Abstract
A packed-bed plug-flow reactor, denoted as the lab-scale liquid-solid (LS)² reactor, has been developed for the assessment of heterogeneous catalyst deactivation in liquid-phase reactions. The possibility to measure intrinsic kinetics was first verified with the model transesterification of ethyl acetate with methanol, catalyzed by the stable commercial resin Lewatit K2629, for which a turnover frequency (TOF) of 6.2 ± 0.4 × 10−3 s−1 was obtained. The absence of temperature and concentration gradients was verified with correlations and experimental tests. The potential for assessing the deactivation of a catalyst was demonstrated by a second intrinsic kinetics evaluation where a methylaminopropyl (MAP)-functionalized mesoporous silica catalyst was used for the aldol reaction of acetone with 4-nitrobenzaldehyde in different solvents. The cooperative MAP catalyst deactivated as a function of time on stream when using hexane as solvent. Yet, the monofunctional MAP catalyst exhibited stable activity for at least 4 h on stream, which resulted in a TOF of 1.2 ± 0.1 × 10−3 s−1. It did, however, deactivate with dry acetone or DMSO as solvent due to the formation of site-blocking species. This deactivation was mitigated by co-feeding 2 wt % of water to DMSO, resulting in stable catalyst activity.
1. Introduction
The activity of heterogeneous catalysts in liquid-phase reactions is often evaluated using a batch type reactor [1,2,3,4,5]. However, not only the activity of the catalyst, but also its stability is an important factor in the assessment for a potential application [6]. This means that the catalyst should exhibit a stable activity throughout its lifetime, or be easily regenerated to a similar activity level. It is thus important to unambiguously characterize the stability of new heterogeneous catalysts. However, due to the nature of a batch reactor, catalyst deactivation phenomena cannot be observed separately from the normal time evolution of a single experiment [7]. Hence, to evaluate heterogeneous catalyst stability in a batch reactor, the spent catalyst should be recycled in a subsequent experiment. Yet, all too often, catalyst stability is concluded from repeated high yields that are either at full reactant conversion or at the thermodynamic equilibrium [8,9,10,11,12,13,14,15,16,17,18,19]. As a result, such experimental information does not provide any indication of the catalyst stability. Deactivation is a kinetic phenomenon for which initial rates, far from thermodynamically controlled conversion levels, should be measured in consecutive batch experiments [20,21,22].
This concept is visualized in Figure 1, where hypothetical catalyst activity curves are drawn for a first run and two consecutive runs of a deactivating catalyst for the reaction A → B with only one sample after a time tsample in a batch reactor. Even though in a second run the reaction also reaches a 100% conversion, reaching this level of conversion takes a much longer time. Yet, this kinetic phenomenon is not visible when a single sample is taken at tsample. Moreover, when a third run of the same catalyst is evaluated, a slightly lower than 100% conversion level is reached which can, mistakenly, be interpreted as minor catalyst deactivation. However, when the turnover frequency (TOF) of the different runs is measured from the slope of the time versus conversion graph in the initial conversion region, a clear drop in catalytic activity can be noted and the extent of deactivation can be quantified by their respective ratios.
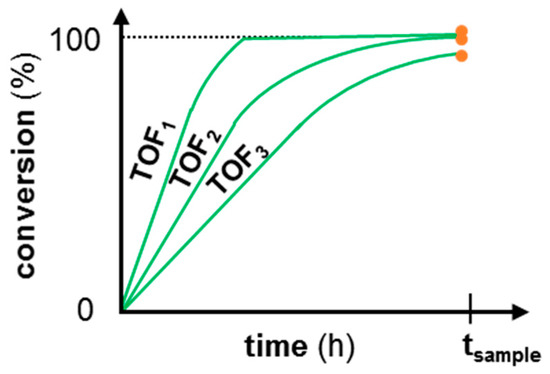
Figure 1.
Hypothetical catalyst activity (turnover frequency—TOF) in a first and two recycle batch experiments for the reaction A → B. Only one sample is taken for each run after a time tsample, and is indicated in orange.
Moreover, investigation of catalyst reusability in a batch reactor typically requires many consecutive manual interventions by the operator, almost unavoidably including catalyst exposure to (often poorly controlled) atmospheric conditions, rendering reproducible operation very challenging. Hence, to avoid having to filter the catalyst from the reactor, a continuous-flow reactor would preferentially be used. As opposed to a batch-type reactor, this type of reactor provides a direct way to observe deactivation phenomena of a heterogeneous catalyst during a single experiment by measuring its performance as a function of the time on stream [23].
1.1. Continuous-Flow Reactor for the Assessment of Catalyst Deactivation
When steady state operation is achieved in terms of hydrodynamics, a stable catalyst in a fixed bed under continuous-flow conditions will exhibit constant activity as a function of the time on stream. A decreasing activity as a function of time on stream at constant operating conditions is then caused by catalyst deactivation. The specific evolution in the catalyst activity can then be used to gain more insight into the mechanism responsible for this deactivation [24]. Hence, the assessment of catalyst stability is preferably done in a continuous-flow reactor as opposed to a batch reactor. Deactivation of a heterogeneous catalyst can occur in various manners such as the loss of surface area due to strong adsorption of components in the feed or produced during reaction, thermal degradation, etc. [25]. Kinetically, this leads to a rate which not only depends on the operating conditions, but also depends on the historical evolution of operating conditions the catalyst has been subjected to. A common approach to deactivation kinetics results in a “separable” rate expression [24,26], written as a time-on-stream independent kinetics term, and a time-on-stream dependent deactivation term. Hence, to determine the catalyst activity free of any deactivation phenomena, a function representative for the deactivation kinetics should be found [24,27,28].
While time-on-stream measurements in continuous-flow reactors are sometimes used to assess the stability of heterogeneous catalysts, often conditions are chosen such that the behavior is thermodynamically controlled, i.e., that complete or equilibrium reactant conversion is obtained [29,30,31,32]. Yet, similarly to batch experiments, it is important to not operate at these high, or equilibrium, conversion levels, because it does not provide the required kinetic information to adequately assess deactivation [21]. This is visualized in Figure 2, where hypothetical catalyst activity curves are drawn as a function of space time (W/F0feed) and time on stream (TOS) for the reaction A → B. The deactivation is, in this example, assumed to be caused by an undesired side-reaction of a reactive intermediate on the catalyst surface, which results in the deactivation moving as a front through the catalyst bed. Nonetheless, the conclusions drawn from this example are generally applicable to any catalyst deactivation behavior in a plug-flow reactor.
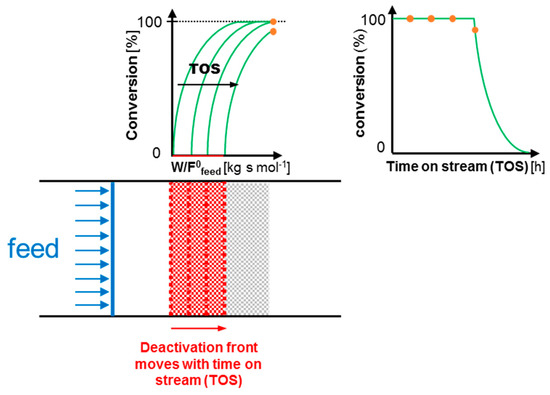
Figure 2.
Hypothetical example of catalyst deactivation by off-cycle species poisoning in a plug-flow reactor for the reaction A → B, operated at full reagent conversion level.
The conversion versus space time (W/ F0feed) curve in Figure 2 indicates that, in the absence of any catalyst deactivation, complete reactant conversion is already reached in the middle of the catalyst bed. When the deactivation is progressing through the catalyst bed as a function of time on stream, the reactant conversion will, at the end of the reactor where samples are drawn, remain at 100%. This causes a constant (100%) level of conversion to be observed as a function of the time on stream for a certain amount of time from which, mistakenly, stable catalyst operation could be concluded.
Even though real catalyst deactivation will inevitably be much more complex, and can present itself in a variety of different ways besides a drop in conversion, such as a shift in selectivity, it remains paramount that also in a continuous-flow reactor the catalyst performance is always measured at intermediate reactant conversion levels.
1.2. Intrinsic Kinetics Assessment
When catalyst performance is evaluated in a continuous-flow setup, typically a packed-bed tubular reactor is used due to its simple design and ease of operation. However, in this kind of setup, as opposed to a batch reactor, it is more challenging to guarantee the “intrinsic” character of the acquired kinetic data, i.e., ensuring that the kinetics that are not affected by mass or heat transport limitations [7,33,34]. Several types of concentration and temperature gradients can occur in such reactors, as illustrated in Figure 3: axial and radial reactor-scale gradients, interphase gradients between the bulk of the liquid phase and the external catalyst pellet surface, and intrapellet gradients within the porous catalyst [7,35]. In batch operation, the reactor-scale axial, radial, and interphase gradients can easily be overcome by employing an adequate mixing rod and a high stirring speed. This ensures that the reactor is homogeneous in both temperature and concentration. Intrapellet gradients can be prevented by employing a sufficiently smaller catalyst particle diameter.
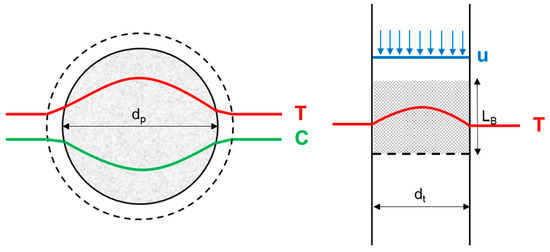
Figure 3.
Possible exothermal temperature and reagent concentration gradients in a packed-bed tubular reactor under plug-flow conditions on the pellet scale (left) and reactor scale (right).
In a packed bed tubular reactor, however, a trade-off needs to be inherently pursued with respect to the pellet size. It should be sufficiently small to obtain ideal plug-flow hydrodynamics as well as for discarding intrapellet gradients [36], while it should not be too small to avoid an increased pressure drop over the catalyst bed. External concentration gradients can be avoided by operating under a sufficiently high linear velocity of the feed over the catalyst bed. However, this will negatively impact the conversion over the catalyst bed. It is thus clear that designing the reactor and determining the reaction conditions for measuring “intrinsic” kinetics of heterogeneous catalysts in the absence of any transport limitations is considerably more challenging in a tubular reactor than in a batch reactor [7].
To aid in the assessment of whether intrinsic kinetics can be attained for a specified liquid-phase reaction in a packed-bed tubular reactor, correlations can be used. To ensure plug-flow conditions with a uniform velocity profile (u) over the catalyst bed such as displayed in Figure 3, ideal mixing in the radial direction and no mixing in the axial direction is required. Both these criteria can be evaluated with correlations from Mears et al. [37,38] which depend on the reactor tube diameter (dt), the catalyst pellet size (dp) and the bed length (LB). The pressure drop (Δp) over the catalyst bed is evaluated with the Ergun equation [39] and should not exceed 20% of the total operating pressure for first order reactions. Radial isothermicity is verified by a criterion derived by Mears et al. [40], and axial temperature gradients can be reduced by diluting the bed with inert particles [40]. This bed dilution, however, should also be limited to avoid an improper catalyst distribution which can affect the conversion [41]. The absence of both radial and axial temperature gradients can also be experimentally evaluated with internal thermocouples in the catalyst bed.
On the catalyst pellet scale, the external mass transfer limitation around the pellet can be expressed with Carberry’s number (Ca) [42], using Wilke and Chang’s correlation for the liquid-phase molecular diffusion coefficient [43]. The intrapellet mass transfer limitation is evaluated by the Weisz‒Prater criterion (φ) [44]. Both the external and intrapellet mass transfer limitations can also be experimentally assessed by, respectively, varying the linear velocity of the feed (u) for a fixed space time, or varying the catalyst particle diameter (dp) [45]. The occurrence of external temperature gradients is assessed by an energy balance over the film around the catalyst pellet [7], with the heat transfer coefficient obtained from the Chilton‒Colburn j-factor [46]. Similarly, intrapellet temperature gradients are assessed by an energy balance over the complete particle [7].
1.3. Aim of this Work
In this work, a lab-scale liquid-solid (LS)² plug-flow reactor with a fixed catalyst bed is designed aiming at assessing deactivation phenomena of heterogeneous catalysts in liquid-phase reactions. The performance of the (LS)² reactor is demonstrated for two frequently used liquid-phase reactions, i.e., a transesterification reaction and an aldol reaction. The intrinsic character of the measurements is verified both with correlations as well as experimental tests. The difference between a stable catalyst and a deactivating catalyst is then straightforwardly observed by evaluation of their performance as a function of time on stream. If stable catalyst operation is obtained, different space time conditions are employed in order to determine the turnover frequency of the catalyst.
2. Results and Discussion
The possibility of acquiring intrinsic kinetics in the (LS)² plug-flow reactor is evaluated for a model transesterification of ethyl acetate and methanol, catalyzed by a stable commercial sulfonic acid functionalized resin catalyst. Next, catalyst deactivation phenomena are assessed with a model aldol reaction between acetone and 4-nitrobenzaldehyde, catalyzed by an aminated mesoporous silica catalyst. Both types of reactions have a potential application in the bio-based industry and are traditionally catalyzed by homogeneous acid or base catalysts, but can be heterogeneously catalyzed in a fixed-bed plug-flow reactor. Aldol reactions can, for example, be used for the production of biofuels from lignocellulosic biomass, by upgrading smaller organic molecules to heavier hydrocarbons in the range desired for internal combustion engines [47]. Transesterification reactions, on the other hand, are already employed in the production of biodiesel [48], but are currently homogeneously catalyzed. The evaluation of new heterogeneous catalysts for this type of reaction is thus currently widely investigated [49,50,51].
2.1. Case Study: Transesterification Reaction
Acidic ion exchange resins are a promising class of heterogeneous acid catalyst for the transesterification reaction because they are ecofriendly, noncorrosive, and reusable [52,53]. The most frequently commercially employed resins are based on polystyrene, cross-linked with a well-chosen amount of divinylbenzene [53,54]. Many of these commercial catalysts have been evaluated in a transesterification or an esterification reaction [1,50,51,55,56,57,58,59,60]. However, these reactions are often performed batch wise. Hence, as an example case study, the Lewatit K2629 catalyst is evaluated in the (LS)² plug-flow reactor for the transesterification of ethyl acetate and methanol towards methyl acetate and ethanol (see also Scheme 1). This catalyst is a strongly acidic macroporous resin with 4.8 mol kg−1 sulfonic acid groups as active sites and is known to be fully recyclable in a batch reactor [61].
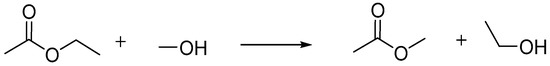
Scheme 1.
Model transesterification of ethyl acetate and methanol towards methyl acetate and ethanol.
2.1.1. Intrinsic Kinetics Verification
Using the experimental TOF determined further in Section 2.1.2, the correlations for the assessment of intrinsic kinetics were evaluated at the lowest employed feed flow rate, where it is expected that attaining intrinsic kinetics is the most challenging. As displayed in Table 1, all correlations are satisfied, which indicates that intrinsic kinetics can indeed be measured for the transesterification reaction under the employed conditions in the (LS)² reactor.

Table 1.
Criteria for intrinsic kinetics of the transesterification between methanol and ethyl acetate in the (LS)² reactor, catalyzed by Lewatit K2629 at 60 °C.
Comparison of the temperature measured by the internal thermocouple and external thermocouple, at the height of the catalyst bed, allowed the absence of a radial temperature gradient throughout all the experiments to be verified. Temperature gradients on the pellet scale, however, are not directly measurable with an experimental observable.
An additional experimental test is performed to ensure the absence of external concentration gradients. As can be seen in Table 2, changing the feed flowrate at a constant site time results in the same conversion, which confirms the absence of external concentration gradients at the lower range of flow rates used for this case study.
Table 2.
Conversion and flow rate for two transesterification experiments with an identical site time.
2.1.2. Catalyst Stability and Activity
The ethyl acetate conversion as a function of time on stream is displayed in Figure 4. The steady conversion from 1 h on stream up to 4 h on stream, indicates that the Lewatit K2629 catalyst is, effectively, stable in this timeframe.
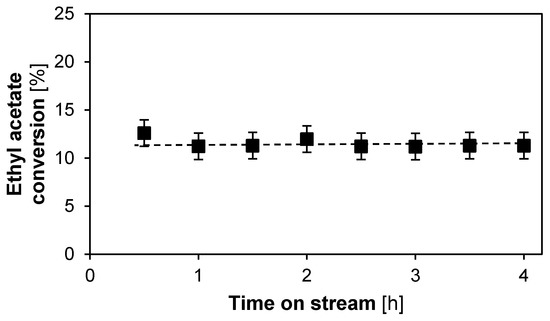
Figure 4.
Conversion in function of time on stream for the transesterification between methanol and ethyl acetate using n-octane as a solvent. Lewatit K 2629 catalyst, T = 60 °C, Ptot = 180 kPa, site time = 15.75 molsite s mol−1, 19.8 wt % ethyl acetate, 71.4 wt % methanol, 8.8 wt % octane (internal standard). Fixed error of 1.4%.
Determining the TOF of a stable catalyst in a plug-flow reactor can straightforwardly be performed by measuring the slope of the initial linear part of the conversion graph at different catalyst site times (see Figure 5). This way, the TOF for the transesterification between methanol and ethyl acetate using octane as a solvent over the Lewatit K2629 catalyst is determined at 6.2 ± 0.4 × 10−3 s−1. This value is in the expected range for this type of transesterification reaction catalyzed by an acidic resin [61,62,63,64]. Only the slope of the first three sample points in Figure 5 is used for the linear fit of the TOF, because the sample at a site time of 31.5 molsite s molethyl acetate−1 is in the so-called integral reactor regime, where the decrease in reactant concentration causes a decrease in the reaction rate [7].
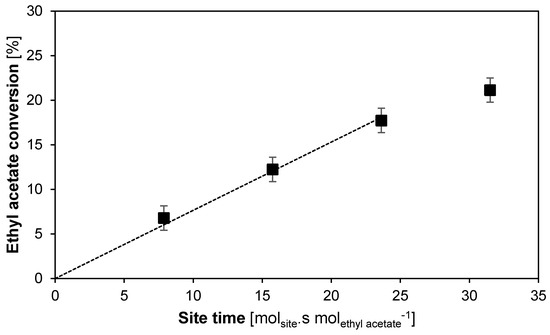
Figure 5.
Conversion in function of site time of the Lewatit K2629 for the transesterification between methanol and ethyl acetate. T = 60 °C, Ptot = 180 kPa, site time = 7.9–31.5 molsite s mol−1, 19.8 wt % ethyl acetate, 71.4 wt % methanol, 8.8 wt % octane (internal standard). Fixed error of 1.4%.
2.2. Case study: Aldol Reaction
The aldol reaction between acetone and 4-nitrobenzaldehyde (see Scheme 2) has often been investigated as a model reaction in batch reactors with heterogeneous catalysts such as amines supported on mesoporous silica [65,66,67]. Methylaminopropyl (MAP) grafted on mesoporous silica is reported in literature as the best performing secondary amine catalyst due to its limited steric hindrance, and reported TOF in the batch reactor amounted up to a maximum of 3.3 ± 0.2 × 10−3 s−1 [68,69,70] for the best performing catalyst. When the amine site is promoted by the residual silanol groups on the surface of the silica support, the catalyst is denoted as “cooperative acid-base”. When the residual silanol groups have been endcapped with trimethylsilyl groups, and are thus no longer available for promotion, the catalyst is called “monofunctional base”. Both types of catalysts are evaluated for their stability in this work, under the same conditions as previously employed in the batch reactor [5,68].
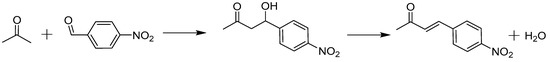
Scheme 2.
Model aldol reaction of acetone with 4-nitrobenzaldehyde, towards the aldol product 4-hydroxy-4-(4-nitrophenyl)butan-2-one, and subsequent dehydration to the enone product 4-(4-nitrophenyl)-3-buten-2-one.
2.2.1. Intrinsic Kinetics Verification
Using the reported TOF from evaluation of the cooperative MAP catalyst in a batch reactor [68], the correlations for the assessment of intrinsic kinetics in the (LS)² reactor were evaluated for either hexane or DMSO as solvent in 50 vol % acetone as excess reagent. Solvent-free operation, i.e., 100 vol % acetone, is also evaluated. In the assessment, the most severe operation possible is analyzed, i.e., the most active catalyst with the lowest feed flow rate, for which it is expected that attaining intrinsic kinetics will be the most challenging. The values reported in Table 3 indicate that intrinsic kinetics can indeed be measured for all the solvents under the given conditions. All obtained values deviate at least an order of magnitude from the critical value, except the Weisz‒Prater number (ϕ) for internal mass diffusion. Because this value is close to the limit value of 8 × 10−2, an additional experimental validation is performed with a catalyst of a different pellet size.
Table 3.
Criteria for measuring intrinsic kinetics using a secondary methylaminopropyl (MAP)-functionalized mesoporous silica catalyst in the aldol reaction between acetone and 4-nitrobenzaldehyde with a 50/50 vol % solvent/acetone liquid mixture or solvent-free in 100 vol % acetone.
To experimentally assess the occurrence of internal concentration gradients, a MAP catalyst with a pellet size of 137 µm is synthesized. The performance of this catalyst using hexane as solvent is compared to that of the catalyst with a pellet diameter of 375 µm in Table 4, and confirms the absence of internal concentration gradients.
Table 4.
Conversion of two aldol reaction experiments with hexane as solvent for different catalyst pellet sizes.
External concentration gradients are evaluated by performing experiments using hexane as solvent with identical site times at two different feed flow rates in the lower range of the flow rates used for this case study. This correlates to a different liquid velocity (u). The absence of external concentration gradients is confirmed when the conversion is identical, which is indeed the case within the experimental error, as can be seen in Table 5.
Table 5.
Conversion of two different aldol reaction experiments using hexane as solvent with an identical site time but different feed flow rates.
2.2.2. Catalyst Stability and Activity
Figure 6 shows the 4-nitrobenzaldehyde conversion as a function of time on stream for both the cooperative acid-base and the monofunctional base catalyst evaluated with hexane as solvent. The cooperative acid-base catalyst appears to continuously deactivate from 85.5% conversion at 30 min on stream, which is below the equilibrium conversion of 99.5%, to 70.6% conversion after 3.5 h on stream. In contrast, the decrease in activity for the monofunctional base catalyst remains limited to 3.0% in a 3.5 h timespan. This indicates that the deactivation of the cooperative acid-base catalyst is mainly caused by the presence of the promoting silanol groups.
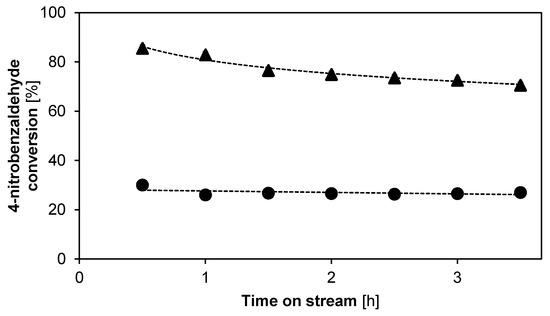
Figure 6.
Conversion as function of time on stream for the aldol reaction between acetone and 4-nitrobenzaldehyde using hexane as a solvent. ●: cooperative acid-base catalyst, ▲: monofunctional base catalyst. (T = 55 °C, Ptot = 180 kPa, site time = 165.1 molsite s mol−1, 0.6 wt % 4-nitrobenzaldehyde, 55.6 wt % acetone, 43.5 wt % hexane, 0.25 wt % methyl-4-nitrobenzoate (internal standard). Fixed error of 1.8%, not visible on the scale). Line is a guide for the eye.
As previously reported [5], when DMSO is used as solvent, the amine sites themselves can be subject to deactivation when the concentration of water is too low. In Figure 7, the stability of the monofunctional base catalyst is evaluated for DMSO as solvent with respectively 0 wt % and 2 wt % of water added. As expected from literature, the catalyst activity is lower when using DMSO as solvent as compared to hexane, due to the increased stabilization of ionic intermediates [5,68,69]. Regarding the stability, the same trend as a function of the added water amount in DMSO is noted as previously reported for repeat batch experiments [5]: without water addition, the catalyst deactivates as a function of time on stream. With 2 wt % of water added, the conversion profile as a function of time on stream is stable for at least 5 h on stream. This can directly be related to an increased hydrolysis of the iminium ion intermediate which would otherwise, under low water concentrations, lead to site-blocking [5].
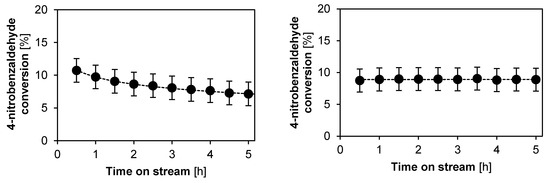
Figure 7.
Conversion as a function of time on stream for the aldol reaction between acetone and 4-nitrobenzaldehyde, catalyzed by the monofunctional base catalyst, using DMSO as a solvent with varying amounts of water. Left: no added water, Right: 2 wt % water added. (T = 55 °C, Ptot = 180 kPa, site time = 165.1 molsite s mol−1, 0.45 wt % 4-nitrobenzaldehyde, 44.6 wt % acetone, 52.5–55.0 wt % DMSO, 0.25 wt % IS). Fixed error of 1.8%. Line is a guide for the eye.
Figure 8 indicates the 4-nitrobenzaldehyde conversion as a function of the time on stream for the monofunctional base catalyst, evaluated without solvent, i.e., in 100 vol % dry acetone. The initial catalytic activity is higher than in the case of DMSO as solvent, but still lower than with hexane. This can be attributed to the polarity of acetone, which is in-between that of DMSO and hexane [69]. Moreover, its activity does not appear to stabilize, even after 8 h on stream and indicates a steady catalyst deactivation. This can be attributed to the stabilization of the iminium ion intermediate which, coupled with a low water concentration, will lead to a similar catalyst deactivation as in the case of DMSO as solvent [5].
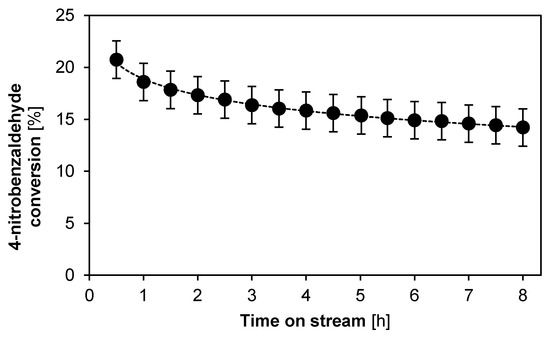
Figure 8.
Conversion as function of time on stream for the aldol reaction between acetone and 4-nitrobenzaldehyde employing the monofunctional base catalyst, solvent-free. (T = 55 °C, Ptot = 180 kPa, site time = 165.1 molsite s mol−1, 0.45 wt % 4-nitrobenzaldehyde, 99.3 wt % dry acetone, 0.25 wt % methyl-4-benzoate (internal standard)). Fixed error of 1.8%.
To determine the TOF of a deactivating catalyst, extrapolation to “time on stream equal to zero” should be performed to ensure that the catalytic performance free of any deactivation is measured. However, the nature of this extrapolation will strongly depend on a model for the kinetics of the deactivation phenomenon and, hence, requires a more detailed investigation. Therefore, in this section only the catalytic activity of the stable monofunctional base catalyst in hexane is evaluated as an example. From the linear fit in Figure 9, a TOF of 1.2 ± 0.1 × 10−3 s−1 is obtained, which is close to the literature reported TOF of 1.0 ± 0.1 × 10−3 s−1 for the monofunctional MAP catalyst in a batch reactor under the same reaction conditions [68].
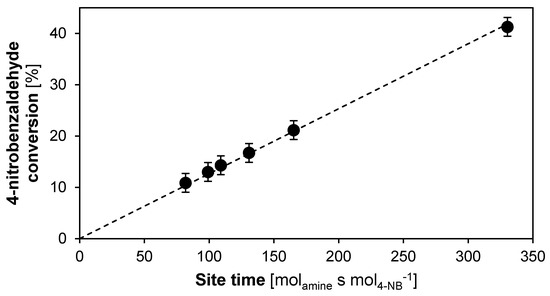
Figure 9.
Conversion as function of site time of a monofunctional base catalyst for the aldol reaction between acetone and 4-nitrobenzaldehyde using hexane as solvent. (T = 55 °C, Ptot = 180 kPa, site time = 81.75–330.3 molsite s mol−1, 0.45 wt % 4-nitrobenzaldehyde, 55.8 wt % acetone, 43.5 wt % hexane, 0.25 wt % IS. Fixed error of 1.8 %).
3. Materials and Methods
3.1. Reactor Design
The developed lab-scale liquid-solid (LS)² plug-flow reactor is schematically represented in Figure 10.
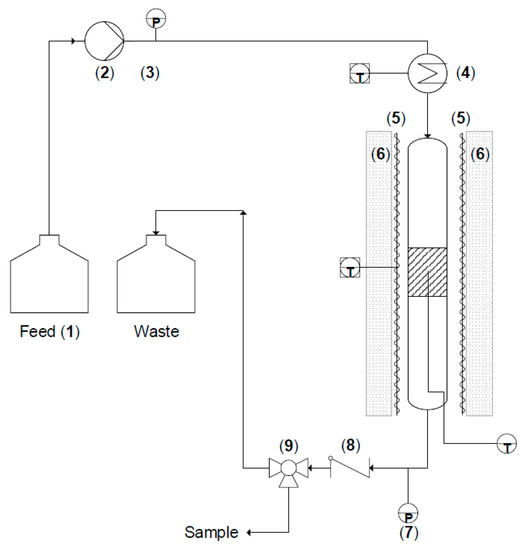
Figure 10.
Schematic of the (LS)² reactor with the feed vessel (1), the positive displacement pump (2) with pulsation damper and pressure readout (3), the pre-heating (4), the reactor heating (5) and insulation (6), the outlet pressure gauge (7), and the check valve (8) and three-way valve (9) for drawing samples.
The feed section essentially comprises a flask with the reactant mixture (1 in Figure 10) connected to an Eldex 2SMP plunger pump (2 in Figure 10). The flask is gently stirred to avoid the development of concentration gradients within the reactant mixture. The positive displacement pump can deliver flow rates between 0.01 and 10 mL/min with discharge pressures up to 400 bar. To reduce flow fluctuations, a pulsation damper is installed on the pump. A manometer (3 in Figure 10) is coupled to this damper, which measures the pump discharge pressure and is able to impose a high-pressure limit above which the pump operation is stopped. The piston is made from sapphire, and the inlet and outlet valves incorporate sapphire seats with ruby balls. The piston seal material is made from graphite fiber reinforced polytetrafluoroethylene (PTFE) and all the tubing is made of type 316 stainless steel. Seals are either a chlorotrifluoroethylene (CTFE) or Kal-Rez® type. All these internal parts are checked for their chemical compatibility with the employed reagents in this work.
To ensure that the reactant mixture is fed at the right temperature upon reaching the catalyst bed for a variety of flow rates, the option for additional pre-heating before the reactor inlet is available. The preheater is an insulated box around the feed tube with a radiation heater of 125 W (4 in Figure 10). The box itself is made of thermally insulating material ‘FACT01 PROMASIL 1100 super’. A thermocouple is placed inside the box in order to measure and control the temperature with a PID controller (Omron e5ck).
The actual reactor is a stainless steel 316 tube with an inner diameter of 0.75 cm and a length of 30 cm. At 10 cm from the bottom of the reactor, a mesh is welded to the reactor wall to prevent the packing and catalyst from leaving the reactor. The reactor tube is surrounded by a heating coat to bring the reactor to the desired reaction temperature, controlled with an Omron e5ck PID controller. The entire reactor and heating wire are insulated in order to keep the reactor isothermal (6 in Figure 10). The external thermocouple is attached to the reactor tube, between the heating wire and the outside of the tube. An internal thermocouple is inserted via the bottom of the reactor tube and is located in the middle of the catalyst bed.
The heated sections of the reactor are at a constant pressure, which is maintained slightly above ambient pressure to avoid vaporization of the liquid phase. The outlet pressure can be monitored on the pressure gauge which is installed immediately after the reactor outlet (7 in Figure 10). To create a back-pressure, a check valve (8 in Figure 10) is installed with a spring that, in this work, is set to compress at 1.8 barg. Higher pressure check valves can be installed for other applications, up to the pressure limit of the feed pump. The difference between this backpressure and the pump outlet pressure gives an indication of the pressure drop over the entire setup.
Samples of the reactor outlet can be drawn by switching the three-way valve (9 in Figure 10). This allows samples to be taken in an easy and safe way. When no sample is being taken, the reactor effluent is collected in a waste vessel that is placed on a balance. This balance is used to verify the mass balance over the set-up.
The catalyst activity is determined from the slope of a conversion versus site time plot. Site time is defined as the space time W Freagent,0−1, multiplied with the active site concentration Csite. With W the catalyst mass (kgcat) and Freagent0 the molar feed flow rate of the limiting reagent (molreagent s−1) and Csite (molsite kgcat−1) the concentration of active sites, site time has units (molsite s molreagent−1).
3.2. Case Study: Transesterification
Lewatit K2629, a commercially available resin, was used as transesterification catalyst. It is a strongly acidic, macroporous, polymer-based resin in spherical bead form with an average pellet size of 550 µm, with 4.8 mol/kg sulfonic acid groups. Before loading the catalyst in the reactor, it is freeze-dried using Alpha 1-2 LDplus apparatus operated at 0.12 mbar, in order to remove all the water from the active sites.
For the transesterification kinetics, a feed mixture comprising 19.8 wt % ethyl acetate, 71.4 wt % methanol, and 8.8 wt % octane (internal standard) was employed similar to literature reported conditions [1]. All transesterification experiments were performed at 60 °C. Sample analysis was performed with gas chromatograph from Agilent Technologies equipped with a FID (flame ionization detector) and a PONA column. The injector temperature was set at 270 °C and the detector temperature was set at 300 °C. The following temperature program was used in the oven: it started at 40 °C for 5 min, then increased at a rate of 10.0 °C s−1 to 150 °C. A split flow of 50:1 was used. The helium flow over the column was 155.4 10−3 Nl min−1. Quantification of the components in the reaction mixture was performed by relating the corresponding peak surface areas to the amount of internal standard that was added in the reaction.
3.3. Case Study: Aldol Reaction
Two types of catalysts were prepared by grafting N-methylaminopropyltrimethoxysilane (MAPTMS, ABCR, 99%) on silicagel 60 with different support pellet sizes (grade 7734, Sigma-Aldrich, average pellet size 375 µm and 137 µm), using the same procedure as previously reported [5,71]. Endcapping of the surface silanol groups was performed similarly as previously reported [5,71] using 1,1,1,3,3,3-hexamethyldisilazane (99%, HMDS, ABCR).
The loading of the amine sites was determined with elemental (CHN) analysis. These measurements were performed on a Thermo Flash 2000 elemental analyzer using V2O5 as an oxidation. The mass percent of nitrogen in the sample was then obtained by referring the peak surface area of nitrogen to a calibration curve that was obtained with methionine (USP, 99%).
The feed mixture when using hexane as solvent consisted of 0.45 wt % 4-nitrobenzaldehyde, 55.8 wt % acetone, 43.5 wt % hexane, and 0.25 wt % methyl 4-nitrobenzoate (internal standard). When using DMSO as solvent, this changed to 0.45 wt % 4-nitrobenzaldehyde, 44.6 wt % acetone, 52.5 wt % or 54.7 wt % DMSO, 0.25 wt % methyl 4-nitrobenzoate, and 0 wt % or 2.19 wt % deionized water. All aldol reactions were performed at 55 °C. The reaction samples were analyzed using a reversed-phase high-performance liquid chromatograph (RP-HPLC), from Agilent (1100 series) on a XDB-C18 column, using the same procedure as reported before [5]. Quantification of the different components in the reaction mixture was performed by relating the surface areas of the corresponding components to the amount of internal standard added in the reaction.
4. Conclusions
A lab-scale continuous-flow reactor was built for assessing heterogeneous catalyst stability in liquid-phase reactions, and is referred to as the (LS)² reactor, i.e., the lab-scale liquid-solid reactor. The reactor, and its possibility to measure intrinsic kinetics and deactivation phenomena, was evaluated in two case studies.
The possibility to measure intrinsic kinetics was verified in a first case study for the transesterification between ethyl acetate and methanol, catalyzed by the stable commercial Lewatit K2629 resin at 60 °C. The related correlations were evaluated, and experimentally, the absence of external concentration gradients and radial temperature gradients were verified. The catalyst indeed exhibited a stable activity, which resulted in a turnover frequency (TOF) of 6.2 ± 0.4 × 10−3 s−1.
In the second case study, the stability of methylaminopropyl (MAP)-functionalized mesoporous silica was evaluated for the aldol reaction of acetone with 4-nitrobenzaldehyde in a variety of solvents. For all the reaction conditions, it was verified with correlations that measurement of intrinsic kinetics was possible. When the amine group was promoted by surface silanol groups, no stable activity as a function of the time on stream was found when using hexane as solvent. However, the unpromoted monofunctional MAP catalyst did exhibit a stable activity for at least 4 h on stream in hexane, but deactivation could not be avoided with dry acetone or DMSO as solvent. Adding 2 wt % of water to DMSO significantly reduced the extent of deactivation and resulted in a stable conversion profile during 4 h on stream. The TOF of the stable monofunctional MAP catalyst in hexane was determined at 1.2 ± 0.1 × 10−3 s−1, which is closely related to the TOF found in a batch reactor under identical conditions [68]. To ensure intrinsic kinetics were measured, two additional experimental tests for the absence of internal and external concentration gradients were successfully performed and the absence of a radial temperature gradient was experimentally confirmed.
This work shows that the developed (LS)² reactor can successfully be used for the intrinsic kinetics assessment of heterogeneous catalysts in liquid-phase reactions and can adequately determine deactivation phenomena of these catalysts.
Author Contributions
Conceptualization, J.L. and J.W.T.; data curation, S.V.A.; formal analysis, A.D.V.; investigation, S.V.A.; methodology, J.D.C.; project administration, J.W.T.; resources, J.W.T.; supervision, A.D.V., J.L., J.D.C. and J.W.T.; validation, A.D.V.; visualization, A.D.V.; writing—original draft, A.D.V.; writing—review and editing, A.D.V., J.L., J.D.C. and J.W.T.
Funding
The authors acknowledge financial support from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) / ERC grant agreement n° 615456. J.L. is a postdoctoral fellow of the Research Foundation—Flanders (12Z2218N).
Acknowledgments
The authors would like to express their gratitude towards Bert Depuydt, Hans Heene, Michaël Lottin, Wim Rogiers, Lambert Peka Tchokam, Erwin Turtelboom, Tom Verspeelt and Brecht Vervust for their technical expertise and help in designing and building the lab-scale liquid-solid (LS)² reactor. Lanxess nv. is acknowledged for providing the Lewatit K2629 catalyst.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- van de Steene, E.; de Clercq, J.; Thybaut, J.W. Ion-Exchange Resin Catalyzed Transesterification of Ethyl Acetate with Methanol: Gel Versus Macroporous Resins. Chem. Eng. J. 2014, 242, 170–179. [Google Scholar] [CrossRef]
- Hora, L.; Kelbichová, V.; Kikhtyanin, O.; Bortnovskiy, O.; Kubička, D. Aldol Condensation of Furfural and Acetone over Mg Al Layered Double Hydroxides and Mixed Oxides. Catal. Today 2014, 223, 138–147. [Google Scholar] [CrossRef]
- Ellebracht, N.C.; Jones, C.W. Amine Functionalization of Cellulose Nanocrystals for Acid-Base Organocatalysis: Surface Chemistry, Cross-Linking, and Solvent Effects. Cellulose 2018, 25, 6495–6512. [Google Scholar] [CrossRef]
- Clerick, S.; de Canck, E.; Hendrickx, K.; Van Speybroeck, V.; van Der Voort, P. Heterogeneous Ru (Iii) Oxidation Catalysts Via ‘Click’ bidentate Ligands on A Periodic Mesoporous Organosilica Support. Green Chem. 2016, 18, 6035–6045. [Google Scholar] [CrossRef]
- De Vylder, A.; Lauwaert, J.; Esquivel, D.; Poelman, D.; De Clercq, J.; Van Der Voort, P.; Thybaut, J.W. The Role of Water in the Reusability of Aminated Silica Catalysts for Aldol Reactions. J. Catal. 2018, 361, 51–61. [Google Scholar] [CrossRef]
- Hammond, C. Intensification Studies of Heterogeneous Catalysts: Probing and Overcoming Catalyst Deactivation During Liquid Phase Operation. Green Chem. 2017, 19, 2711–2728. [Google Scholar] [CrossRef]
- Marin, G.B.; Yablonsky, G.S.; Constales, D. Kinetics of Chemical Reactions: Decoding Complexity; Wiley-Vch: Weinheim, Germany, 2019. [Google Scholar]
- Li, X.; Wang, S.; Wang, K.; Jia, X.; Hu, Z. Polymer Ionic Liquid Network: A Highly Effective Reusable Catalyst for One-Pot Synthesis of Heterocyclic Compounds. RSC Adv. 2018, 8, 42292–42299. [Google Scholar] [CrossRef]
- Li, S.; Chen, F.; Li, N.; Wang, W.; Sheng, X.; Wang, A.; Cong, Y.; Wang, X.; Zhang, T. Synthesis of Renewable Triketones, Diketones, and Jet-Fuel Range Cycloalkanes with 5-Hydroxymethylfurfural and Ketones. Chemsuschem 2017, 10, 711–719. [Google Scholar] [CrossRef]
- Du, J.; Tao, M.; Zhang, W. Fiber-Supported Acid-Base Bifunctional Catalysts for Efficient Nucleophilic Addition in Water. ACS Sustain. Chem. Eng. 2016, 4, 4296–4304. [Google Scholar] [CrossRef]
- Wach, A.; Drozdek, M.; Dudek, B.; Łątka, P.; Kuśtrowski, P. Control of Availability of Poly (Vinylamine)-Derived Basic Sites, Catalytically Active in Knoevenagel Condensation, by Deposition on Various Mesoporous Silicas. Microporous Mesoporous Mater. 2016, 226, 433–443. [Google Scholar] [CrossRef]
- Touchy, A.S.; Kon, K.; Onodera, W.; Shimizu, K.I. Unprecedented Reductive Esterification of Carboxylic Acids Under Hydrogen by Reusable Heterogeneous Platinum Catalysts. Adv. Synth. Catal. 2015, 357, 1499–1506. [Google Scholar] [CrossRef]
- Pamuk, H.; Aday, B.; Şen, F.; Kaya, M. Pt Nps@ Go As A Highly Efficient And Reusable Catalyst for One-Pot Synthesis of Acridinedione Derivatives. RSC Adv. 2015, 5, 49295–49300. [Google Scholar] [CrossRef]
- Narkhede, N.; Patel, A. Efficient Synthesis of Biodiesel over A Recyclable Catalyst Comprising A Monolacunary Silicotungstate and Zeolite Hβ. RSC Adv. 2014, 4, 64379–64387. [Google Scholar] [CrossRef]
- Srivastava, V. Recyclable Hydrotalcite Clay Catalysed Baylis—Hillman Reaction. J. Chem. Sci. 2013, 125, 1207–1212. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Leung, D.Y.; Toy, P.H. Rasta Resin-Tbd as A Reusable Catalyst for Transesterification Reactions. Synlett 2013, 24, 1870–1874. [Google Scholar]
- Malyaadri, M.; Jagadeeswaraiah, K.; Prasad, P.S.; Lingaiah, N. Synthesis of Glycerol Carbonate By Transesterification of Glycerol with Dimethyl Carbonate Over Mg/Al/Zr Catalysts. Appl. Catal. A Gen. 2011, 401, 153–157. [Google Scholar] [CrossRef]
- Shanmuganathan, S.; Greiner, L.; De María, P.D. Silica-Immobilized Piperazine: A Sustainable Organocatalyst for Aldol and Knoevenagel Reactions. Tetrahedron Lett. 2010, 51, 6670–6672. [Google Scholar] [CrossRef]
- Li, H.; Pérez-Trujillo, M.; Cattoën, X.; Pleixats, R. Recyclable Mesoporous Organosilica Nanoparticles Derived from Proline-Valinol Amides for Asymmetric Organocatalysis. ACS Sustain. Chem. Eng. 2019, 7, 14815–14828. [Google Scholar] [CrossRef]
- Jones, C.W. On the Stability and Recyclability of Supported Metal-Ligand Complex Catalysts: Myths, Misconceptions and Critical Research Needs. Top. Catal. 2010, 53, 942–952. [Google Scholar] [CrossRef]
- SchüTh, F.; Ward, M.D.; Buriak, J.M. Common Pitfalls of Catalysis Manuscripts Submitted to Chemistry of Materials. Chem. Mater. 2018, 30, 3599–3600. [Google Scholar] [CrossRef]
- Scott, S.L. A Matter of Life (Time) and Death. ACS Catal. 2018, 8, 8597–8599. [Google Scholar] [CrossRef]
- Perego, C.; Peratello, S. Experimental Methods In Catalytic Kinetics. Catal. Today 1999, 52, 133–145. [Google Scholar] [CrossRef]
- Rajkhowa, T.; Marin, G.B.; Thybaut, J.W. Quantifying The Dominant Factors in Cu Catalyst Deactivation During Glycerol Hydrogenolysis. J. Ind. And Chem. 2017, 54, 270–277. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering. Ind. Eng. Chem. Res. 1999, 38, 4140–4143. [Google Scholar] [CrossRef]
- Forzatti, P.; Borghesi, M.; Pasquon, I.; Tronconi, E. Experimental Study on The Separability of Reaction-Deactivation Kinetics: Thermal Desorption of Alcohols from Fresh and Na-Poisoned Γ-Al2o3. AIChE J. 1986, 32, 87–95. [Google Scholar] [CrossRef]
- Janssens, T.V. A New Approach to the Modeling of Deactivation in the Conversion of Methanol on Zeolite Catalysts. J. Catal. 2009, 264, 130–137. [Google Scholar] [CrossRef]
- Govender, A.; Mahomed, A.; Friedrich, H. Water: Friend or Foe in Catalytic Hydrogenation? A Case Study Using Copper Catalysts. Catalysts 2018, 8, 474. [Google Scholar] [CrossRef]
- Ayats, C.; Henseler, A.H.; Pericàs, M.A. A Solid-Supported Organocatalyst for Continuous-Flow Enantioselective Aldol Reactions. ChemSusChem 2012, 5, 320–325. [Google Scholar] [CrossRef]
- Burguete, M.I.; Erythropel, H.; Garcia-Verdugo, E.; Luis, S.V.; Sans, V. Base Supported Ionic Liquid-Like Phases as Catalysts for The Batch and Continuous-Flow Henry Reaction. Green Chem. 2008, 10, 401–407. [Google Scholar] [CrossRef]
- Bortolini, O.; Caciolli, L.; Cavazzini, A.; Costa, V.; Greco, R.; Massi, A.; Pasti, L. Silica-Supported 5-(Pyrrolidin-2-Yl) Tetrazole: Development of Organocatalytic Processes from Batch to Continuous-Flow Conditions. Green Chem. 2012, 14, 992–1000. [Google Scholar] [CrossRef]
- Forni, L. Mass And Heat Transfer in Catalytic Reactions. Catal. Today 1999, 52, 147–152. [Google Scholar] [CrossRef]
- Dautzenberg, F.M. Ten Guidelines for Catalyst Testing. ACS Symp. Ser. 1989, 411, 99–119. [Google Scholar]
- Berger, R.J.; Stitt, E.H.; Marin, G.B.; Kapteijn, F.; Moulijn, J.A. Eurokin. Chemical Reaction Kinetics in Practice. Cattech 2001, 5, 36–60. [Google Scholar] [CrossRef]
- Froment, G. Fixed Bed Catalytic Reactors—Current Design Status. Ind. Eng. Chem. 1967, 59, 18–27. [Google Scholar] [CrossRef]
- Mears, D.E. The Role of Axial Dispersion in Trickle-Flow Laboratory Reactors. Chem. Eng. Sci. 1971, 26, 1361–1366. [Google Scholar] [CrossRef]
- Ancheyta, J. Modeling and Simulation of Catalytic Reactors for Petroleum Refining; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Ergun, S. Fluid Flow Through Packed Columns. Chem. Eng. Prog. 1952, 48, 89–94. [Google Scholar]
- Mears, D.E. Diagnostic Criteria for Heat Transport Limitations in Fixed Bed Reactors. J. Catal. 1971, 20, 127–131. [Google Scholar] [CrossRef]
- Van Den Bleek, C.; Van Der Wiele, K.; Van Den Berg, P. The Effect of Dilution on the Degree of Conversion in Fixed Bed Catalytic Reactors. Chem. Eng. Sci. 1969, 24, 681–694. [Google Scholar] [CrossRef]
- Carberry, J.J. Chemical and Catalytic Reaction Engineering; Courier Corporation: Chelmsford, MA, USA, 2001. [Google Scholar]
- Wilke, C.; Chang, P. Correlation of Diffusion Coefficients In Dilute Solutions. AIChE J. 1955, 1, 264–270. [Google Scholar] [CrossRef]
- Weisz, P.; Prater, C. Interpretation of Measurements In Experimental Catalysis. In Advances in Catalysis; Elsevier: Amsterdam, The Netherlands, 1954; Volume 6, pp. 143–196. [Google Scholar]
- Madon, R.J.; Boudart, M. Experimental Criterion for the Absence of Artifacts in the Measurement of Rates of Heterogeneous Catalytic Reactions. Ind. Eng. Chem. Fundam. 1982, 21, 438–447. [Google Scholar] [CrossRef]
- Gupta, A.S.; Thodos, G. Mass And Heat Transfer in the Flow of Fluids Through Fixed and Fluidized Beds of Spherical Particles. AIChE J. 1962, 8, 608–610. [Google Scholar] [CrossRef]
- Barrett, C.; Chheda, J.; Huber, G.; Dumesic, J. Single-Reactor Process for Sequential Aldol-Condensation and Hydrogenation of Biomass-Derived Compounds in Water. Appl. Catal. B Environ. 2006, 66, 111–118. [Google Scholar] [CrossRef]
- Dossin, T.F.; Reyniers, M.-F.; Berger, R.J.; Marin, G.B. Simulation of Heterogeneously Mgo-Catalyzed Transesterification for Fine-Chemical and Biodiesel Industrial Production. Appl. Catal. B Environ. 2006, 67, 136–148. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q. Solid Acid Catalysis Using Ion-Exchange Resins. Appl. Catal. A Gen. 2001, 221, 45–62. [Google Scholar] [CrossRef]
- Ali, S.H.; Al-Rashed, O.; Azeez, F.A.; Merchant, S.Q. Potential Biofuel Additive from Renewable Sources—Kinetic Study of Formation of Butyl Acetate by Heterogeneously Catalyzed Transesterification of Ethyl Acetate with Butanol. Bioresour. Technol. 2011, 102, 10094–10103. [Google Scholar] [CrossRef] [PubMed]
- Zielinska-Nadolska, I.; Warmuzinski, K.; Richter, J. Zeolite and other Heterogeneous Catalysts for the Transesterification Reaction of Dimethyl Carbonate with Ethanol. Catal. Today 2006, 114, 226–230. [Google Scholar] [CrossRef]
- Tesser, R.; Casale, L.; Verde, D.; Di Serio, M.; Santacesaria, E. Kinetics and Modeling of Fatty Acids Esterification on Acid Exchange Resins. Chem. Eng. J. 2010, 157, 539–550. [Google Scholar] [CrossRef]
- Bhandari, V.; Sorokhaibam, L.; Ranade, V. Ion Exchange Resin Catalyzed Reactions—An Overview. In Industrial Catalytic Processes for Fine and Specialty Chemicals; Elsevier: Amsterdam, The Netherlands, 2016; pp. 393–426. [Google Scholar]
- Chakrabarti, A.; Sharma, M. Cationic Ion Exchange Resins As Catalyst. React. Polym. 1993, 20, 1–45. [Google Scholar] [CrossRef]
- Van De Steene, E.; De Clercq, J.; Thybaut, J.W. Adsorption and Reaction in the Transesterification of Ethyl Acetate with Methanol on Lewatit K1221. J. Mol. Catal. A Chem. 2012, 359, 57–68. [Google Scholar] [CrossRef]
- Saha, B.; Streat, M. Transesterification of Cyclohexyl Acrylate with N-Butanol and 2-Ethylhexanol: Acid-Treated Clay, Ion Exchange Resins and Tetrabutyl Titanate as Catalysts. React. Funct. Polym. 1999, 40, 13–27. [Google Scholar] [CrossRef]
- Alonso, D.M.; Granados, M.L.; Mariscal, R.; Douhal, A. Polarity of the Acid Chain of Esters and Transesterification Activity of Acid Catalysts. J. Catal. 2009, 262, 18–26. [Google Scholar] [CrossRef]
- Pappu, V.K.; Yanez, A.J.; Peereboom, L.; Muller, E.; Lira, C.T.; Miller, D.J. A Kinetic Model of the Amberlyst-15 Catalyzed Transesterification of Methyl Stearate with N-Butanol. Bioresour. Technol. 2011, 102, 4270–4272. [Google Scholar] [CrossRef] [PubMed]
- Bożek-Winkler, E.; Gmehling, J. Transesterification of Methyl Acetate and N-Butanol Catalyzed by Amberlyst 15. Ind. Eng. Chem. Res. 2006, 45, 6648–6654. [Google Scholar] [CrossRef]
- Jalilnejad Falizi, N.; Güngören Madenoğlu, T.; Yüksel, M.; Kabay, N. Biodiesel Production Using Gel-Type Cation Exchange Resin at Different Ionic Forms. Int. J. Energy Res. 2019, 43, 2188–2199. [Google Scholar] [CrossRef]
- Van De Steene, E. Kinetic Study of the (Trans) Esterification Catalyzed by Gel and Macroporous Resins; Ghent University, Faculty of Engineering and Architecture: Ghent, Belgium, 2014; p. 173. [Google Scholar]
- Alsalme, A.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Heteropoly Acids as Catalysts for Liquid-Phase Esterification and Transesterification. Appl. Catal. A Gen. 2008, 349, 170–176. [Google Scholar] [CrossRef]
- Lopez, D.E.; Goodwin, J.G., Jr.; Bruce, D.A.; Lotero, E. Transesterification of Triacetin with Methanol on Solid Acid and Base Catalysts. Appl. Catal. A Gen. 2005, 295, 97–105. [Google Scholar] [CrossRef]
- Russbueldt, B.M.; Hoelderich, W.F. New Sulfonic Acid Ion-Exchange Resins for the Preesterification of Different Oils and Fats with High Content of Free Fatty Acids. Appl. Catal. A Gen. 2009, 362, 47–57. [Google Scholar] [CrossRef]
- Brunelli, N.A.; Jones, C.W. Tuning Acid-Base Cooperativity to Create next Generation Silica-Supported Organocatalysts. J. Catal. 2013, 308, 60–72. [Google Scholar] [CrossRef]
- Kandel, K.; Althaus, S.M.; Peeraphatdit, C.; Kobayashi, T.; Trewyn, B.G.; Pruski, M.; Slowing, I.I. Substrate Inhibition in the Heterogeneous Catalyzed Aldol Condensation: A Mechanistic Study of Supported Organocatalysts. J. Catal. 2012, 291, 63–68. [Google Scholar] [CrossRef]
- Singappuli-Arachchige, D.; Kobayashi, T.; Wang, Z.; Burkhow, S.J.; Smith, E.A.; Pruski, M.; Slowing, I.I. Interfacial Control of Catalytic Activity in the Aldol Condensation: Combining the Effects of Hydrophobic Environments and Water. ACS Catal. 2019. [Google Scholar] [CrossRef]
- Lauwaert, J.; De Canck, E.; Esquivel, D.; Van Der Voort, P.; Thybaut, J.W.; Marin, G.B. Effects of Amine Structure and Base Strength on Acid-Base Cooperative Aldol Condensation. Catal. Today 2015, 246, 35–45. [Google Scholar] [CrossRef]
- Kandel, K.; Althaus, S.M.; Peeraphatdit, C.; Kobayashi, T.; Trewyn, B.G.; Pruski, M.; Slowing, I.I. Solvent-Induced Reversal of Activities between Two Closely Related Heterogeneous Catalysts in the Aldol Reaction. ACS Catal. 2013, 3, 265–271. [Google Scholar] [CrossRef]
- De Vylder, A.; Lauwaert, J.; Sabbe, M.K.; Reyniers, M.-F.; De Clercq, J.; Van Der Voort, P.; Thybaut, J.W. Rational Design of Nucleophilic Amine Sites Via Computational Probing of Steric and Electronic Effects. Catal. Today 2019, 334, 96–103. [Google Scholar] [CrossRef]
- Lauwaert, J.; De Canck, E.; Esquivel, D.; Thybaut, J.W.; Van Der Voort, P.; Marin, G.B. Silanol-Assisted Aldol Condensation on Aminated Silica: Understanding the Arrangement of Functional Groups. Chemcatchem 2014, 6, 255–264. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).