, S.V. Resolving the halogen vs. hydrogen bonding dichotomy in solutions: Intermolecular complexes of trihalomethanes with halide and pseudohalide anions. Phys. Chem. Chem. Phys. 2018, 20, 21999–22007. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen Bonding versus Hydrogen Bonding: A Molecular Orbital Perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualising crystal structures. Acta Cryst. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.; Thorn, J. Equilibrium nuclear quadrupole coupling constants from the rotational spectrum of BrCl: A source of the electric quadrupole moment ratios Q (79 Br)/ Q (81 Br) and Q (35 Cl)/ Q (37 Cl). Chem. Phys. Lett. 1993, 215, 554–560. [Google Scholar] [CrossRef]
- Nizhnik, Y.P.; Sons, A.; Zeller, M.; Rosokha, S.V. Effect of supramolecular architecture on halogen bonding between diiodine and heteroaromatic N-oxides. Cryst. Growth Des. 2018, 18, 1198–1207. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Yannacone, S.; Oliveira, V.P.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Dolgushin, F.M.; Skabitsky, I.V.; Popova, A.E.; Skabitskiy, I.V. Isomorphic substitution in molecular crystals and geometry of hypervalent tellurium: Comments inspired by a case study of RMeTeI2 and [RMe2Te]+I− (R = Ph, Fc). New J. Chem. 2019, 43, 12225–12232. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Sokalski, W.A.; Dyguda, E.; Leszczyński, J. Quantitative classification of covalent and noncovalent H-bonds. J. Phys. Chem. B 2006, 110, 6444–6446. [Google Scholar] [CrossRef]
- Kertesz, M. Pancake Bonding: An Uunusual pi-stacking interaction. Chem. Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef]
- Molčanov, K.; Jelsch, C.; Landeros, B.; Hernández-Trujillo, J.; Wenger, E.; Stilinović, V.; Kojić-Prodić, B.; Escudero-Adán, E.C. Partially Covalent Two-Electron/Multicentric Bonding between Semiquinone Radicals. Cryst. Growth Des. 2018, 19, 391–402. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).