Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock


Abstract
1. Introduction
2. Electrochemical Reduction of CO2 on Molecular Transition-Metal Catalysts
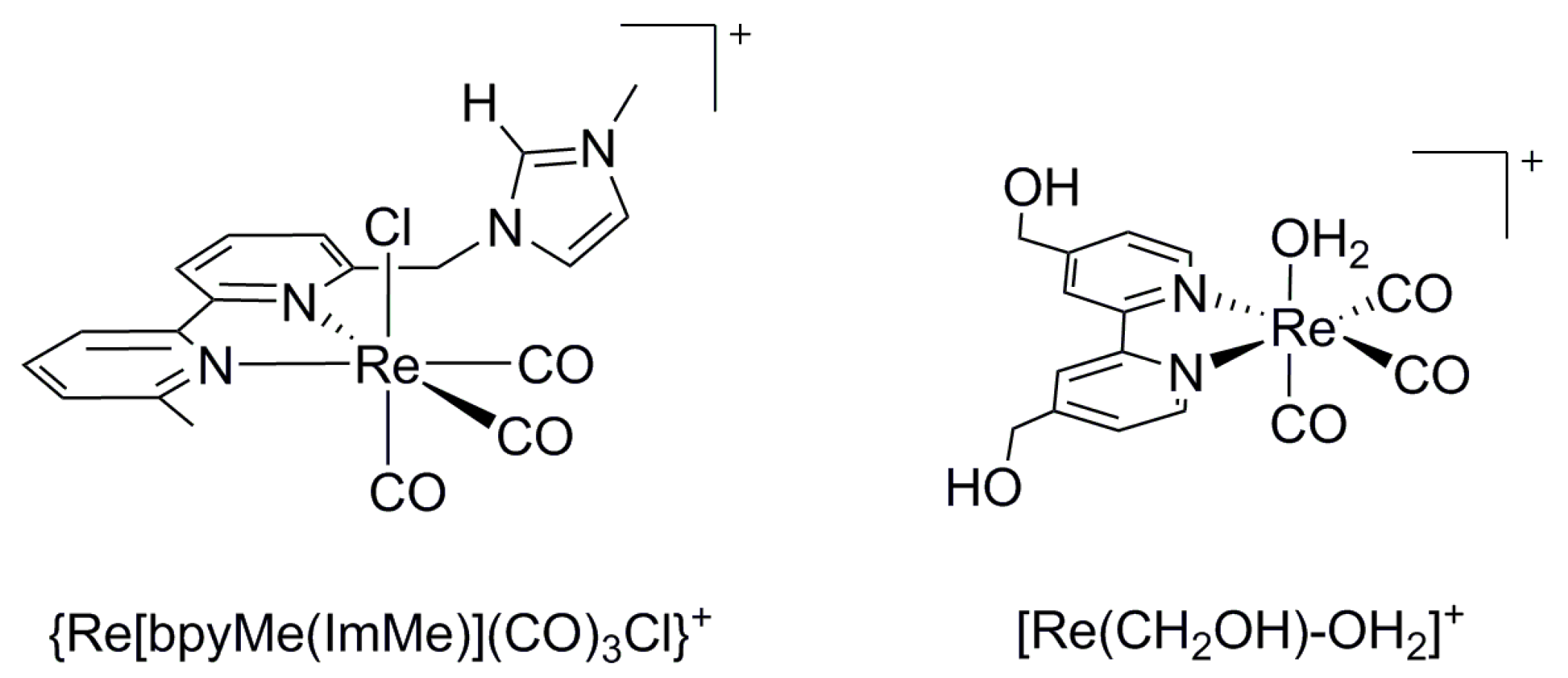
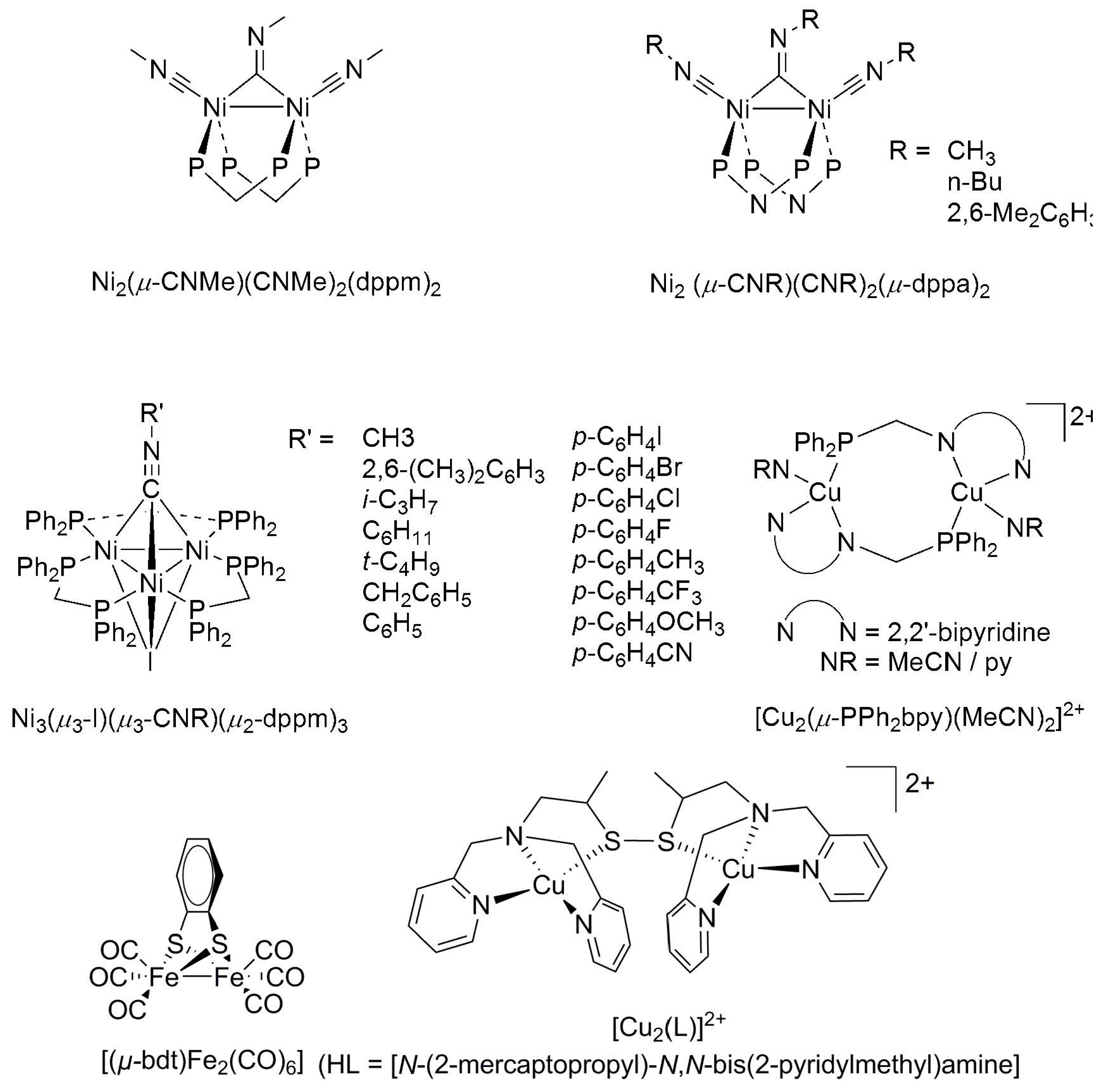
2.1. 4d and 5d Metal CO2 Reduction Electrocatalysts
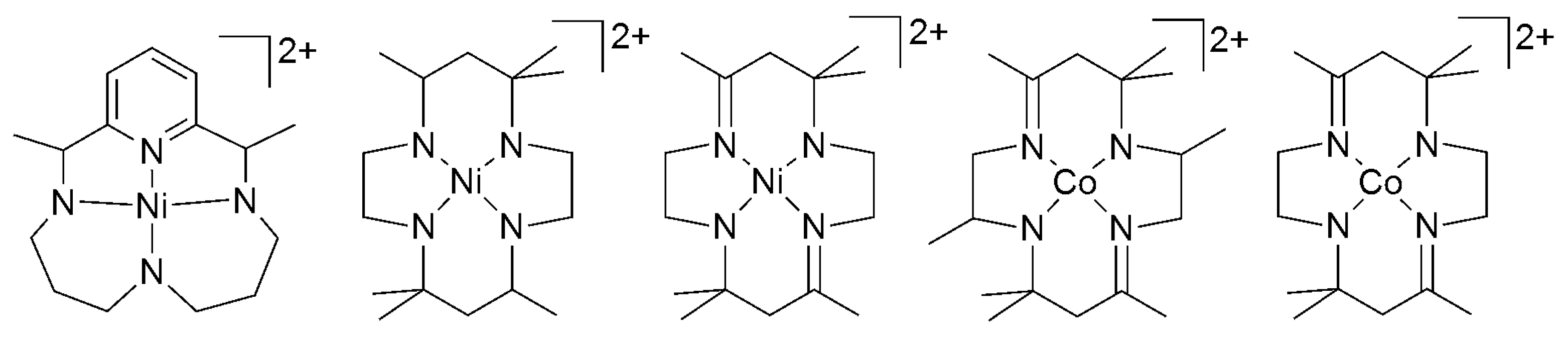
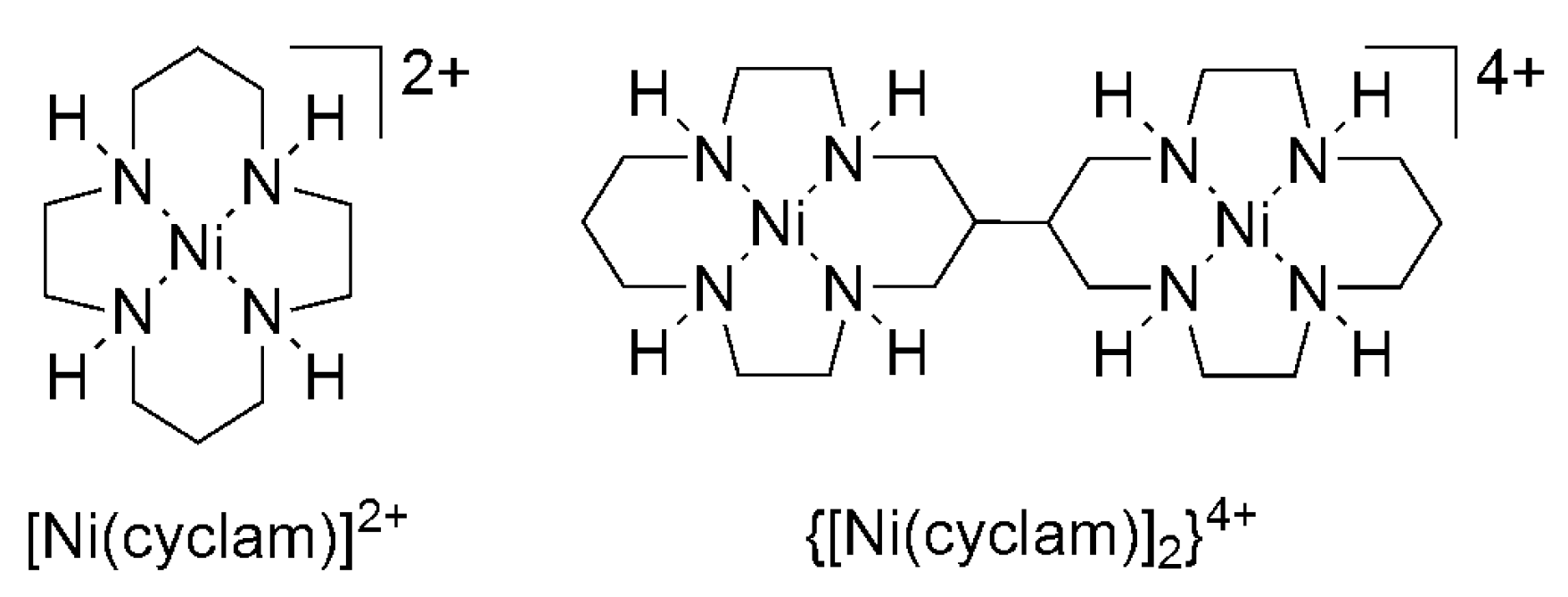
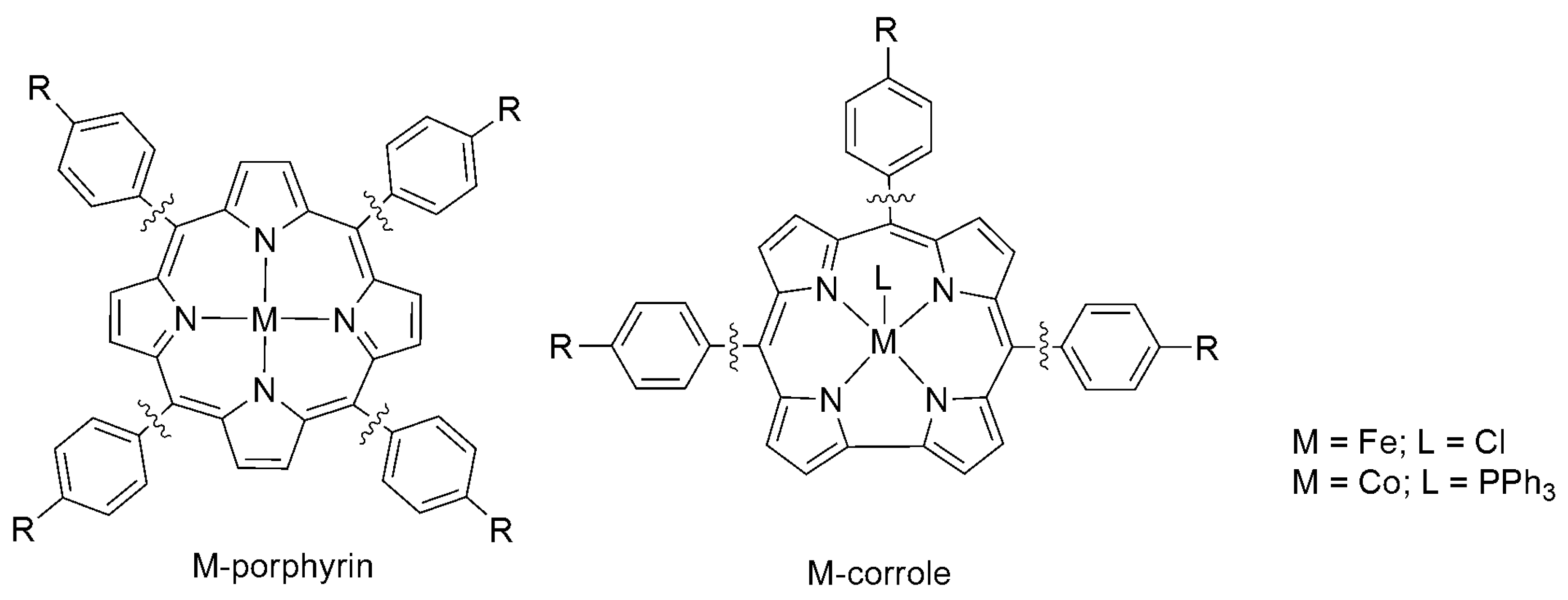
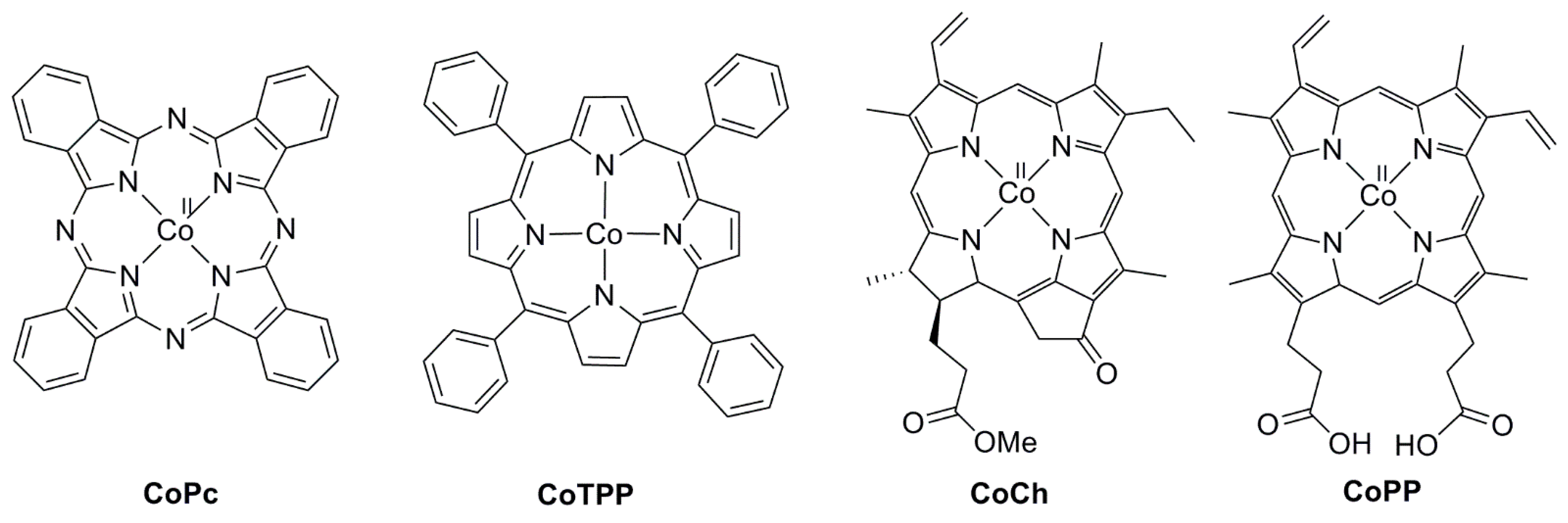
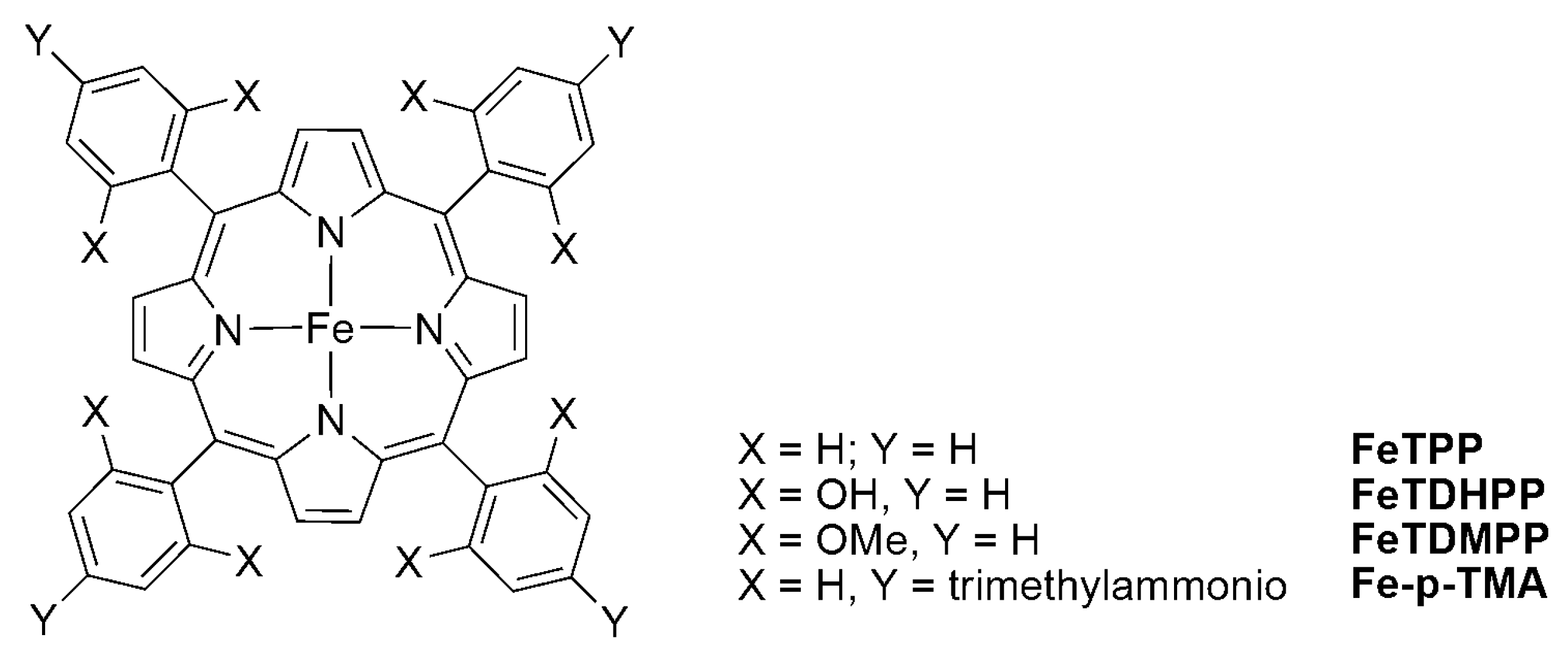
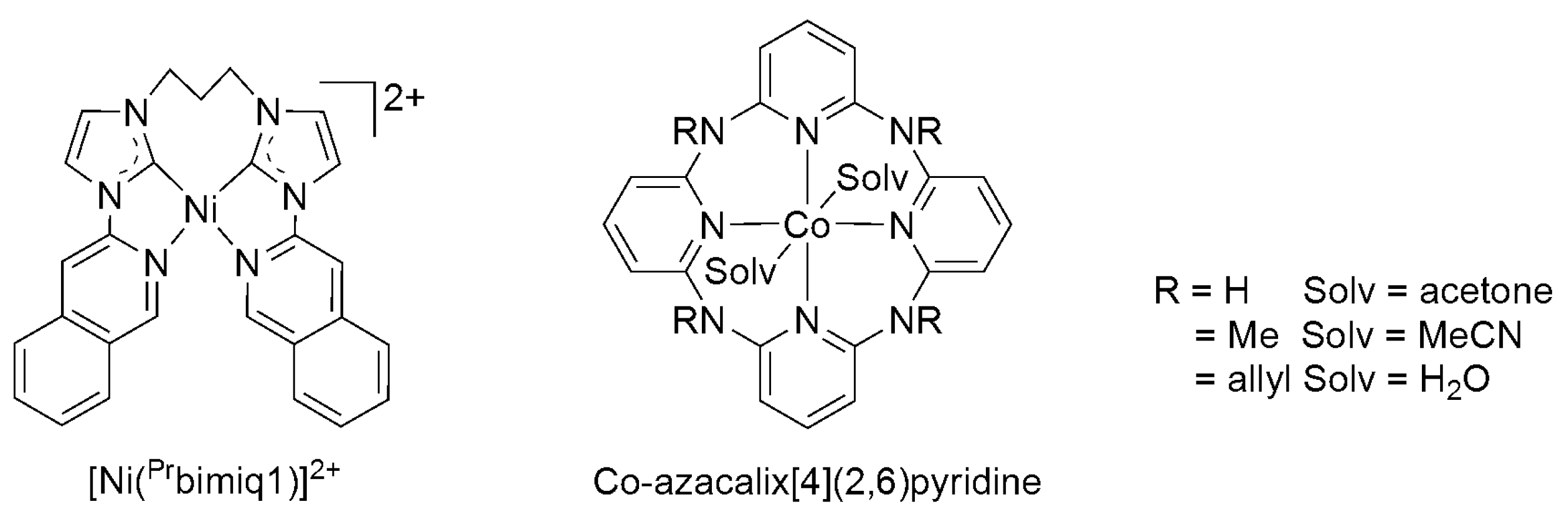
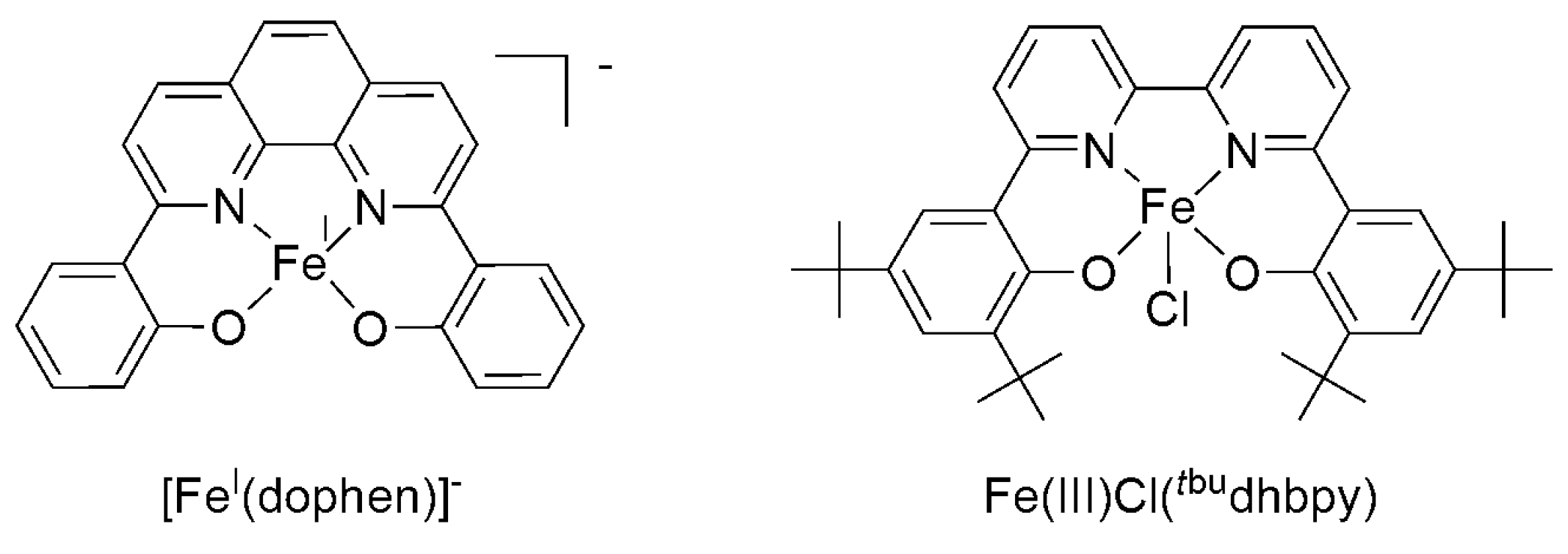
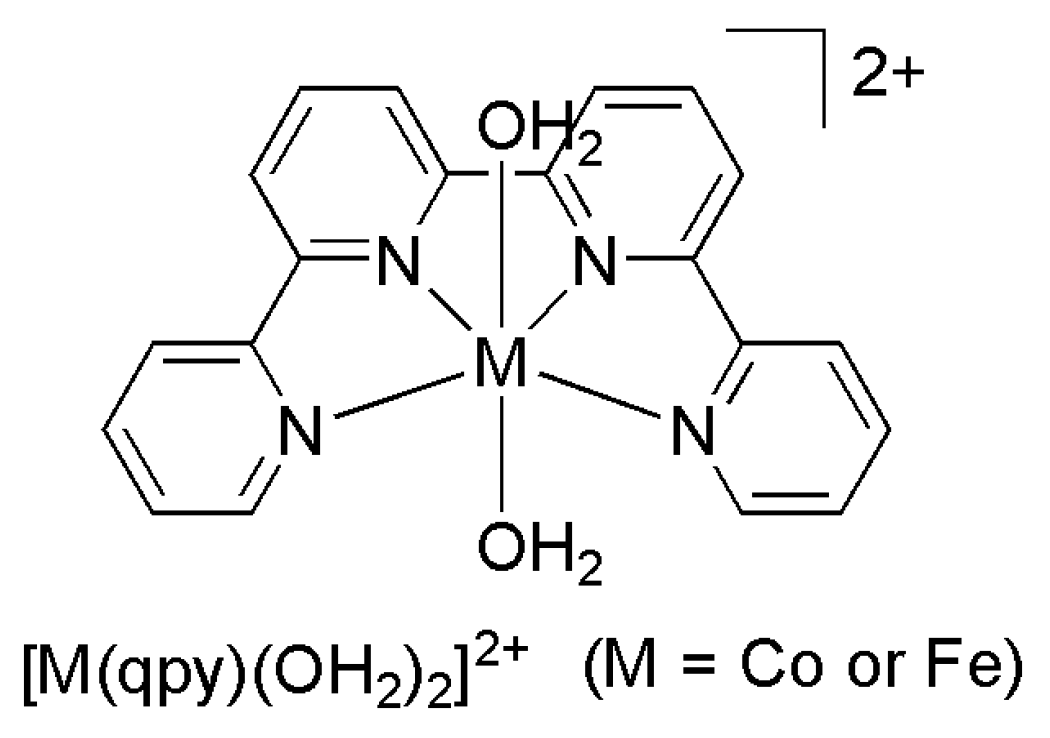
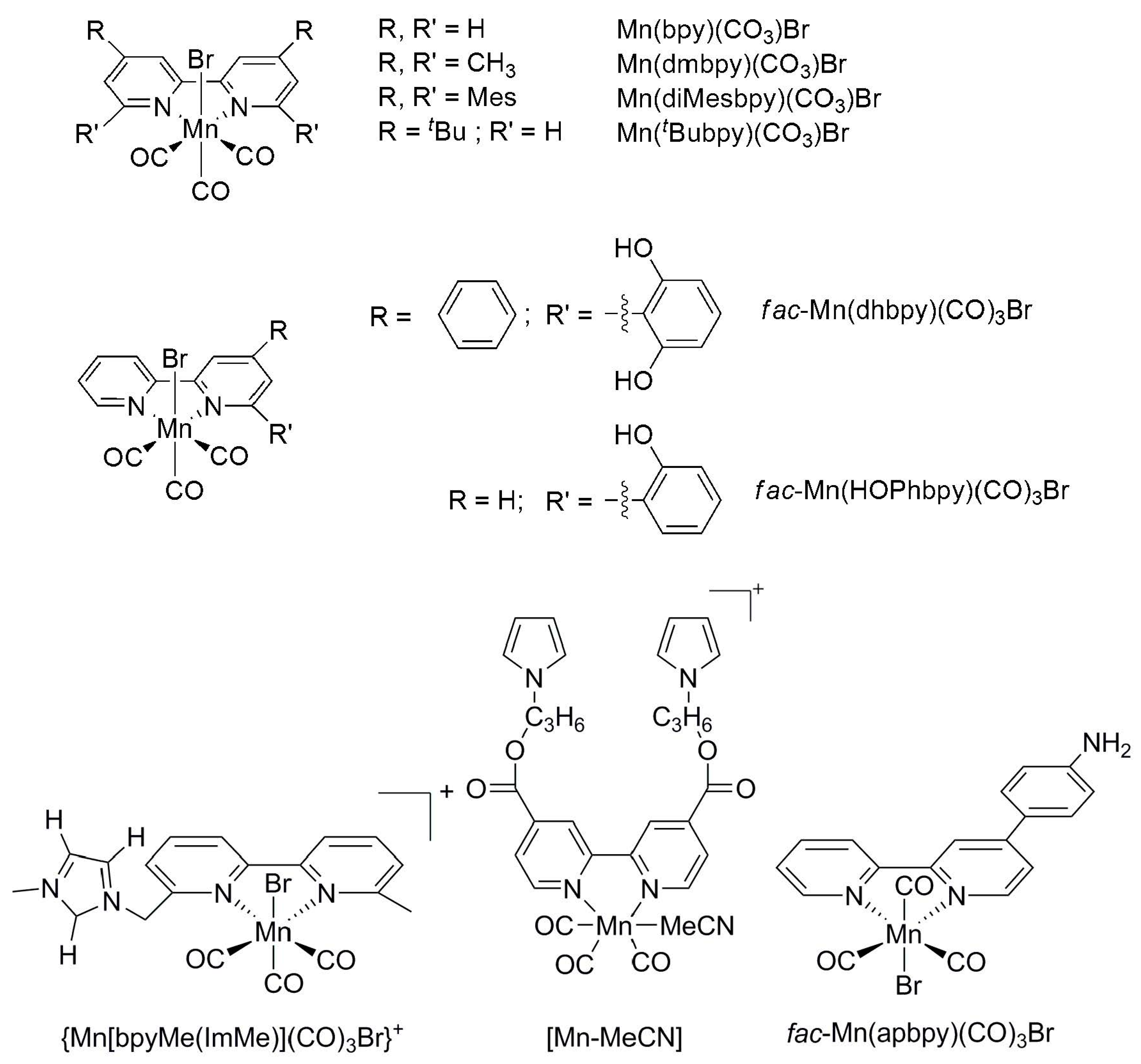
2.2. 3d Metal CO2 Reduction Electrocatalysts
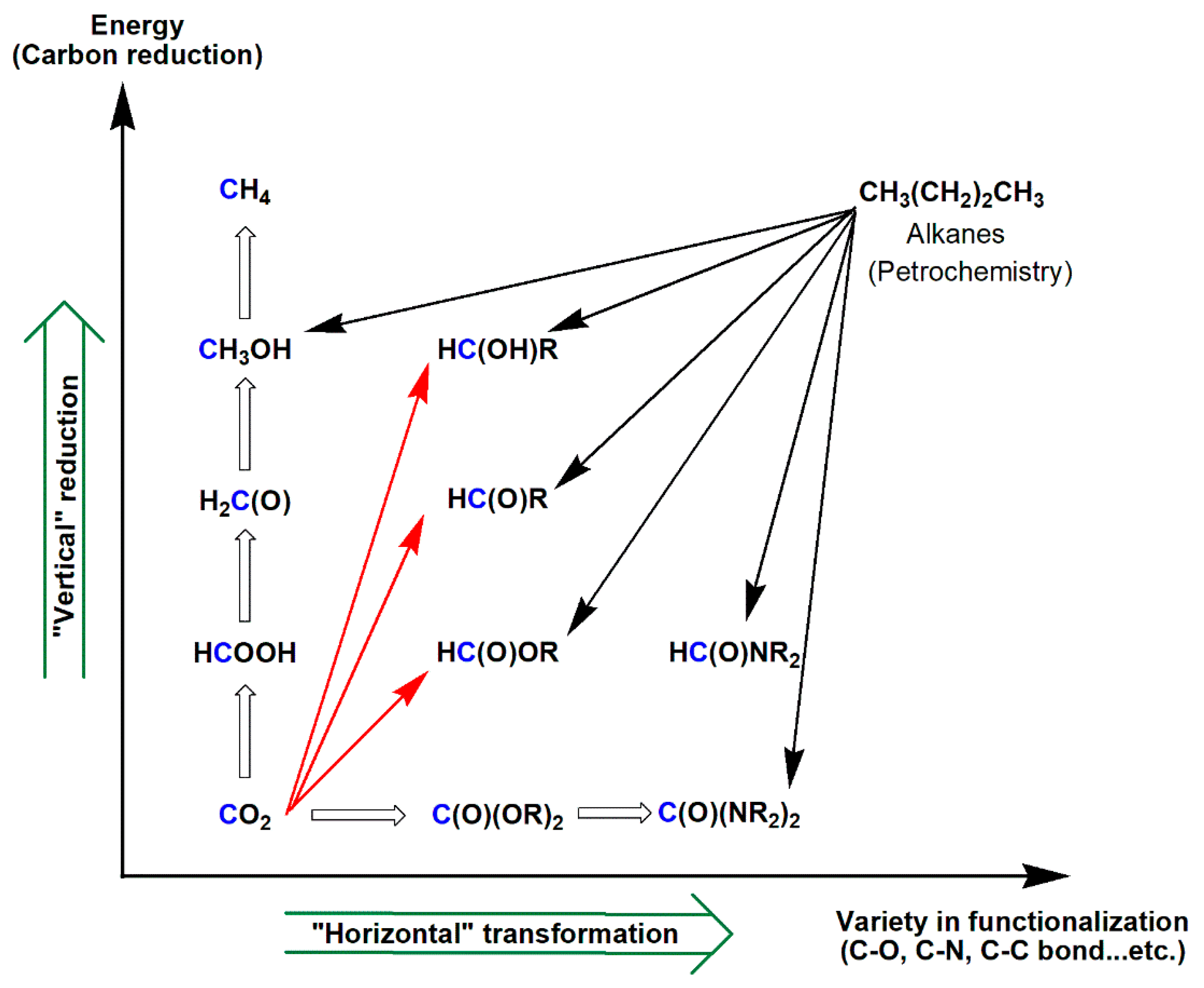
3. Functionalization of Carbon Dioxide
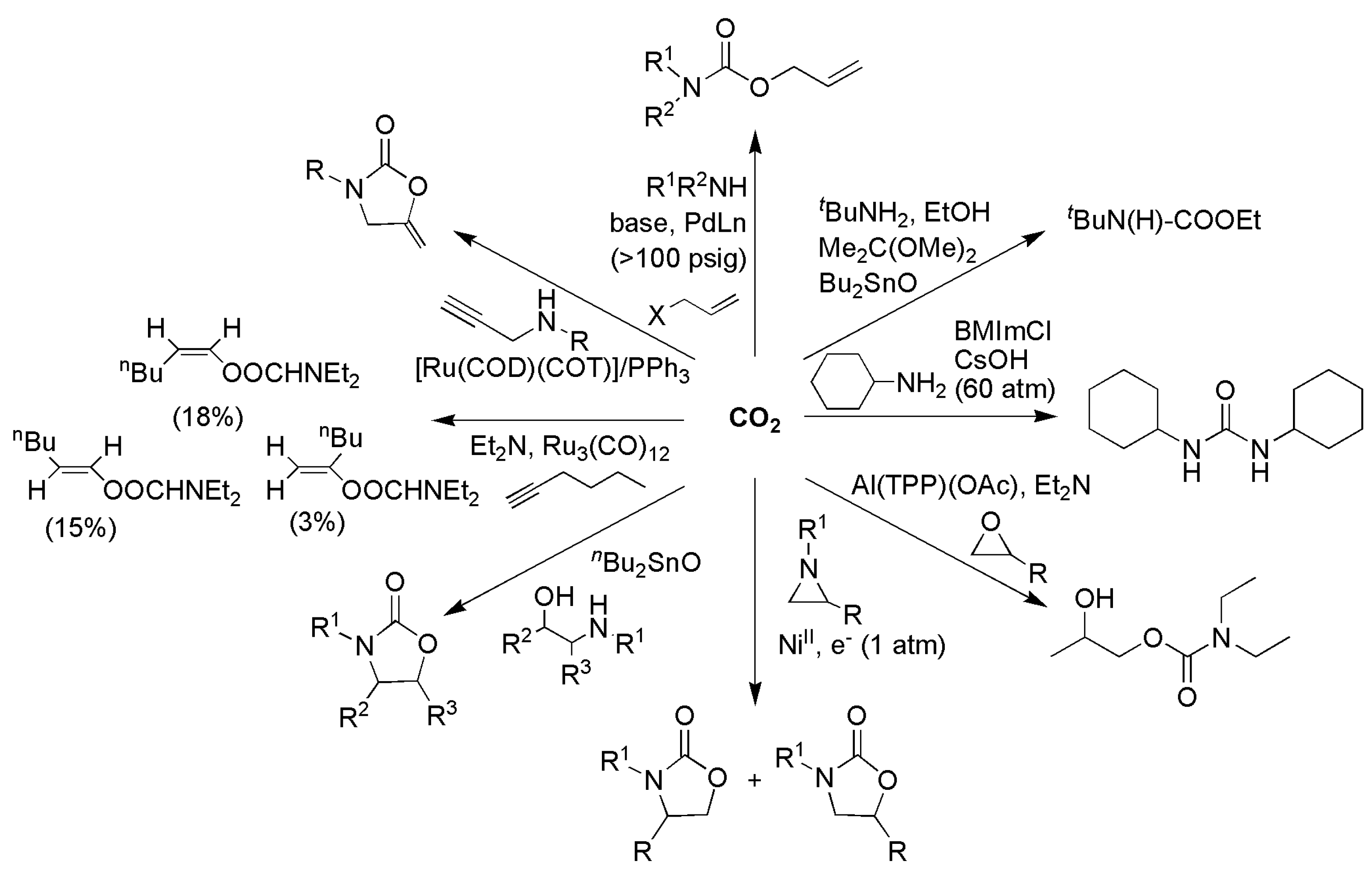
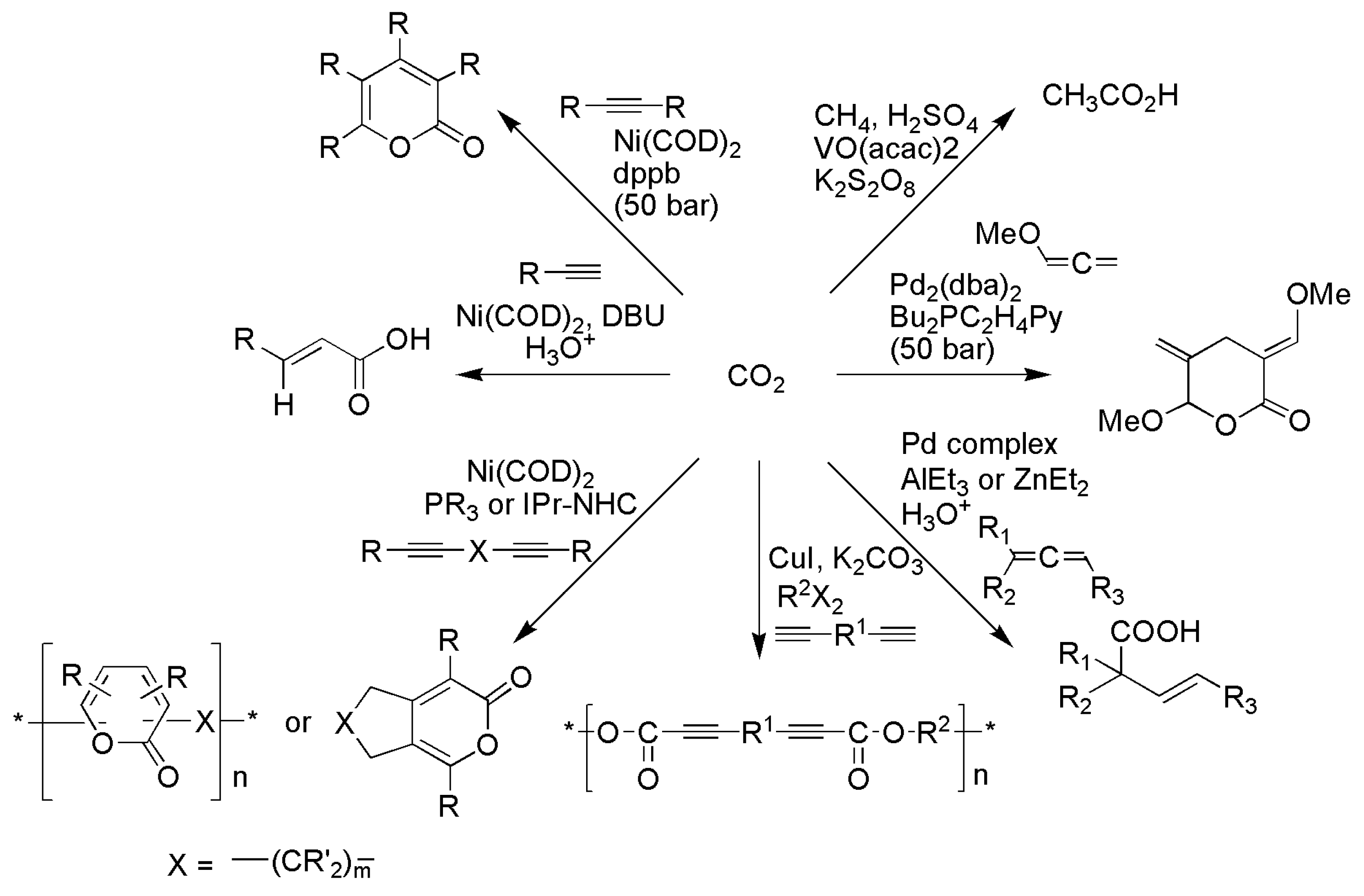
3.1. Horizontal Functionalization of CO2
3.2. Electro- and Photocatalytic CO2 Functionalization
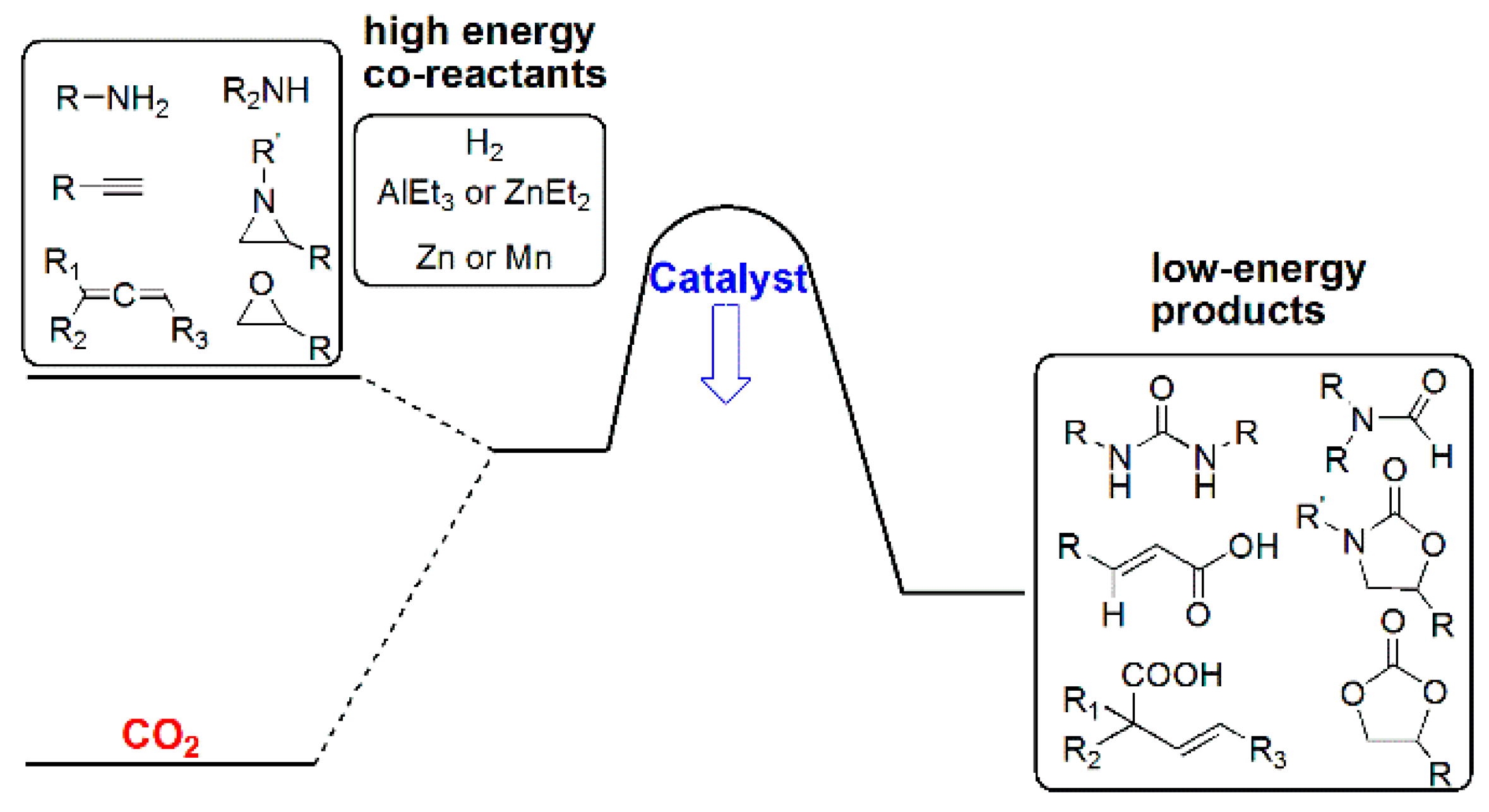
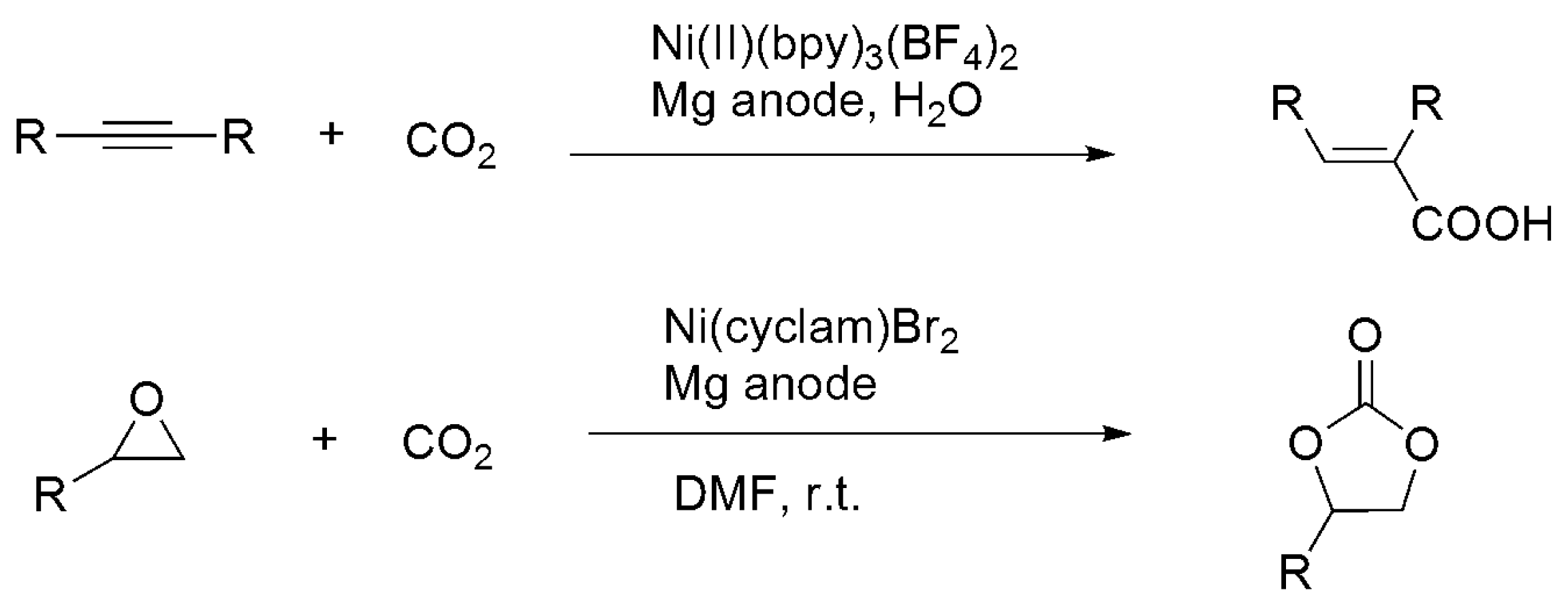
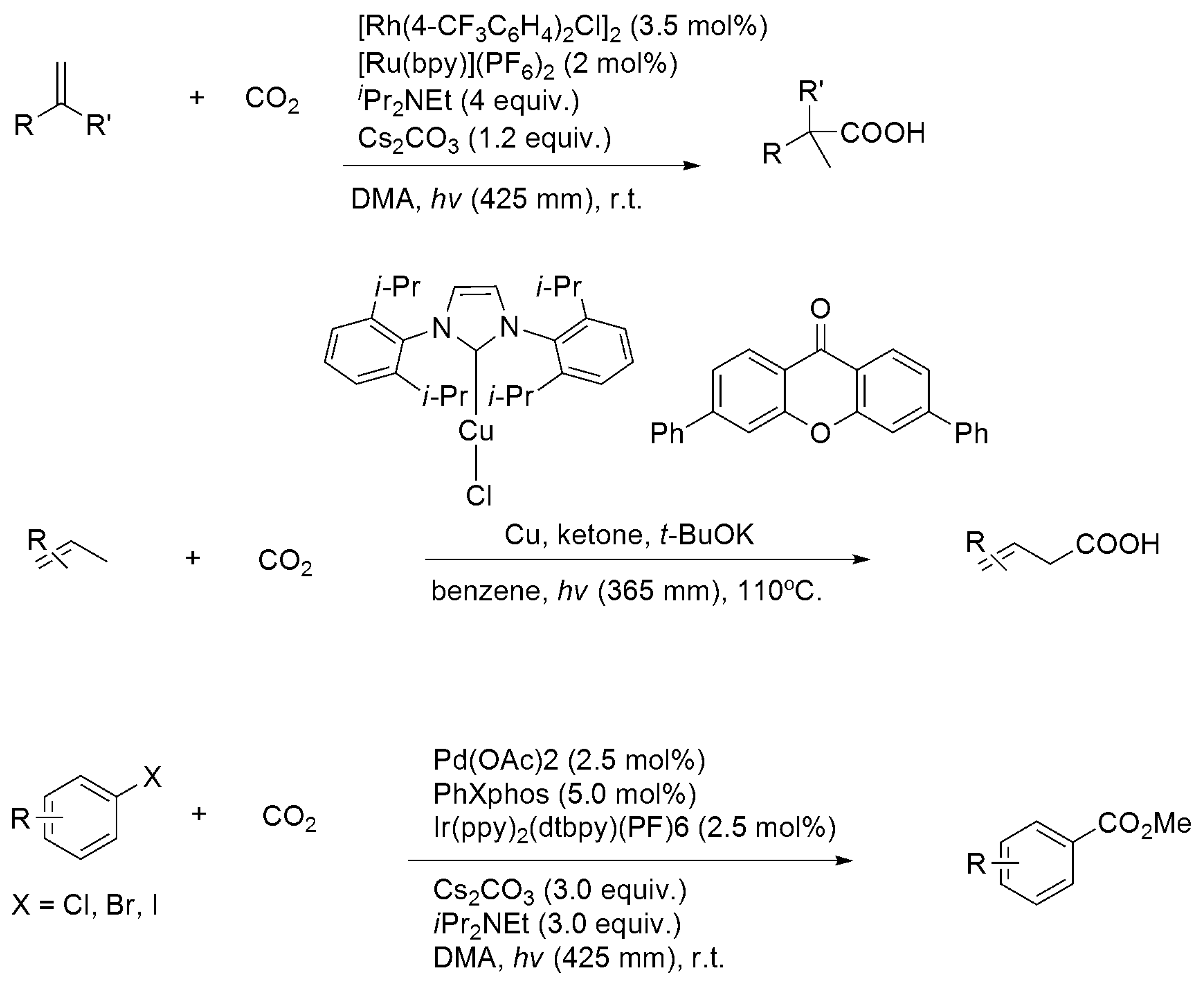
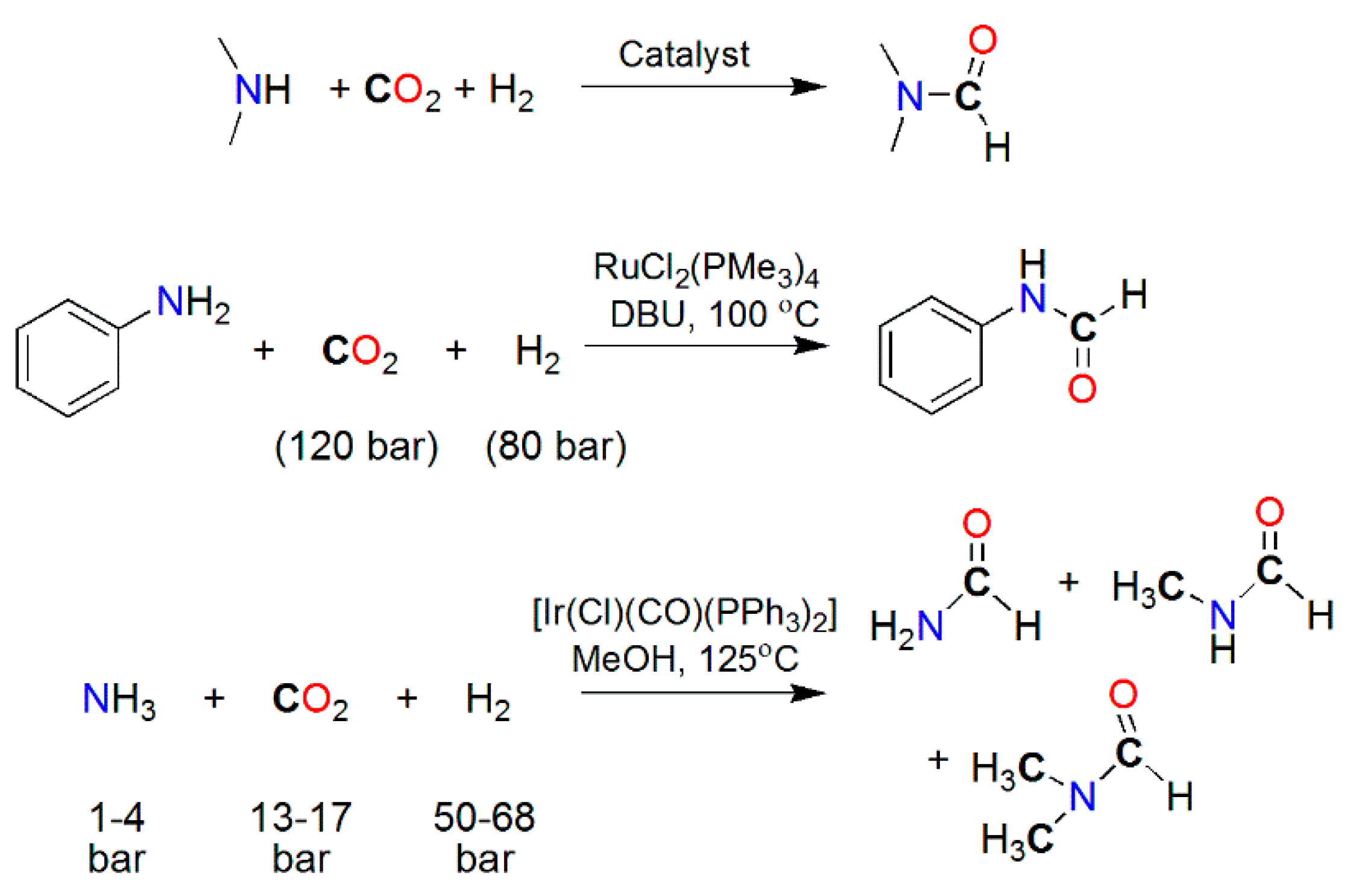
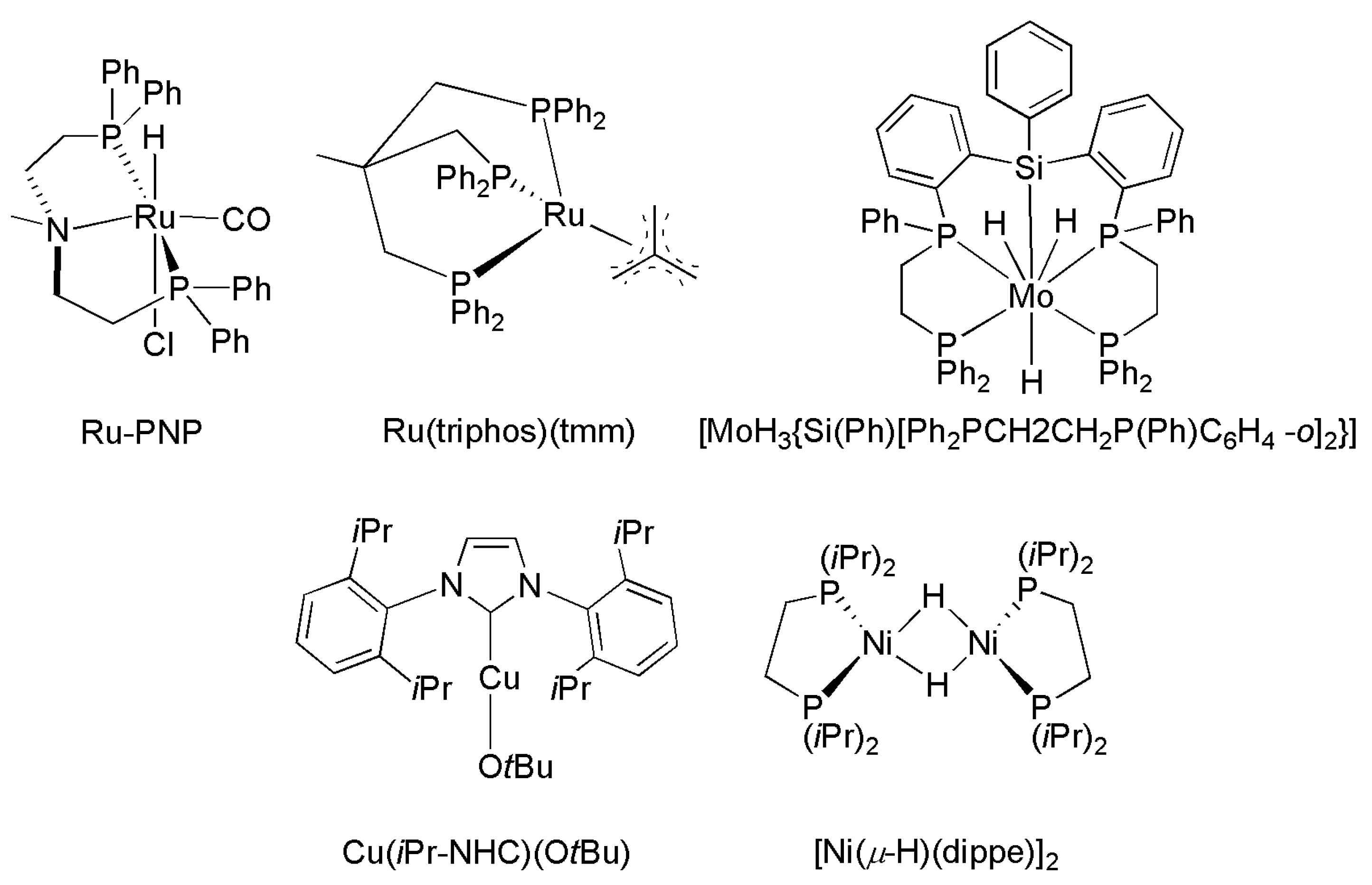
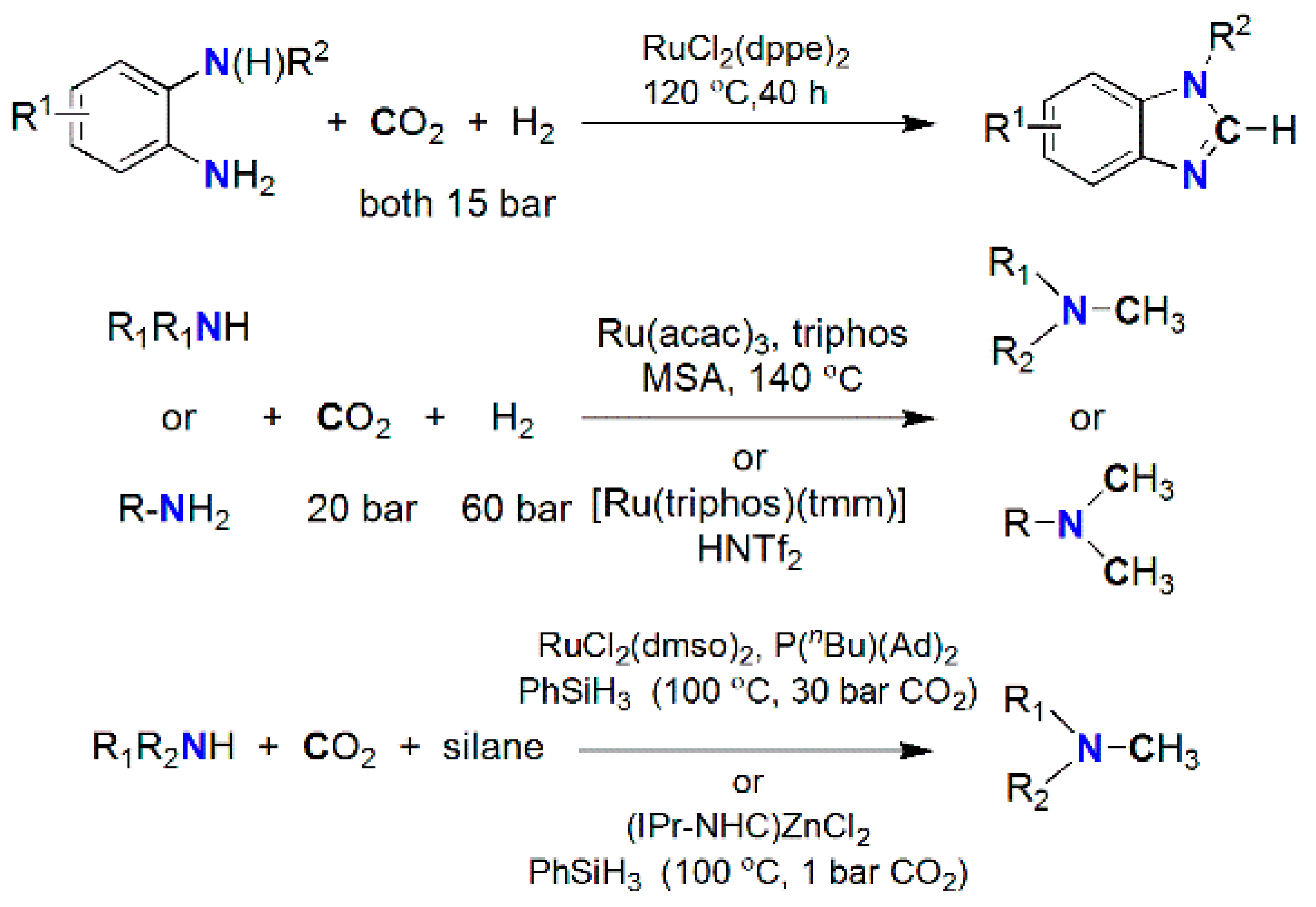
3.3. Diagonal (Reductive) CO2 Functionalization
4. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Ghaib, K.; Nitz, K.; Ben-Fares, F.-Z. Chemical Methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Yu, K.M.K.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent Advances in CO2 Capture and Utilization. ChemSusChem Chem. Sustain. Energy Mater. 2008, 1, 893–899. [Google Scholar] [CrossRef]
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent Progress in the Electrochemical Conversion and Utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Wilcox, E.M.; Roberts, G.W.; Spivey, J.J. Direct Catalytic Formation of Acetic Acid from CO2 and Methane. Catal. Today 2003, 88, 83–90. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A Short Review of Catalysis for CO2 Conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Bian, Z.; Das, S.; Wai, M.H.; Hongmanorom, P.; Kawi, S. A Review on Bimetallic Nickel-Based Catalysts for CO2 Reforming of Methane. ChemPhysChem 2017, 18, 3117–3134. [Google Scholar] [CrossRef]
- Buelens, L.C.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Super-Dry Reforming of Methane Intensifies CO2 Utilization via Le Chatelier’s Principle. Science 2016, 354, 449–452. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- Schreier, M.; Héroguel, F.; Steier, L.; Ahmad, S.; Luterbacher, J.S.; Mayer, M.T.; Luo, J.; Grätzel, M. Solar Conversion of CO2 to CO Using Earth-Abundant Electrocatalysts Prepared by Atomic Layer Modification of CuO. Nat. Energy 2017, 2, 17087. [Google Scholar] [CrossRef]
- Sorcar, S.; Hwang, Y.; Grimes, C.A.; In, S.-I. Highly Enhanced and Stable Activity of Defect-Induced Titania Nanoparticles for Solar Light-Driven CO2 Reduction into CH4. Mater. Today 2017, 20, 507–515. [Google Scholar] [CrossRef]
- Park, H.; Ou, H.-H.; Colussi, A.J.; Hoffmann, M.R. Artificial Photosynthesis of C1--C3 Hydrocarbons from Water and CO2 on Titanate Nanotubes Decorated with Nanoparticle Elemental Copper and CdS Quantum Dots. J. Phys. Chem. A 2015, 119, 4658–4666. [Google Scholar] [CrossRef] [PubMed]
- Sorcar, S.; Thompson, J.; Hwang, Y.; Park, Y.H.; Majima, T.; Grimes, C.A.; Durrant, J.R.; In, S.-I. High-Rate Solar-Light Photoconversion of CO2 to Fuel: Controllable Transformation from C1 to C2 Products. Energy Environ. Sci. 2018, 11, 3183–3193. [Google Scholar] [CrossRef]
- Li, N.; Wang, B.; Si, Y.; Xue, F.; Zhou, J.; Lu, Y.; Liu, M. Toward High-Value Hydrocarbon Generation by Photocatalytic Reduction of CO2 in Water Vapor. ACS Catal. 2019, 9, 5590–5602. [Google Scholar] [CrossRef]
- Thampi, K.R.; Lucarelli, L.; Kiwi, J. Characterization of a Ruthenium/Titania Catalyst for Selective Methanation at Room Temperature and Atmospheric Pressure. Langmuir 1991, 7, 2642–2648. [Google Scholar] [CrossRef]
- Yang, X.; Fugate, E.A.; Mueanngern, Y.; Baker, L.R. Photoelectrochemical CO2 Reduction to Acetate on Iron--Copper Oxide Catalysts. ACS Catal. 2016, 7, 177–180. [Google Scholar] [CrossRef]
- Jeon, H.S.; Kunze, S.; Scholten, F.; Roldan Cuenya, B. Prism-Shaped Cu Nanocatalysts for Electrochemical CO2 Reduction to Ethylene. ACS Catal. 2017, 8, 531–535. [Google Scholar] [CrossRef]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 2009, 42, 1983–1994. [Google Scholar] [CrossRef]
- Windle, C.D.; Perutz, R.N. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction. ACS Catal. 2016, 7, 70–88. [Google Scholar] [CrossRef]
- Hunt, A.J.; Sin, E.H.K.; Marriott, R.; Clark, J.H. Generation, capture, and utilization of industrial carbon dioxide. ChemSusChem Chem. Sustain. Energy Mater. 2010, 3, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M.A.; Müller, T.E.; Leitner, W.; Gürtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.D.; Grills, D.C.; Muckerman, J.T.; Polyansky, D.E.; Fujita, E. Toward more efficient photochemical CO2 reduction: Use of scCO2 or photogenerated hydrides. Coord. Chem. Rev. 2010, 254, 2472–2482. [Google Scholar] [CrossRef]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T.M. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef]
- Liu, X.; Inagaki, S.; Gong, J. Heterogeneous molecular systems for photocatalytic CO2 reduction with water oxidation. Angew. Chem. Int. Ed. 2016, 55, 14924–14950. [Google Scholar] [CrossRef] [PubMed]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re (bipy)(CO)3Cl (bipy= 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 328–330. [Google Scholar] [CrossRef]
- Clark, M.L.; Cheung, P.L.; Lessio, M.; Carter, E.A.; Kubiak, C.P. Kinetic and Mechanistic Effects of Bipyridine (bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 8, 2021–2029. [Google Scholar] [CrossRef]
- Bolinger, C.M.; Story, N.; Sullivan, B.P.; Meyer, T.J. Electrocatalytic reduction of carbon dioxide by 2, 2′-bipyridine complexes of rhodium and iridium. Inorg. Chem. 1988, 27, 4582–4587. [Google Scholar] [CrossRef]
- Sung, S.; Kumar, D.; Gil-Sepulcre, M.; Nippe, M. Electrocatalytic CO2 Reduction by Imidazolium-Functionalized Molecular Catalysts. J. Am. Chem. Soc. 2017, 139, 13993–13996. [Google Scholar] [CrossRef]
- Nakada, A.; Ishitani, O. Selective Electrocatalysis of a Water-Soluble Rhenium (I) Complex for CO2 Reduction Using Water as an Electron Donor. ACS Catal. 2017, 8, 354–363. [Google Scholar] [CrossRef]
- Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. The effect of pH on the formation of CO and HCOO−. Organometallics 1987, 6, 181–186. [Google Scholar] [CrossRef]
- Bruce, M.R.M.; Megehee, E.; Sullivan, B.P.; Thorp, H.; O’Toole, T.R.; Downard, A.; Meyer, T.J. Electrocatalytic reduction of carbon dioxide by associative activation. Organometallics 1988, 7, 238–240. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A molecular ruthenium electrocatalyst for the reduction of carbon dioxide to CO and formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef] [PubMed]
- Slater, S.; Wagenknecht, J.H. Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J. Am. Chem. Soc. 1984, 106, 5367–5368. [Google Scholar] [CrossRef]
- DuBois, D.L.; Miedaner, A.; Haltiwanger, R.C. Electrochemical reduction of CO2 catalyzed by [Pd(triphosphine)(solvent)](BF4)2 complexes: Synthetic and mechanistic studies. J. Am. Chem. Soc. 1991, 113, 8753–8764. [Google Scholar] [CrossRef]
- Raebiger, J.W.; Turner, J.W.; Noll, B.C.; Curtis, C.J.; Miedaner, A.; Cox, B.; DuBois, D.L. Electrochemical reduction of CO2 to CO catalyzed by a bimetallic palladium complex. Organometallics 2006, 25, 3345–3351. [Google Scholar] [CrossRef]
- Fisher, B.J.; Eisenberg, R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102, 7361–7363. [Google Scholar] [CrossRef]
- Fujita, E. Photochemical carbon dioxide reduction with metal complexes. Coord. Chem. Rev. 1999, 185, 373–384. [Google Scholar] [CrossRef]
- Fujita, E.; Szalda, D.J.; Creutz, C.; Sutin, N. Carbon dioxide activation: Thermodynamics of carbon dioxide binding and the involvement of two cobalt centers in the reduction of carbon dioxide by a cobalt (I) macrocycle. J. Am. Chem. Soc. 1988, 110, 4870–4871. [Google Scholar] [CrossRef]
- Fujita, E.; Creutz, C.; Sutin, N.; Szalda, D.J. Carbon dioxide activation by cobalt (I) macrocycles: Factors affecting carbon dioxide and carbon monoxide binding. J. Am. Chem. Soc. 1991, 113, 343–353. [Google Scholar] [CrossRef]
- Creutz, C.; Schwarz, H.A.; Wishart, J.F.; Fujita, E.; Sutin, N. Thermodynamics and kinetics of carbon dioxide binding to two stereoisomers of a cobalt (I) macrocycle in aqueous solution. J. Am. Chem. Soc. 1991, 113, 3361–3371. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.P.; Ruppert, R.; Sauvage, J.P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: Study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461–7467. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: An extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar] [CrossRef]
- Fujita, E.; Haff, J.; Sanzenbacher, R.; Elias, H. High Electrocatalytic Activity of RRSS-[NiIIHTIM](ClO4)2 and [NiIIDMC](ClO4)2 for Carbon Dioxide Reduction (HTIM= 2,3,9,10-Tetramethyl-1,4,8,11-tetraazacyclotetradecane, DMC = C-meso-5,12-Dimethyl-1,4,8,11-tetraazacyclotetradecane). Inorg. Chem. 1994, 33, 4627–4628. [Google Scholar] [CrossRef]
- Froehlich, J.D.; Kubiak, C.P. Homogeneous CO2 reduction by Ni(cyclam) at a glassy carbon electrode. Inorg. Chem. 2012, 51, 3932–3934. [Google Scholar] [CrossRef]
- Schneider, J.; Jia, H.; Kobiro, K.; Cabelli, D.E.; Muckerman, J.T.; Fujita, E. Nickel(II) macrocycles: Highly efficient electrocatalysts for the selective reduction of CO2 to CO. Energy Environ. Sci. 2012, 5, 9502–9510. [Google Scholar] [CrossRef]
- Collin, J.P.; Jouaiti, A.; Sauvage, J.P. Electrocatalytic properties of Ni(cyclam)2+ and Ni2(biscyclam)4+ with respect to CO2 and H2O reduction. Inorg. Chem. 1988, 27, 1986–1990. [Google Scholar] [CrossRef]
- Tinnemans, A.H.A.; Koster, T.P.M.; Thewissen, D.; Mackor, A. Tetraaza-macrocyclic cobalt(II) and nickel(II) complexes as electron-transfer agents in the photo(electro)chemical and electrochemical reduction of carbon dioxide. Recl. Trav. Chim. Pays Bas 1984, 103, 288–295. [Google Scholar] [CrossRef]
- Che, C.-M.; Mak, S.-T.; Lee, W.-O.; Fung, K.-W.; Mak, T.C.W. Electrochemical studies of nickel(II) and cobalt(II) complexes of tetra-azamacrocycles bearing a pyridine functional group and X-ray structures of [NiII(L3)Cl]ClO4 and [NiII(L3)][ClO4]2·H2O {L3 = meso-2,3,7,11,12-pentamethyl-3,7,11,17-tetra-azabicyclo[11.3.1]heptadeca-1,13,15-triene}. J. Chem. Soc. Dalt. Trans. 1988, 2153–2159. [Google Scholar]
- Chen, L.; Guo, Z.; Wei, X.-G.; Gallenkamp, C.; Bonin, J.; Anxolabéhère-Mallart, E.; Lau, K.-C.; Lau, T.-C.; Robert, M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Earth-abundant metal complexes. Selective production of CO vs. HCOOH by switching of the metal center. J. Am. Chem. Soc. 2015, 137, 10918–10921. [Google Scholar] [CrossRef] [PubMed]
- Savéant, J.-M. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A local proton source enhances CO2 electroreduction to CO by a molecular Fe catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Current issues in molecular catalysis illustrated by iron porphyrins as catalysts of the CO2-to-CO electrochemical conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Hammouche, M.; Lexa, D.; Momenteau, M.; Saveant, J.M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. Role of the addition of magnesium cations. J. Am. Chem. Soc. 1991, 113, 8455–8466. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins: Synergystic effect of weak Brönsted acids. J. Am. Chem. Soc. 1996, 118, 1769–1776. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins. Synergistic effect of Lewis acid cations. J. Phys. Chem. 1996, 100, 19981–19985. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Yanagida, S.; Brunschwig, B.S.; Fujita, E. Mechanistic and kinetic studies of cobalt macrocycles in a photochemical CO2 reduction system: Evidence of Co-CO2 adducts as intermediates. J. Am. Chem. Soc. 1995, 117, 6708–6716. [Google Scholar] [CrossRef]
- Grodkowski, J.; Neta, P.; Fujita, E.; Mahammed, A.; Simkhovich, L.; Gross, Z. Reduction of cobalt and iron corroles and catalyzed reduction of CO2. J. Phys. Chem. A 2002, 106, 4772–4778. [Google Scholar] [CrossRef]
- Meshitsuka, S.; Ichikawa, M.; Tamaru, K. Electrocatalysis by metal phthalocyanines in the reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 1974, 158–159. [Google Scholar] [CrossRef]
- Furuya, N.; Matsui, K. Electroreduction of carbon dioxide on gas-diffusion electrodes modified by metal phthalocyanines. J. Electroanal. Chem. Interfacial Electrochem. 1989, 271, 181–191. [Google Scholar] [CrossRef]
- Atoguchi, T.; Aramata, A.; Kazusaka, A.; Enyo, M. Cobalt(II)--tetraphenylporphyrin--pyridine complex fixed on a glassy carbon electrode and its prominent catalytic activity for reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 1991, 156–157. [Google Scholar] [CrossRef]
- Sonoyama, N.; Kirii, M.; Sakata, T. Electrochemical reduction of CO2 at metal-porphyrin supported gas diffusion electrodes under high pressure CO2. Electrochem. Commun. 1999, 1, 213–216. [Google Scholar] [CrossRef]
- Aoi, S.; Mase, K.; Ohkubo, K.; Fukuzumi, S. Selective electrochemical reduction of CO2 to CO with a cobalt chlorin complex adsorbed on multi-walled carbon nanotubes in water. Chem. Commun. 2015, 51, 10226–10228. [Google Scholar] [CrossRef]
- Bonin, J.; Chaussemier, M.; Robert, M.; Routier, M. Homogeneous photocatalytic reduction of CO2 to CO using iron(0) porphyrin catalysts: Mechanism and intrinsic limitations. ChemCatChem 2014, 6, 3200–3207. [Google Scholar] [CrossRef]
- Rao, H.; Schmidt, L.C.; Bonin, J.; Robert, M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 2017, 548, 74. [Google Scholar] [CrossRef]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef]
- Smith, P.T.; Benke, B.P.; Cao, Z.; Kim, Y.; Nichols, E.M.; Kim, K.; Chang, C.J. Iron Porphyrins Embedded into a Supramolecular Porous Organic Cage for Electrochemical CO2 Reduction in Water. Angew. Chem. Int. Ed. 2018, 57, 9684–9688. [Google Scholar] [CrossRef]
- Weng, Z.; Jiang, J.; Wu, Y.; Wu, Z.; Guo, X.; Materna, K.L.; Liu, W.; Batista, V.S.; Brudvig, G.W.; Wang, H. Electrochemical CO2 Reduction to Hydrocarbons on a Heterogeneous Molecular Cu Catalyst in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 8076–8079. [Google Scholar] [CrossRef] [PubMed]
- Thoi, V.S.; Kornienko, N.; Margarit, C.G.; Yang, P.; Chang, C.J. Visible-light photoredox catalysis: Selective reduction of carbon dioxide to carbon monoxide by a nickel N-heterocyclic carbene-isoquinoline complex. J. Am. Chem. Soc. 2013, 135, 14413–14424. [Google Scholar] [CrossRef] [PubMed]
- Chapovetsky, A.; Do, T.H.; Haiges, R.; Takase, M.K.; Marinescu, S.C. Proton-assisted reduction of CO2 by cobalt aminopyridine macrocycles. J. Am. Chem. Soc. 2016, 138, 5765–5768. [Google Scholar] [CrossRef] [PubMed]
- Pun, S.-N.; Chung, W.-H.; Lam, K.-M.; Guo, P.; Chan, P.-H.; Wong, K.-Y.; Che, C.-M.; Chen, T.-Y.; Peng, S.-M. Iron(I) complexes of 2,9-bis(2-hydroxyphenyl)-1,10-phenanthroline (H2dophen) as electrocatalysts for carbon dioxide reduction. X-ray crystal structures of [Fe(dophen)Cl]2·2HCON(CH3)2 and [Fe (dophen)(N-MeIm)2]ClO4 (N-MeIm = 1-methylimidazole). J. Chem. Soc. Dalton Trans. 2002, 575–583. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Cometto, C.; Chen, L.; Lo, P.-K.; Guo, Z.; Lau, K.-C.; Anxolabéhère-Mallart, E.; Fave, C.; Lau, T.-C.; Robert, M. Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs. Co Quaterpyridine Complexes upon Mechanistic Studies. ACS Catal. 2018, 8, 3411–3417. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Orio, M.; Molton, F.; Vezin, H.; Duboc, C.; Deronzier, A.; Chardon-Noblat, S. Pulsed-EPR Evidence of a Manganese(II) Hydroxycarbonyl Intermediate in the Electrocatalytic Reduction of Carbon Dioxide by a Manganese Bipyridyl Derivative. Angew. Chem. Int. Ed. 2014, 53, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.D.; Kubiak, C.P. Manganese electrocatalysts with bulky bipyridine ligands: Utilizing Lewis acids to promote carbon dioxide reduction at low overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a substitute for rhenium in CO2 reduction catalysts: The importance of acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Vallana, F.F.; Sordello, F.; Priola, E.; Minero, C.; Nervi, C.; Gobetto, R. A Local Proton Source in a [Mn(bpy-R)(CO)3Br]-Type Redox Catalyst Enables CO2 Reduction Even in the Absence of Brønsted Acids. Chem. Commun. 2014, 50, 14670–14673. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Shaw, T.W.; Schaefer III, H.F.; Bocarsly, A.B. Design of a catalytic active site for electrochemical CO2 reduction with Mn(I)-tricarbonyl species. Inorg. Chem. 2015, 54, 5285–5294. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy-R)(CO)3Br] Complexes. Chem. Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Li, X.; Wolf, L.M.; Meeder, J.R.; Bhuvanesh, N.S.; Grice, K.A.; Panetier, J.A.; Nippe, M. Synergistic Effects of Imidazolium-Functionalization on Fac-Mn(CO)3 Bipyridine Catalyst Platforms for Electrocatalytic Carbon Dioxide Reduction. J. Am. Chem. Soc. 2019, 141, 6569–6582. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Saita, K.; Sekizawa, K.; Maeda, S.; Morikawa, T. Low-Energy Electrocatalytic CO2 Reduction in Water over Mn-Complex Catalyst Electrode Aided by a Nanocarbon Support and K+ Cations. ACS Catal. 2018, 8, 4452–4458. [Google Scholar] [CrossRef]
- Sun, C.; Rotundo, L.; Garino, C.; Nencini, L.; Yoon, S.S.; Gobetto, R.; Nervi, C. Electrochemical CO2 Reduction at Glassy Carbon Electrodes Functionalized by MnI and ReI Organometallic Complexes. ChemPhysChem 2017, 18, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Filippi, J.; Gobetto, R.; Miller, H.A.; Rocca, R.; Nervi, C.; Vizza, F. Electrochemical CO2 Reduction in Water at Carbon Cloth Electrodes Functionalized with a Fac-Mn (apbpy)(CO)3Br Complex. Chem. Commun. 2019, 55, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Simón-Manso, E.; Kubiak, C.P. Dinuclear nickel complexes as catalysts for electrochemical reduction of carbon dioxide. Organometallics 2005, 24, 96–102. [Google Scholar] [CrossRef]
- DeLaet, D.L.; DelRosario, R.; Fanwick, P.E.; Kubiak, C.P. Carbon dioxide chemistry and electrochemistry of a binuclear “Cradle” complex of Ni, Ni2(µ-CNMe)(CNMe)2(PPh2CH2PPh2)2. J. Am. Chem. Soc. 1987, 109, 754–758. [Google Scholar] [CrossRef]
- Ratliff, K.S.; Lentz, R.E.; Kubiak, C.P. Carbon dioxide chemistry of the trinuclear complex [Ni3 (µ3-CNMe)( µ3-I)(dppm)3][PF6]. Electrocatalytic reduction of carbon dioxide. Organometallics 1992, 11, 1986–1988. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Ferrence, G.M.; Washington, J.; Henderson, J.I.; Rosenhein, L.; Heise, J.D.; Fanwick, P.E.; Kubiak, C.P. A Class of Halide-Supported Trinuclear Nickel Clusters [Ni3(µ3-L)(µ3-X)(µ2-dppm)3]n+ (L = I−, Br−, CO, CNR.; X = I−, Br−; n = 0, 1; dppm= Ph2PCH2PPh2): Novel Physical Properties and the Fermi Resonance of Symmetric µ3-η1 Bound Isocyanide Ligands. J. Am. Chem. Soc. 1996, 118, 2198–2207. [Google Scholar] [CrossRef]
- Haines, R.J.; Wittrig, R.E.; Kubiak, C.P. Electrocatalytic Reduction of Carbon Dioxide by the Binuclear Copper Complex [Cu2(6-(diphenylphosphino-2,2′-bipyridyl)2(MeCN)2][PF6]2. Inorg. Chem. 1994, 33, 4723–4728. [Google Scholar] [CrossRef]
- Cheng, M.; Yu, Y.; Zhou, X.; Luo, Y.; Wang, M. Chemical Versatility of [FeFe]-Hydrogenase Models: Distinctive Activity of [μ-C6H4-1,2-(κ2-S)2][Fe2(CO)6] for Electrocatalytic CO2 Reduction. ACS Catal. 2018, 9, 768–774. [Google Scholar] [CrossRef]
- Angamuthu, R.; Byers, P.; Lutz, M.; Spek, A.L.; Bouwman, E. Electrocatalytic CO2 Conversion to Oxalate by a Copper Complex. Science 2010, 327, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Maeda, C.; Miyazaki, Y.; Ema, T. Recent progress in catalytic conversions of carbon dioxide. Catal. Sci. Technol. 2014, 4, 1482–1497. [Google Scholar] [CrossRef]
- Tlili, A.; Blondiaux, E.; Frogneux, X.; Cantat, T. Reductive functionalization of CO2 with amines: An entry to formamide, formamidine and methylamine derivatives. Green Chem. 2015, 17, 157–168. [Google Scholar] [CrossRef]
- Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Efficient, selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- Fiorani, G.; Guo, W.; Kleij, A.W. Sustainable conversion of carbon dioxide: The advent of organocatalysis. Green Chem. 2015, 17, 1375–1389. [Google Scholar] [CrossRef]
- Borjesson, M.; Moragas, T.; Gallego, D.; Martin, R. Metal-catalyzed carboxylation of organic (pseudo)halides with CO2. ACS Catal. 2016, 6, 6739–6749. [Google Scholar] [CrossRef]
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-metal-catalyzed C-C bond formation through the fixation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Kleij, A.W. Myth or reality? Fixation of carbon dioxide into complex organic matter under mild conditions. ChemSusChem 2011, 4, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Köhler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Müller, T.E. Chemical technologies for exploiting and recycling carbon dioxide into the value chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef] [PubMed]
- Juliá-Hernández, F.; Gaydou, M.; Serrano, E.; van Gemmeren, M.; Martin, R. Ni- and Fe-catalyzed carboxylation of unsaturated hydrocarbons with CO2. In Ni-and Fe-Based Cross-Coupling Reactions; Springer: Dodrecht, The Netherlands, 2017; pp. 91–128.CO2. In Ni-and Fe-Based Cross-Coupling Reactions; Springer: Dodrecht, The Netherlands, 2017; pp. 91–128. [Google Scholar]
- Aida, T.; Inoue, S. Activation of carbon dioxide with aluminum porphyrin and reaction with epoxide. Studies on (tetraphenylporphinato) aluminum alkoxide having a long oxyalkylene chain as the alkoxide group. J. Am. Chem. Soc. 1983, 105, 1304–1309. [Google Scholar] [CrossRef]
- Kruper, W.J.; Dellar, D.D. Catalytic formation of cyclic carbonates from epoxides and CO2 with chromium metalloporphyrinates. J. Org. Chem. 1995, 60, 725–727. [Google Scholar] [CrossRef]
- Paddock, R.L.; Hiyama, Y.; McKay, J.M.; Nguyen, S.T. Co(III) porphyrin/DMAP: An efficient catalyst system for the synthesis of cyclic carbonates from CO2 and epoxides. Tetrahedron Lett. 2004, 45, 2023–2026. [Google Scholar] [CrossRef]
- Jin, L.; Jing, H.; Chang, T.; Bu, X.; Wang, L.; Liu, Z. Metal porphyrin/phenyltrimethylammonium tribromide: High efficient catalysts for coupling reaction of CO2 and epoxides. J. Mol. Catal. A Chem. 2007, 261, 262–266. [Google Scholar] [CrossRef]
- Bai, D.; Wang, Q.; Song, Y.; Li, B.; Jing, H. Synthesis of cyclic carbonate from epoxide and CO2 catalyzed by magnetic nanoparticle-supported porphyrin. Catal. Commun. 2011, 12, 684–688. [Google Scholar] [CrossRef]
- Ahmadi, F.; Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Khosropour, A.R. Highly efficient chemical fixation of carbon dioxide catalyzed by high-valent tetraphenylporphyrinatotin(IV) triflate. Inorg. Chem. Commun. 2011, 14, 1489–1493. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Koyama, S.; Yano, Y.; Sakai, T. A bifunctional catalyst for carbon dioxide fixation: Cooperative double activation of epoxides for the synthesis of cyclic carbonates. Chem. Commun. 2012, 48, 4489–4491. [Google Scholar] [CrossRef] [PubMed]
- Melendez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Escárcega-Bobadilla, M.V.; Martinez Belmonte, M.; Martin, E.; Escudero-Adán, E.C.; Kleij, A.W. A Recyclable Trinuclear Bifunctional Catalyst Derived from a Tetraoxo Bis-Zn(salphen) Metalloligand. Chem. Eur. J. 2013, 19, 2641–2648. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Nakamura, M.; Nozaki, K. Alternating copolymerization of cyclohexene oxide with carbon dioxide catalyzed by (salalen)CrCl complexes. Macromolecules 2009, 42, 6972–6980. [Google Scholar] [CrossRef]
- Wu, G.-P.; Wei, S.-H.; Ren, W.-M.; Lu, X.-B.; Xu, T.-Q.; Darensbourg, D.J. Perfectly alternating copolymerization of CO2 and epichlorohydrin using cobalt(III)-based catalyst systems. J. Am. Chem. Soc. 2011, 133, 15191–15199. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Niu, Y. Bifunctional cobalt Salen complex: A highly selective catalyst for the coupling of CO2 and epoxides under mild conditions. Appl. Organomet. Chem. 2011, 25, 424–428. [Google Scholar] [CrossRef]
- Vagin, S.I.; Reichardt, R.; Klaus, S.; Rieger, B. Conformationally flexible dimeric salphen complexes for bifunctional catalysis. J. Am. Chem. Soc. 2010, 132, 14367–14369. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Hashimoto, S.; Nozaki, K. Bimetallic mechanism operating in the copolymerization of propylene oxide with carbon dioxide catalyzed by cobalt-salen complexes. Chem. Sci. 2010, 1, 369–373. [Google Scholar] [CrossRef]
- Ishida, T.; Kikuchi, S.; Yamada, T. Efficient preparation of 4-hydroxyquinolin-2(1H)-one derivatives with silver-catalyzed carbon dioxide incorporation and intramolecular rearrangement. Org. Lett. 2013, 15, 3710–3713. [Google Scholar] [CrossRef]
- Ishida, T.; Kikuchi, S.; Tsubo, T.; Yamada, T. Silver-catalyzed incorporation of carbon dioxide into o-alkynylaniline derivatives. Org. Lett. 2013, 15, 848–851. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous hydrogenation of carbon dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
- McGhee, W.D.; Riley, D.P.; Christ, M.E.; Christ, K.M. Palladium-catalyzed generation of O-allylic urethanes and carbonates from amines/alcohols, carbon dioxide, and allylic chlorides. Organometallics 1993, 12, 1429–1433. [Google Scholar] [CrossRef]
- Kayaki, Y.; Mori, N.; Ikariya, T. Palladium-catalyzed carboxylative cyclization of α-allenyl amines in dense carbon dioxide. Tetrahedron Lett. 2009, 50, 6491–6493. [Google Scholar] [CrossRef]
- Sasaki, Y.; Dixneuf, P.H. A novel catlytic synthesis of vinyl carboamtes from carbon dioxide, diethylamine, and alkynes in the presence of Ru3(CO)12. J. Chem. Soc. Chem. Commun. 1986, 790–791. [Google Scholar] [CrossRef]
- Mitsudo, T.; Hori, Y.; Yamakawa, Y.; Watanabe, Y. Ruthenium catalyzed selective synthesis of enol carbamates by fixation of carbon dioxide. Tetrahedron Lett. 1987, 28, 4417–4418. [Google Scholar] [CrossRef]
- Tominaga, K.; Sasaki, Y. Synthesis of 2-oxazolidinones from CO2 and 1,2-aminoalcohols catalyzed by n-Bu2SnO. Synlett 2002, 2002, 307–309. [Google Scholar] [CrossRef]
- Shi, F.; Deng, Y.; SiMa, T.; Peng, J.; Gu, Y.; Qiao, B. Alternatives to phosgene and carbon monoxide: Synthesis of symmetric urea derivatives with carbon dioxide in ionic liquids. Angew. Chem. 2003, 115, 3379–3382. [Google Scholar] [CrossRef]
- Kawanami, H.; Matsumoto, H.; Ikushima, Y. Effective scCO2-ionic liquid reaction system based on symmetric aliphatic ammonium salts for the rapid CO2 fixation with aziridine to 2-oxazolidinone. Chem. Lett. 2004, 34, 60–61. [Google Scholar] [CrossRef]
- Mahe, R.; Sasaki, Y.; Bruneau, C.; Dixneuf, P.H. Catalytic synthesis of vinyl carbamates from carbon dioxide and alkynes with ruthenium complexes. J. Org. Chem. 1989, 54, 1518–1523. [Google Scholar] [CrossRef]
- Abla, M.; Choi, J.-C.; Sakakura, T. Halogen-free process for the conversion of carbon dioxide to urethanes by homogeneous catalysis. Chem. Commun. 2001, 2238–2239. [Google Scholar] [CrossRef]
- Ohmiya, H.; Tanabe, M.; Sawamura, M. Copper-catalyzed carboxylation of alkylboranes with carbon dioxide: Formal reductive carboxylation of terminal alkenes. Org. Lett. 2011, 13, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Z.; Li, W.-J.; Zhang, X.; Zhou, H.; Lu, X.-B. Cu(I)-catalyzed carboxylative coupling of terminal alkynes, allylic chlorides, and CO2. Org. Lett. 2010, 12, 4748–4751. [Google Scholar] [CrossRef] [PubMed]
- Leon, T.; Correa, A.; Martin, R. Ni-catalyzed direct carboxylation of benzyl halides with CO2. J. Am. Chem. Soc. 2013, 135, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.R.; Santos, R.H.A.; Gambardella, M.T.P.; DeMacedo, L.G.M.; Rodrigues-Filho, U.P.; Launay, J.-C.; Franco, D.W. Activation of carbon dioxide by bicyclic amidines. J. Org. Chem. 2004, 69, 8005–8011. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nakagawa, S.; Koizumi, T.; Hirayama, K.; Yamamoto, Y. Nickel-mediated regio- and chemoselective carboxylation of alkynes in the presence of carbon dioxide. J. Org. Chem. 1999, 64, 3975–3978. [Google Scholar] [CrossRef]
- Inoue, Y.; Itoh, Y.; Kazama, H.; Hashimoto, H. Reaction of dialkyl-substituted alkynes with carbon dioxide catalyzed by nickel(0) complexes. Incorporation of carbon dioxide in alkyne dimers and novel cyclotrimerization of the alkynes. Bull. Chem. Soc. Jpn. 1980, 53, 3329–3333. [Google Scholar] [CrossRef]
- Tsuda, T.; Maruta, K.; Kitaike, Y. Nickel(0)-catalyzed alternating copolymerization of carbon dioxide with diynes to poly(2-pyrones). J. Am. Chem. Soc. 1992, 114, 1498–1499. [Google Scholar] [CrossRef]
- Fujihara, T.; Nogi, K.; Xu, T.; Terao, J.; Tsuji, Y. Nickel-catalyzed carboxylation of aryl and vinyl chlorides employing carbon dioxide. J. Am. Chem. Soc. 2012, 134, 9106–9109. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Martin, R. Palladium-catalyzed direct carboxylation of aryl bromides with carbon dioxide. J. Am. Chem. Soc. 2009, 131, 15974–15975. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Iwasawa, N. Hydrocarboxylation of allenes with CO2 catalyzed by silyl pincer-type palladium complex. J. Am. Chem. Soc. 2008, 130, 15254–15255. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Brehme, V.A. Homogeneous and heterogeneous catalyzed three-step synthesis of 2-ethylheptanoic acid from carbon dioxide, butadiene and hydrogen. J. Mol. Catal. A Chem. 2002, 187, 69–80. [Google Scholar] [CrossRef]
- Feng, X.; Sun, A.; Zhang, S.; Yu, X.; Bao, M. Palladium-catalyzed carboxylative coupling of benzyl chlorides with allyltributylstannane: Remarkable effect of Palladium nanoparticles. Org. Lett. 2012, 15, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Sasano, K.; Takaya, J.; Iwasawa, N. Palladium(II)-catalyzed direct carboxylation of alkenyl C-H bonds with CO2. J. Am. Chem. Soc. 2013, 135, 10954–10957. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Yamamoto, T.; Saegusa, T. Palladium-catalyzed cycloaddition of carbon dioxide with methoxyallene. J. Organomet. Chem. 1992, 429, C46–C48. [Google Scholar] [CrossRef]
- Greenhalgh, M.D.; Thomas, S.P. Iron-catalyzed, highly regioselective synthesis of α-aryl carboxylic acids from styrene derivatives and CO2. J. Am. Chem. Soc. 2012, 134, 11900–11903. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Takaya, J.; Iwasawa, N. Rhodium(I)-catalyzed direct carboxylation of arenes with CO2 via chelation-assisted C-H bond activation. J. Am. Chem. Soc. 2010, 133, 1251–1253. [Google Scholar] [CrossRef]
- Mita, T.; Michigami, K.; Sato, Y. Sequential Protocol for C(sp3)-H Carboxylation with CO2: Transition-Metal-Catalyzed Benzylic C-H Silylation and Fluoride-Mediated Carboxylation. Org. Lett. 2012, 14, 3462–3465. [Google Scholar] [CrossRef]
- Dai, Y.; Feng, X.; Wang, B.; He, R.; Bao, M. Preparation and application of air-stable P,N-bidentate ligands for the selective synthesis of δ-lactone via the palladium-catalyzed telomerization of 1,3-butadiene with carbon dioxide. J. Organomet. Chem. 2012, 696, 4309–4314. [Google Scholar] [CrossRef]
- Saylik, D.; Horvath, M.J.; Elmes, P.S.; Jackson, W.R.; Lovel, C.G.; Moody, K. Preparation of Isocyanates from Primary Amines and Carbon Dioxide Using Mitsunobu Chemistry1. J. Org. Chem. 1999, 64, 3940–3946. [Google Scholar] [CrossRef]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- Dinsmore, C.J.; Mercer, S.P. Carboxylation and Mitsunobu reaction of amines to give carbamates: Retention vs. inversion of configuration is substituent-dependent. Org. Lett. 2004, 6, 2885–2888. [Google Scholar] [CrossRef] [PubMed]
- Haruki, E.; Hara, T.; Inoue, H. Syntheses of Tricyclo (5.2. 1.02, 6) deca-3, 8-diene-1, 8-dicarboxylic Acid and Its Derivatives from Cyclopentadiene and Carbon Dioxide. Chem. Express. 1990, 5, 493. [Google Scholar]
- Ashley, A.E.; Thompson, A.L.; O’Hare, D. Non-metal-mediated homogeneous hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2009, 48, 9839–9843. [Google Scholar] [CrossRef] [PubMed]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. N-Heterocyclic Carbenes as Efficient Organocatalysts for CO2 Fixation Reactions. Angew. Chem. 2009, 121, 4258–4261. [Google Scholar] [CrossRef]
- Ueno, A.; Kayaki, Y.; Ikariya, T. Cycloaddition of tertiary aziridines and carbon dioxide using a recyclable organocatalyst, 1,3-di-tert-butylimidazolium-2-carboxylate: Straightforward access to 3-substituted 2-oxazolidones. Green Chem. 2013, 15, 425–430. [Google Scholar] [CrossRef]
- He, Q.; O’Brien, J.W.; Kitselman, K.A.; Tompkins, L.E.; Curtis, G.C.T.; Kerton, F.M. Synthesis of cyclic carbonates from CO2 and epoxides using ionic liquids and related catalysts including choline chloride-metal halide mixtures. Catal. Sci. Technol. 2014, 4, 1513–1528. [Google Scholar] [CrossRef]
- Calo, V.; Nacci, A.; Monopoli, A.; Fanizzi, A. Cyclic carbonate formation from carbon dioxide and oxiranes in tetrabutylammonium halides as solvents and catalysts. Org. Lett. 2002, 4, 2561–2563. [Google Scholar] [CrossRef]
- Derien, S.; Clinet, J.C.; Dunach, E.; Perichon, J. Activation of carbon dioxide: Nickel-catalyzed electrochemical carboxylation of diynes. J. Org. Chem. 1993, 58, 2578–2588. [Google Scholar] [CrossRef]
- Tascedda, P.; Dunach, E. Electrosynthesis of cyclic carbamates from aziridines and carbon dioxide. Chem. Commun. 2000, 449–450. [Google Scholar] [CrossRef]
- Derien, S.; Dunach, E.; Perichon, J. From stoichiometry to catalysis: Electroreductive coupling of alkynes and carbon dioxide with nickel-bipyridine complexes. Magnesium ions as the key for catalysis. J. Am. Chem. Soc. 1991, 113, 8447–8454. [Google Scholar] [CrossRef]
- Tascedda, P.; Dunãch, E. Novel electrochemical reactivity of Ni(cyclam)Br2: Catalytic carbon dioxide incorporation into epoxides. J. Chem. Soc. Chem. Commun. 1995, 43–44. [Google Scholar] [CrossRef]
- Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. First example of direct carbon dioxide incorporation into 1,3-diynes: A highly regio-and stereo-selective nickel-catalysed electrochemical reaction. J. Chem. Soc. Chem. Commun. 1991, 549–550. [Google Scholar] [CrossRef]
- Kröcher, O.; Köppel, R.A.; Baiker, A. Highly active ruthenium complexes with bidentate phosphine ligands for the solvent-free catalytic synthesis of N,N-dimethylformamide and methyl formate. Chem. Commun. 1997, 453–454. [Google Scholar] [CrossRef]
- Shimomaki, K.; Murata, K.; Martin, R.; Iwasawa, N. Visible-light-driven carboxylation of aryl halides by the combined use of palladium and photoredox catalysts. J. Am. Chem. Soc. 2017, 139, 9467–9470. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Numasawa, N.; Shimomaki, K.; Takaya, J.; Iwasawa, N. Construction of a visible light-driven hydrocarboxylation cycle of alkenes by the combined use of Rh(I) and photoredox catalysts. Chem. Commun. 2017, 53, 3098–3101. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Katcher, M.H.; Jamison, T.F. Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow. Nat. Chem. 2017, 9, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Masuda, Y.; Uemoto, S.; Murakami, M. A Light/Ketone/Copper System for Carboxylation of Allylic C-H Bonds of Alkenes with CO2. Chem. Eur. J. 2016, 22, 6524–6527. [Google Scholar] [CrossRef]
- DasNeves Gomes, C.; Jacquet, O.; Villiers, C.; Thuéry, P.; Ephritikhine, M.; Cantat, T. A diagonal approach to chemical recycling of carbon dioxide: Organocatalytic transformation for the reductive functionalization of CO2. Angew. Chem. Int. Ed. 2012, 51, 187–190. [Google Scholar] [CrossRef]
- Li, Y.; Cui, X.; Dong, K.; Junge, K.; Beller, M. Utilization of CO2 as a C1 Building Block for Catalytic Methylation Reactions. ACS Catal. 2017, 7, 1077–1086. [Google Scholar] [CrossRef]
- Haynes, P.; Slaugh, L.H.; Kohnle, J.F. Formamides from carbon dioxide, amines and hydrogen in the presence of metal complexes. Tetrahedron Lett. 1970, 11, 365–368. [Google Scholar] [CrossRef]
- Kudo, K.; Phala, H.; Sugita, N.; Takezaki, Y. Synthesis of dimethyl formamide from carbon dioxide, hydrogen and dimethyl amine catalyzed by palladium (II) chloride. Chem. Lett. 1977, 6, 1495–1496. [Google Scholar] [CrossRef]
- Schreiner, S.; Yu, J.Y.; Vaska, L. Reversible homogeneous catalysis of carbon dioxide hydrogenation/reduction at room temperature and low pressures. J. Chem. Soc. Chem. Commun. 1988, 602–603. [Google Scholar] [CrossRef]
- Morimoto, Y.; Fujiwara, Y.; Taniguchi, H.; Hori, Y.; Nagano, Y. PdCl2(MeCN)2-catalyzed carbonylation of diethylamine with carbon dioxide: Selective synthesis of tetraethylurea and diethylformamide. Tetrahedron Lett. 1986, 27, 1809–1810. [Google Scholar] [CrossRef]
- Süss-Fink, G.; Langenbahn, M.; Jenke, T. Rutheniumcluster als Katalysatoren für die Carbonylierung von cyclischen Aminen. J. Organomet. Chem. 1989, 368, 103–109. [Google Scholar] [CrossRef]
- Zhang, L.; Han, Z.; Zhao, X.; Wang, Z.; Ding, K. Highly Efficient Ruthenium-Catalyzed N-Formylation of Amines with H2 and CO2. Angew. Chem. 2015, 127, 6284–6287. [Google Scholar] [CrossRef]
- Schreiner, S.; Yu, J.Y.; Vaska, L. Carbon dioxide reduction via homogeneous catalytic synthesis and hydrogenation of N,N-dimethylformamide. Inorg. Chim. Acta 1988, 147, 139–141. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Catalytic production of dimethylformamide from supercritical carbon dioxide. J. Am. Chem. Soc. 1994, 116, 8851–8852. [Google Scholar] [CrossRef]
- Liu, F.; Abrams, M.B.; Baker, R.T.; Tumas, W. Phase-separable catalysis using room temperature ionic liquids and supercritical carbon dioxide. Chem. Commun. 2001, 433–434. [Google Scholar] [CrossRef]
- Affan, M.A.; Jessop, P.G. Catalytic Formylation of Primary and Secondary Amines with CO2 and H2 Using Abundant-Metal Catalysts. Inorg. Chem. 2017, 56, 7301–7305. [Google Scholar] [CrossRef]
- Federsel, C.; Boddien, A.; Jackstell, R.; Jennerjahn, R.; Dyson, P.J.; Scopelliti, R.; Laurenczy, G.; Beller, M. A Well-Defined Iron Catalyst for the Reduction of Bicarbonates and Carbon Dioxide to Formates, Alkyl Formates, and Formamides. Angew. Chem. Int. Ed. 2010, 49, 9777–9780. [Google Scholar] [CrossRef]
- Federsel, C.; Ziebart, C.; Jackstell, R.; Baumann, W.; Beller, M. Catalytic Hydrogenation of Carbon Dioxide and Bicarbonates with a Well-Defined Cobalt Dihydrogen Complex. Chem. Eur. J. 2012, 18, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Ziebart, C.; Federsel, C.; Anbarasan, P.; Jackstell, R.; Baumann, W.; Spannenberg, A.; Beller, M. Well-Defined Iron Catalyst for Improved Hydrogenation of Carbon Dioxide and Bicarbonate. J. Am. Chem. Soc. 2012, 134, 20701–20704. [Google Scholar] [CrossRef] [PubMed]
- Minato, M.; Zhou, D.-Y.; Sumiura, K.; Hirabayashi, R.; Yamaguchi, Y.; Ito, T. Reactions of quadruply chelated silyl– and germyl–molybdenum hydrido complexes with carboxylic acids and carbon dioxide: A first example of carbon dioxide fixation utilizing the trans effect of a silyl ligand. Chem. Commun. 2001, 2654–2655. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Homogeneous Catalysis in Supercritical Fluids: Hydrogenation of Supercritical Carbon Dioxide to Formic Acid, Alkyl Formates, and Formamides. J. Am. Chem. Soc. 1996, 118, 344–355. [Google Scholar] [CrossRef]
- Munshi, P.; Heldebrant, D.J.; McKoon, E.P.; Kelly, P.A.; Tai, C.-C.; Jessop, P.G. Formanilide and carbanilide from aniline and carbon dioxide. Tetrahedron Lett. 2003, 44, 2725–2727. [Google Scholar] [CrossRef]
- Beydoun, K.; vomStein, T.; Klankermayer, J.; Leitner, W. Ruthenium-Catalyzed Direct Methylation of Primary and Secondary Aromatic Amines Using Carbon Dioxide and Molecular Hydrogen. Angew. Chem. 2013, 125, 9733–9736. [Google Scholar] [CrossRef]
- Vaska, L.; Schreiner, S.; Felty, R.A.; Yu, J.Y. Catalytic Reduction of Carbon Dioxide to Methane and Other Species via Formamide Intermediation: Synthesis and Hydrogenation of HC(O)NH2 in the Presence of (Ir(Cl)CO)(Ph3P)2. J. Mol. Catal. 1988, 52, L11–L16. [Google Scholar] [CrossRef]
- Motokura, K.; Takahashi, N.; Kashiwame, D.; Yamaguchi, S.; Miyaji, A.; Baba, T. Copper-diphosphine complex catalysts for N-formylation of amines under 1 atm of carbon dioxide with polymethylhydrosiloxane. Catal. Sci. Technol. 2013, 3, 2392–2396. [Google Scholar] [CrossRef]
- Frogneux, X.; Jacquet, O.; Cantat, T. Iron-catalyzed hydrosilylation of CO2: CO2 conversion to formamides and methylamines. Catal. Sci. Technol. 2014, 4, 1529–1533. [Google Scholar] [CrossRef]
- Itagaki, S.; Yamaguchi, K.; Mizuno, N. Catalytic synthesis of silyl formates with 1 atm of CO2 and their utilization for synthesis of formyl compounds and formic acid. J. Mol. Catal. A Chem. 2013, 366, 347–352. [Google Scholar] [CrossRef]
- González-Sebastián, L.; Flores-Alamo, M.; García, J.J. Nickel-catalyzed hydrosilylation of CO2 in the presence of Et3B for the synthesis of formic acid and related formates. Organometallics 2013, 32, 7186–7194. [Google Scholar] [CrossRef]
- Shintani, R.; Nozaki, K. Copper-catalyzed hydroboration of carbon dioxide. Organometallics 2013, 32, 2459–2462. [Google Scholar] [CrossRef]
- Yu, B.; Zhang, H.; Zhao, Y.; Chen, S.; Xu, J.; Huang, C.; Liu, Z. Cyclization of o-phenylenediamines by CO2 in the presence of H2 for the synthesis of benzimidazoles. Green Chem. 2013, 15, 95–99. [Google Scholar] [CrossRef]
- Li, Y.; Sorribes, I.; Yan, T.; Junge, K.; Beller, M. Selective methylation of amines with carbon dioxide and H2. Angew. Chem. 2013, 125, 12378–12382. [Google Scholar] [CrossRef]
- Jacquet, O.; Frogneux, X.; Gomes, C.D.N.; Cantat, T. CO2 as a C1-building block for the catalytic methylation of amines. Chem. Sci. 2013, 4, 2127–2131. [Google Scholar] [CrossRef]
- Li, Y.; Fang, X.; Junge, K.; Beller, M. A general catalytic methylation of amines using carbon dioxide. Angew. Chem. 2013, 125, 9747–9750. [Google Scholar] [CrossRef]
- Frogneux, X.; Blondiaux, E.; Thuéry, P.; Cantat, T. Bridging amines with CO2: Organocatalyzed reduction of CO2 to aminals. ACS Catal. 2015, 5, 3983–3987. [Google Scholar] [CrossRef]
- Jacquet, O.; DasNeves Gomes, C.; Ephritikhine, M.; Cantat, T. Recycling of carbon and silicon wastes: Room temperature formylation of N-H bonds using carbon dioxide and polymethylhydrosiloxane. J. Am. Chem. Soc. 2012, 134, 2934–2937. [Google Scholar] [CrossRef]
- Jacquet, O.; DasNeves Gomes, C.; Ephritikhine, M.; Cantat, T. Complete catalytic deoxygenation of CO2 into formamidine derivatives. ChemCatChem 2013, 5, 117–120. [Google Scholar] [CrossRef]
- Blondiaux, E.; Pouessel, J.; Cantat, T. Carbon Dioxide Reduction to Methylamines under Metal-Free Conditions. Angew. Chem. Int. Ed. 2014, 53, 12186–12190. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
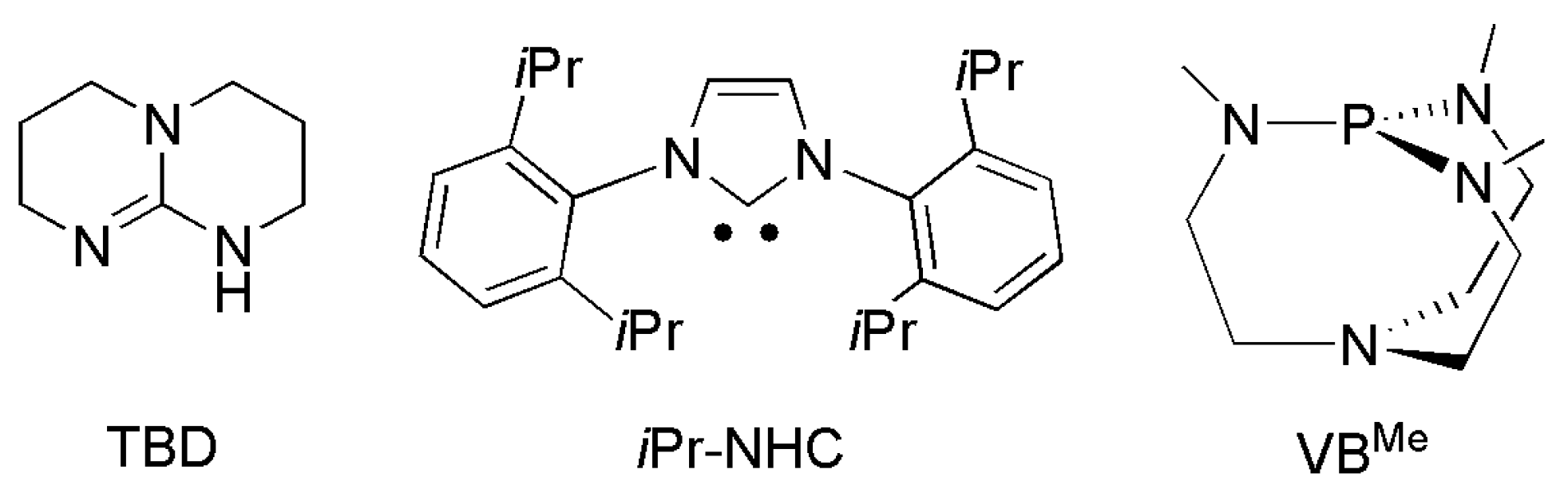{kind=link}
| Catalyst | E1/2 vs. SCE | TON | Solvent | Faradaic Efficiency | Reference | ||
|---|---|---|---|---|---|---|---|
| CO | HCOO− | H2 | |||||
| Re(bpy)(CO)3Cl | −1.49 V | 300 | - b | - a | DMF/H2O | 98% | [27] |
| fac-Re(4,4′-OCH3-bpy)(CO)3X | −2.32 V c | 3.9 | - b | - b | MeCN | 59% | [28] |
| cis-[Rh(bpy)2OTf2]+ | −1.55 V | - a | 6.8–12.3 | 8.5–28.5 | MeCN | 64% | [29] |
| [Ru(bpy)2(CO)Cl]+ | −1.50 V | 10.7–25.5 | 7.8–10.1 | 2.1–21.7 | DMF/H2O | - b | [29] |
| [Ru(bpy)2(CO)2]2+ | −1.50 V | 8.8–26.2 | 18.2–19.9 | 0.2–19.2 | DMF/H2O | - b | [29] |
| cis-[Os(bpy)2(CO)H]+ | −1.50 V | 5.5 a | - a | - b | MeCN | 90% | [30] |
| cis-[Os(bpy)2(CO)H]+ | −1.50 V | - b | 1.8 | - b | MeCN/H2O | 25% | [30] |
| trans-Cl-Ru(mesbpy)(CO)2Cl2 | −2.2 V c | 5.2 | - b | - b | MeCN | 95% | [31] |
| Catalyst | TON | Solvent | Faradaic Efficiency | Reference | ||
|---|---|---|---|---|---|---|
| CO | HCOO− | H2 | ||||
| Rh(dppe)2Cl | - | 1.58 | - | MeCN | 42% | [35] |
| [Pd(etpC)(DMF)](BF4)2 | 130 | - | 154 | DMF | 85% | [36] |
| {m-(triphos)2- [Pd(CH3CN)]2}(BF4)4} | 190 | - | - | DMF | 80% | [37] |
| Catalyst | Amine | P CO2/H2 (T) Bar (°C) | Reductant | TON/Yield (%) | Reference |
|---|---|---|---|---|---|
| IrCl(CO)(PPh3)2 | Me2NH | 27/27 (125) | H2 | 1200 | [188] |
| PdCl2 | Me2NH | 40/80 (170) | H2 | 34 | [172] |
| [Pt2(μ-dppm)3] | Me2NH | 12/94 (75) | H2 | 1460 | [173,177] |
| RuCl2(dppe)2 | Me2NH | 130/85 (100) | H2 | 74,000 | [164] |
| RuCl2(PMe3)4 | Me2NH | 130/80 (100) | H2 | 370,000 | [178] |
| Ru(PNP)(CO)(H)Cl | Morpholine | 35/35 a (120) | H2 | 1,940,000 | [176] |
| NiX2/dmpe(X = CH2COO− or acac) | Morpholine | 100 b (100–135) | H2 | 18,000 | [180] |
| (PPh3)3CuCl | Me2NH | 27/27 (125) | H2 | 900 | [171] |
| [MoH3{Si(Ph)[Ph2PCH2CH2P(Ph)C6H4-o]2}] | Me2NH | 30/20 a (110) | H2 | 115 | [184] |
| Fe(BF4)2⋅6H2O/PP3 | Me2NH | 30/60 | H2 | 727 | [181] |
| Co(BF4)2⋅6H2O/PP3 | Me2NH | 30/60 | H2 | 1308 | [182] |
| Fe(BF4)2⋅6H2O/PAr3 | Me2NH | 30/60 | H2 | 5104 | [183] |
| Rh2(OAc)4/K2CO3 | PhCH2NH2 | 1 a (50) | PhMe2SiH | 41% | [191] |
| piperidine | 43% | ||||
| PhNH2 | 34% | ||||
| [Ni(μ-H)(dippe)]2/BEt3 | PhCH2NH2 | 1 a (80) | Et3SiH | 85% | [192] |
| (PhCH2)2NH | 47% | ||||
| piperidine | 52% | ||||
| Cu(OAc)2⋅H2O/Bz(PR2)2 | Piperidine | 1 a (80) | PMHS | 11,700/94% | [189] |
| Fe(acac)2/P(C2H4PPh2)3 | (Ph)(Me)NH | 1 (RT) | PhSiH3 | 95% | [190] |
| Cu(iPrNHC)(OtBu) | (Ph)(Me)NH | 1 a (35/65) | H-Bpin | 81% | [193] |
| Ph(CH2)2NH2 | 98% | ||||
| (PhCH2)2NH | 90% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, C.-F.; Ho, P.-Y. Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock. Catalysts 2019, 9, 760. https://doi.org/10.3390/catal9090760
Leung C-F, Ho P-Y. Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock. Catalysts. 2019; 9(9):760. https://doi.org/10.3390/catal9090760
Chicago/Turabian StyleLeung, Chi-Fai, and Pui-Yu Ho. 2019. "Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock" Catalysts 9, no. 9: 760. https://doi.org/10.3390/catal9090760
APA StyleLeung, C.-F., & Ho, P.-Y. (2019). Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock. Catalysts, 9(9), 760. https://doi.org/10.3390/catal9090760