Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions


Abstract
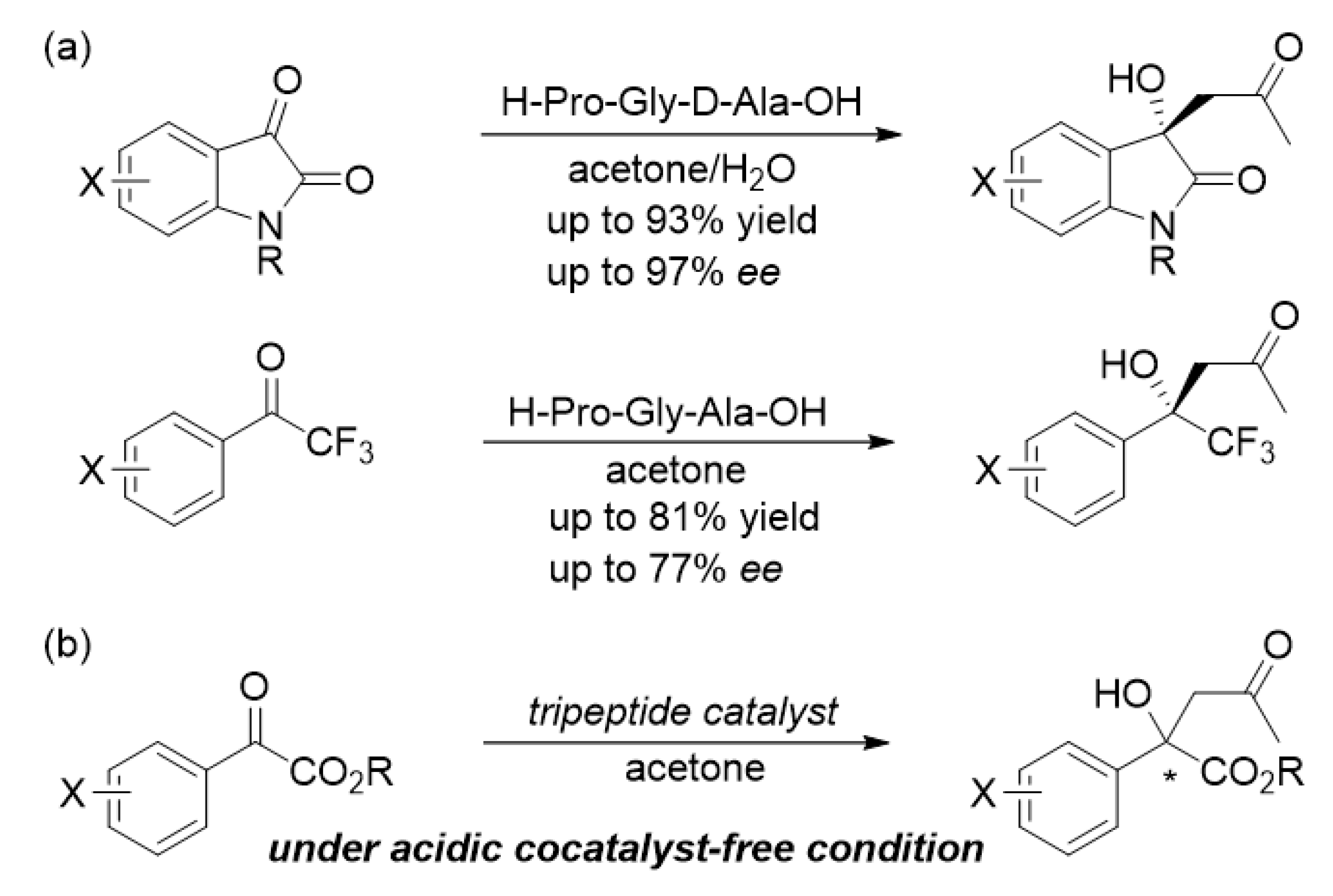
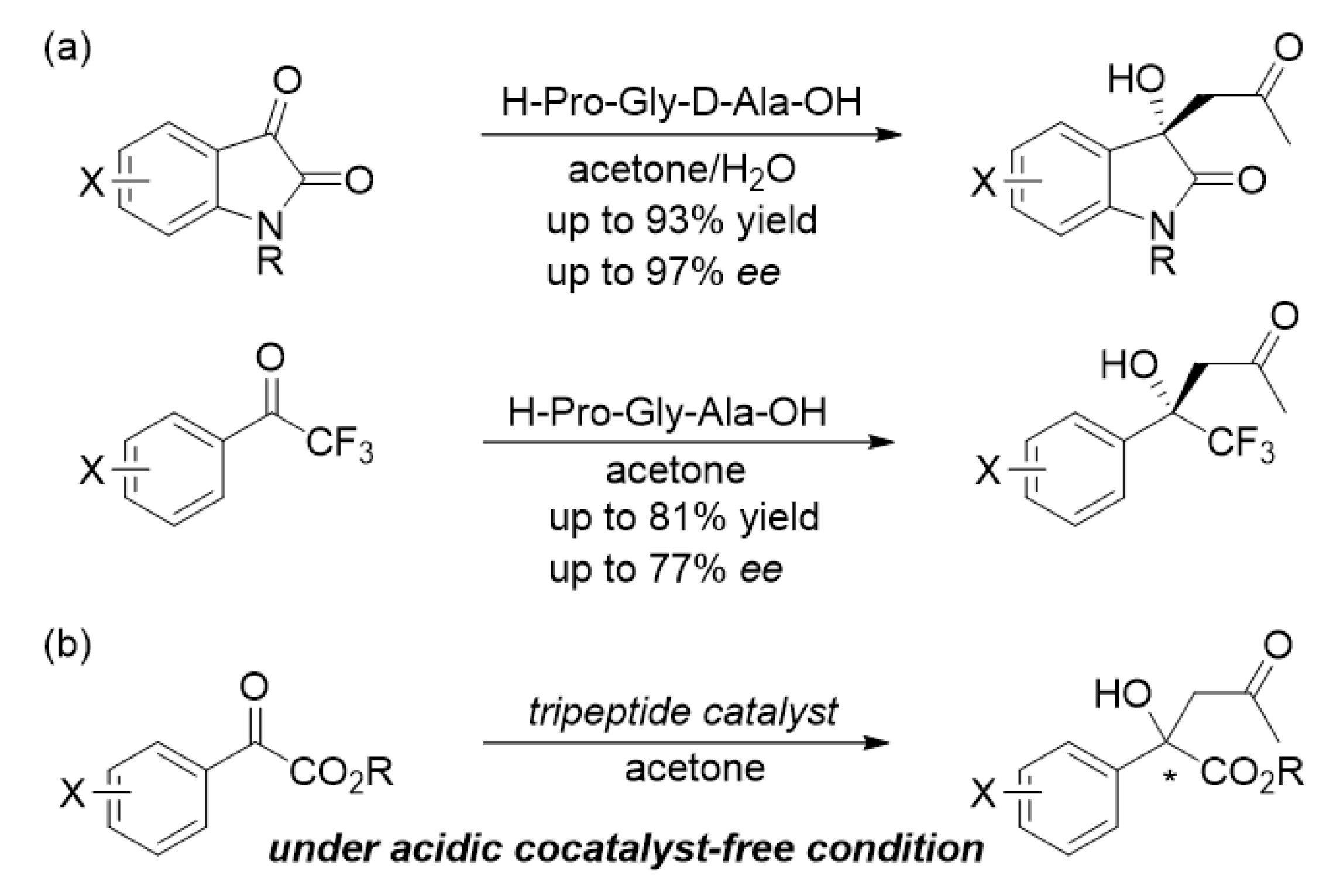
:1. Introduction
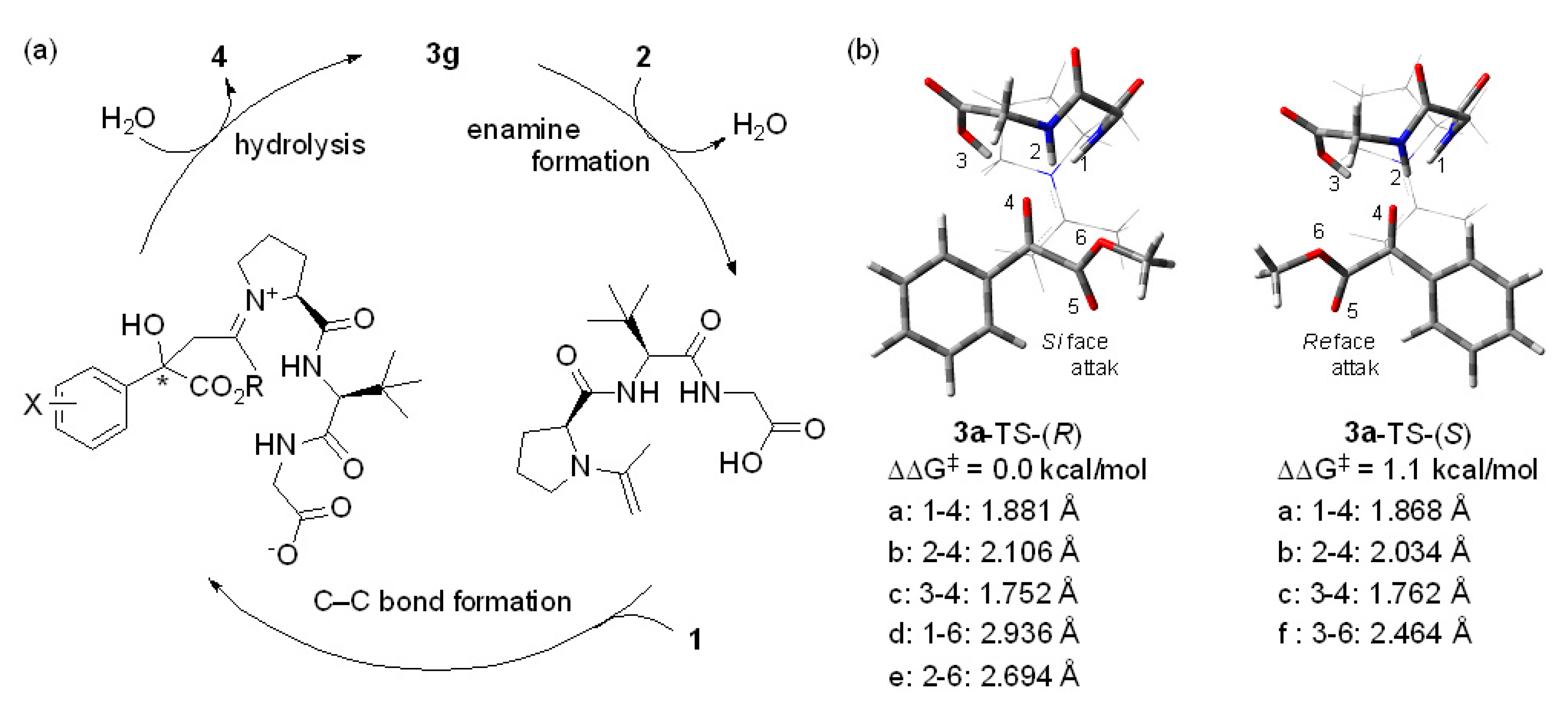
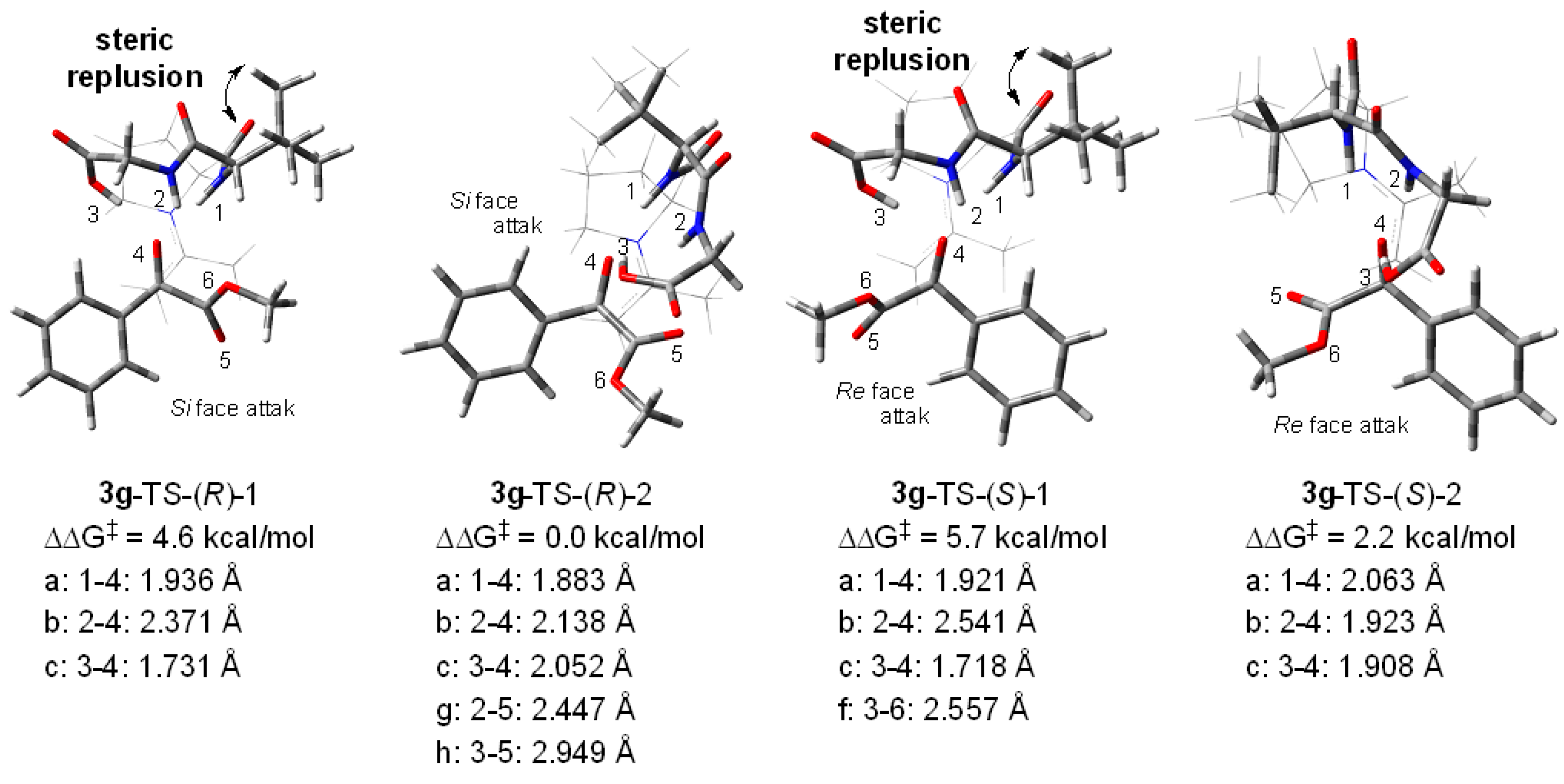
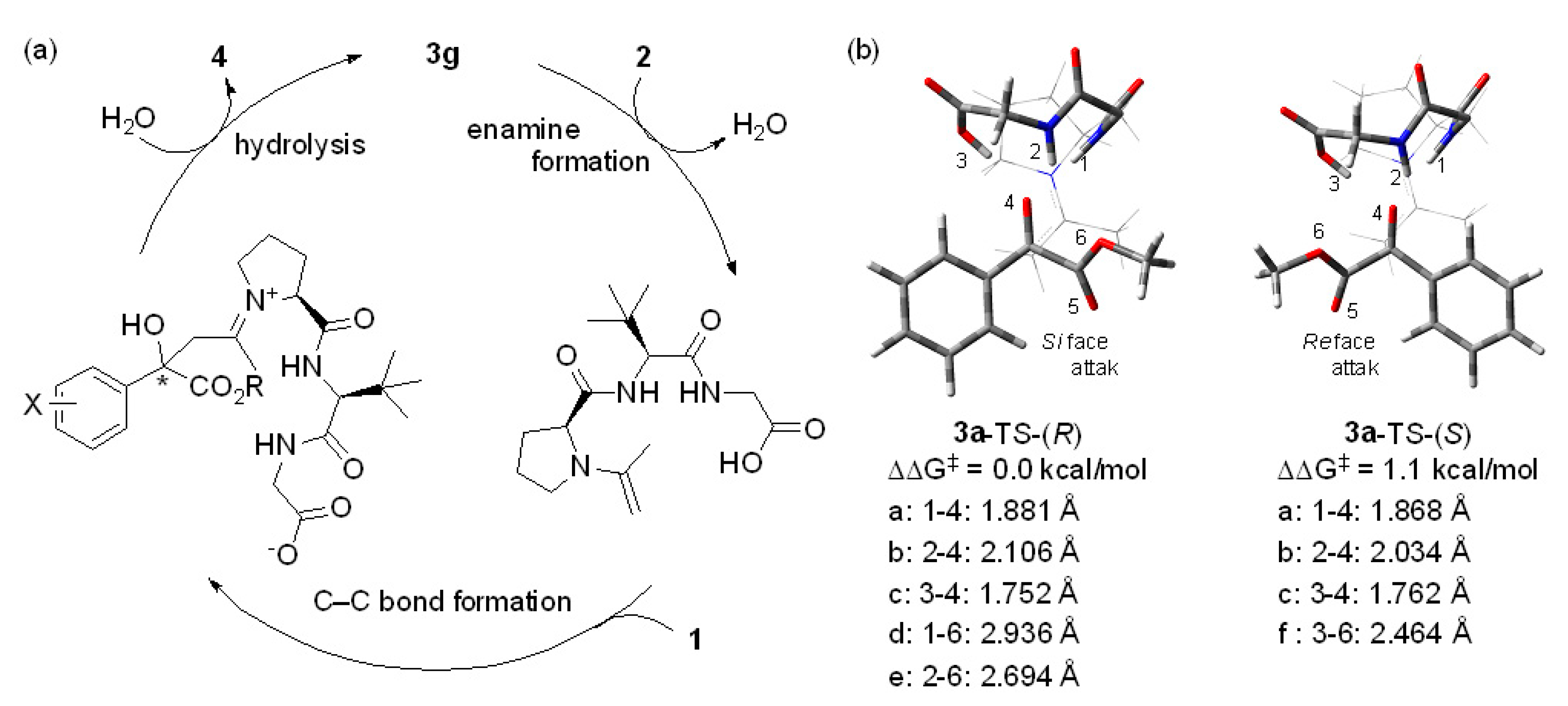
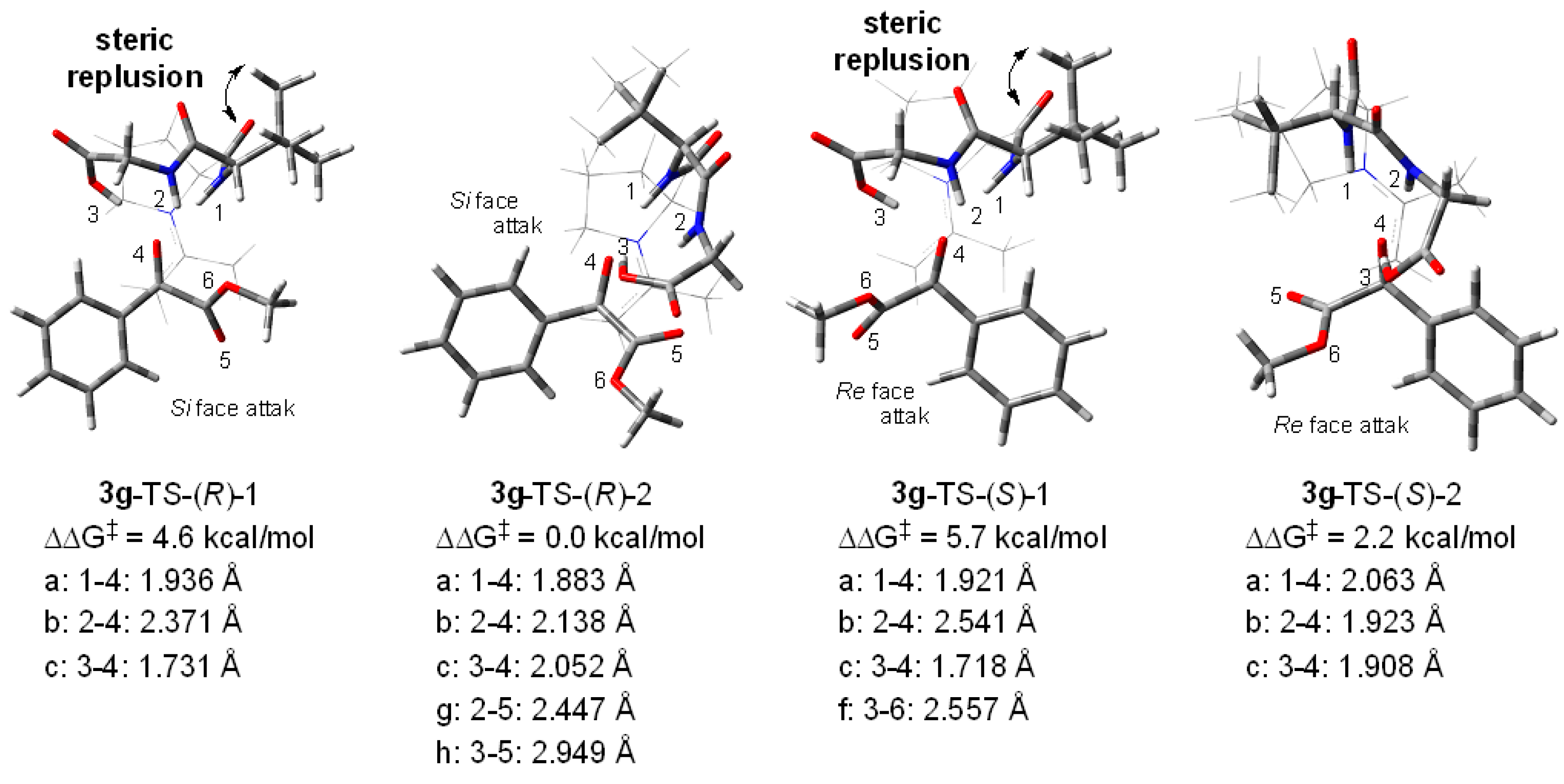
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for the Asymmetric Aldol Reaction between α-Ketoesters and Acetone
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trost, B.M.; Dirat, O.; Gunzner, J.L. Callipeltoside A: Assignment of Absolute and Relative Configuration by Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 841–843. [Google Scholar] [CrossRef]
- Maki, K.; Motoki, R.; Fujii, K.; Kanai, M.; Kobayashi, T.; Tamura, S.; Shibasaki, M. Catalyst-Controlled Asymmetric Synthesis of Fostriecin and 8-epi-Fostriecin. J. Am. Chem. Soc. 2005, 127, 17111–17117. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Porco, J.A. Total Synthesis of the Diazobenzofluorene Antibiotic (−)-Kinamycin C1. J. Am. Chem. Soc. 2006, 128, 14790–14791. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Li, H.; Nold, A.L.; Pappo, D.; Lenzen, A. Total Synthesis of Kinamycins C, F, and J. J. Am. Chem. Soc. 2007, 129, 10356–10357. [Google Scholar] [CrossRef]
- Carpenter, J.; Northrup, A.B.; Chung, D.; Wiener, J.J.M.; Kim, S.-G.; MacMillan, D.W.C. Total Synthesis and Structural Revision of Callipeltoside C. Angew. Chem. Int. Ed. 2008, 47, 3568–3572. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, O.; Kano, T.; Gao, W.-G.; Ikemoto, T.; Maruoka, K. A Practical Synthesis of (S)-2-Cyclohexyl-2-phenylglycolic Acid via Organocatalytic Asymmetric Construction of a Tetrasubstituted Carbon Center. Org. Lett. 2005, 7, 5103–5105. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-Y.; Tang, Z.; Wang, Y.-Z.; Luo, S.-W.; Cun, L.-F.; Gong, L.-Z. Asymmetric Organocatalytic Direct Aldol Reactions of Ketones with α-Keto Acids and Their Application to the Synthesis of 2-Hydroxy-γ-butyrolactones. J. Org. Chem. 2007, 72, 9905–9913. [Google Scholar] [CrossRef]
- Zheng, C.; Wu, Y.; Wang, X.; Zhao, G. Highly Enantioselective Organocatalyzed Construction of Quaternary Carbon Centers via Cross-Aldol Reaction of Ketones in Water. Adv. Synth. Catal. 2008, 350, 2690–2694. [Google Scholar] [CrossRef]
- Wang, F.; Xiong, Y.; Liu, X.; Feng, X. Asymmetric Direct Aldol Reaction of α-Keto Esters and Acetone Catalyzed by Bifunctional Organocatalysts. Adv. Synth. Catal. 2007, 349, 2665–2668. [Google Scholar] [CrossRef]
- Liu, J.; Yang, Z.; Wang, Z.; Wang, F.; Chen, X.; Liu, X.; Feng, X.; Su, Z.; Hu, C. Asymmetric Direct Aldol Reaction of Functionalized Ketones Catalyzed by Amine Organocatalysts Based on Bispidine. J. Am. Chem. Soc. 2008, 130, 5654–5655. [Google Scholar] [CrossRef]
- Liu, Q.-Z.; Wang, X.-L.; Luo, S.-W.; Zheng, B.-L.; Qiu, D.-B. Facile Preparation of Optically Pure Diamines and Their Applications in Asymmetric Aldol Reactions. Tetrahedron Lett. 2008, 49, 7434–7437. [Google Scholar] [CrossRef]
- Raj, M.; Parashari, G.S.; Singh, V.K. Highly Enantioselective Organocatalytic syn- and anti-Aldol Reactions in Aqueous Medium. Adv. Synth. Catal. 2009, 351, 1284–1288. [Google Scholar] [CrossRef]
- Jiang, Z.; Lu, Y. Direct asymmetric aldol reaction of acetone with α-ketoesters catalyzed by primary–tertiary diamine organocatalysts. Tetrahedron Lett. 2010, 51, 1884–1886. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, A.; Fang, L.; Li, W.; Zhu, C.; Cheng, Y. In Situ Formed Bifunctional Primary Amine–Imine Catalyst for Asymmetric Aldol Reactions of α-Keto Esters. Chem. Eur. J. 2011, 17, 8281–8284. [Google Scholar] [CrossRef] [PubMed]
- Viózz, S.F.; Bañón-Caballero, A.; Guillena, G.; NáJera, C.; Gómez-Bengoa, E. Enantioselective Direct Aldol Reaction of α-keto esters Catalyzed by (Sa)-binam-D-prolinamide under Quasi Solvent-Free Conditions. Org. Biomol. Chem. 2012, 10, 4029–4035. [Google Scholar]
- Lisnyak, V.G.; Kucherenko, A.S.; Valeev, E.F.; Zlotin, S.G. (1,2-Diaminoethane-1,2-diyl)bis(N-methylpyridinium) Salts as a Prospective Platform for Designing Recyclable Prolinamide-Based Organocatalysts. J. Org. Chem. 2015, 80, 9570–9577. [Google Scholar] [CrossRef] [PubMed]
- Kochetkov, S.V.; Kucherenko, A.S.; Zlotin, S.G. Asymmetric Aldol Reactions in Ketone/ketone Systems Catalyzed by Ionic Liquid-Supported C2-symmetrical Organocatalyst. Mendeleev Commun. 2015, 25, 168–170. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Shen, Z.-X.; Li, B.; Zhang, Y.-W. Proline Catalyzed Asymmetric Aldol Reaction between Methyl Ketones and α-Ketoesters. Chin. J. Chem. 2006, 24, 1196–1199. [Google Scholar] [CrossRef]
- List, B.; Lerner, A.R.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Bisai, V.; Bisai, A.; Singh, V.K. Enantioselective Organocatalytic Aldol Reaction Using Small Organic Molecules. Tetrahedron 2012, 68, 4541–4580. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V.; Dehghani, M.; Hosseintash, N. Current Applications of Organocatalysts in Asymmetric Aldol Reactions: An Update. Tetrahedron: Asymmetry 2017, 28, 587–707. [Google Scholar] [CrossRef]
- Shi, L.-X.; Sun, Q.; Ge, Z.-M.; Zhu, Y.-Q.; Cheng, T.-M.; Li, R.-T. Dipeptide-Catalyzed Direct Asymmetric Aldol Reaction. Synlett 2004, 12, 2215–2217. [Google Scholar] [CrossRef]
- Krattiger, P.; Kovàsy, R.; Revell, J.D.; Wennemers, H. Using Catalyst–Substrate Coimmobilization for the Discovery of Catalysts for Asymmetric Aldol Reactions in Split-and-Mix Libraries. QSAR Comb. Sci. 2005, 24, 1158–1163. [Google Scholar] [CrossRef]
- Krattiger, P.; Kovasy, R.; Revell, J.D.; Ivan, S.; Wennermers, H. Increased Structural Complexity Leads to Higher Activity: Peptides as Efficient and Versatile Catalysts for Asymmetric Aldol Reactions. Org. Lett. 2005, 7, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Revell, J.D.; Gantenbein, D.; Krattiger, P.; Wennemers, H. Solid-Supported and Pegylated H–Pro–Pro–Asp–NHR as Catalysts for Asymmetric Aldol Reactions. Biopolymers (Pept. Sci.). 2006, 84, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Dodda, R.; Zhao, C.-G. Organocatalytic Enantioselective Synthesis of Secondary α-Hydroxycarboxylates. Synlett 2007, 10, 1605–1609. [Google Scholar] [CrossRef]
- D’Elia, V.; Zwicknagl, H.; Reiser, O. Short α/β-Peptides as Catalysts for Intra- and Intermolecular Aldol Reactions. J. Org. Chem. 2008, 73, 3262–3265. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.-C.; Da, C.-S.; Du, Z.-X.; Guo, Q.-P.; Li, W.-P.; Yi, L.; Jia, Y.-N.; Ma, X. N-Primary-Amine-Terminal β-Turn Tetrapeptides as Organocatalysts for Highly Enantioselective Aldol Reaction. J. Org. Chem. 2009, 74, 4812–4818. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.J.; Panda, S. N-Prolinylanthranilamide Pseudopeptides as Bifunctional Organocatalysts for Asymmetric Aldol Reactions. Org. Lett. 2011, 13, 5548–5551. [Google Scholar] [CrossRef] [PubMed]
- Barrulas, P.; Benaglia, M.; Burke, A.J. Synthesis of Novel Cinchona-Amino Acid Hybrid Organocatalysts for Asymmetric Catalysis. Tetrahedron: Asymmetry 2014, 25, 923–935. [Google Scholar] [CrossRef]
- Szöllösi, G.; Csámpai, A.; Somlai, C.; Fekete, M.; Bartók, M. Unusual enantioselectivities in heterogeneous organocatalyzed reactions: Reversal of direction using proline di- versus tri-peptides in the aldol addition. J. Mol. Catal. A: Chem. 2014, 382, 86–92. [Google Scholar] [CrossRef]
- Milbeo, P.; Maurent, K.; Moulat, L.; Lebrun, A.; Didierjean, C.; Aubert, E.; Martinez, J.; Calmès, M. N-Pyrrolidine-Based α/β-peptides Incorporating ABOC, A Constrained Bicyclic β-Amino Acid, for Asymmetric Aldol Reaction Catalysis. Tetrahedron 2016, 72, 1706–1715. [Google Scholar] [CrossRef]
- Vlasserou, I.; Sfetsa, M.; Gerokonstantis, D.-T.; Kokotos, C.G.; Moutevelis-Minakakis, P. Combining Prolinamides with 2-pyrrolidinone: Novel Organocatalysts for The Asymmetric Aldol Reaction. Tetrahedron 2018, 74, 2338–2349. [Google Scholar] [CrossRef]
- Luppi, G.; Cozzi, P.G.; Monari, M.; Kaptein, B.; Broxterman, Q.B.; Tomasini, C. Dipeptide-Catalyzed Asymmetric Aldol Condensation of Acetone with (N-Alkylated) Isatins. J. Org. Chem. 2005, 70, 7418–7421. [Google Scholar] [CrossRef]
- Córdova, A.; Zou, W.; Dziedzic, P.; Ibrahem, I.; Reyes, E.; Xu, Y. Direct Asymmetric Intermolecular Aldol Reactions Catalyzed by Amino Acids and Small Peptides. Chem. Eur. J. 2006, 12, 5383–5397. [Google Scholar] [CrossRef]
- Luppi, G.; Monari, M.; Corrêa, R.J.; Violante, F.A.; Pinto, A.C.; Kaptein, B.; Broxterman, Q.B.; Garden, S.J.; Tomasini, C. The First Total Synthesis of (R)-convolutamydine A. Tetrahedron 2006, 62, 12017–12024. [Google Scholar] [CrossRef]
- Kon, K.; Kohari, Y.; Murata, M. Unnatural Tripeptide as Highly Enantioselective Organocatalyst for Asymmetric Aldol Reaction of Isatins. Tetrahedron Lett. 2019, 60, 415–418. [Google Scholar] [CrossRef]
- Kon, K.; Kohari, Y.; Murata, M. Tripeptide-Catalyzed Asymmetric Aldol Reaction of Trifluoromethylated Aromatic Ketones with Acetone. Heterocycles 2019, 99, 841–847. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02. Gaussian, Inc.: Wallingford, CT, USA, 2004. Available online: https://gaussian.com/g03citation/ (accessed on 8 June 2019).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01. Gaussian, Inc.: Wallingford, CT, USA, 2009. Available online: https://gaussian.com/g09citation/ (accessed on 8 June 2019).
- Szőri, K.; Balázsik, K.; Felfӧldi, K.; Bartók, M. Study of Enantioselective Hydrogenation of Bulky Esters of Phenylglyoxylic Acid on Pt-CD and Pt-β-ICN Chiral Catalysts: Steric Effect of Ester Groups and Inversion of Enantioselectivity. J. Catal. 2006, 241, 149–154. [Google Scholar] [CrossRef]
- Creary, X. Reaction of Oganometallic Reagents with Ethyl Trifluoroacetate and Diethyl Oxalate. Formation of Trifluoromethyl Ketones and α-Keto Esters via Stable Tetrahedral Adducts. J. Org. Chem. 1987, 52, 5026–5030. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Solvent | Yield (%) a | ee (%) b |
|---|---|---|---|---|
| 1 | H-Pro-Gly-Gly-OH 3a | neat | 61 | 31 |
| 2 | H-Pro-Gly-Ala-OH 3b | neat | 81 | 30 |
| 3 | H-Pro-Gly-D-Ala-OH 3c | neat | 70 | 26 |
| 4 | H-Pro-Ala-Gly-OH 3d | neat | 50 | 38 |
| 5 | H-Pro-D-Ala-Gly-OH 3e | neat | 49 | 34 |
| 6 | H-Pro-Val-Gly-OH 3f | neat | 49 | 50 |
| 7 | H-Pro-Tle-Gly-OH 3g | neat | 90 | 65 |
| 8 | 3g | MeOH | 40 | 30 |
| 9 | 3g | MeCN | 62 | 54 |
| 10 | 3g | CHCl3 | 81 | 68 |
| 11 | 3g | PhMe | 44 | 70 |
| 12 | 3g | THF | 82 | 79 |
| 13 | 3g | Et2O | 74 | 82 |
| 14 c | 3g | THF | 76 | 88 |
| 15 c | 3g | Et2O | 45 | 88 |
| 16 d | 3g | THF | — | — |
| 17 c,e | 3g | THF | 39 | 89 |
| 18 c,f | 3g | THF | 56 | 87 |
| 19 c,g | 3g | THF | 57 | 89 |
| 20 c,h | 3g | THF | 34 | 89 |
| 21 c,i | 3g | THF | 56 | 83 |

| Entry | R1 | R2 | 1 | 4 | Yield (%) a | ee (%) b |
|---|---|---|---|---|---|---|
| 1 | Ph | Me | 1a | 4a | 76 | 88(R) c |
| 2 | Ph | Et | 1b | 4b | 63 | 82 |
| 3 | Ph | iPr | 1c | 4c | 33 | 86 |
| 4 | 4-ClPh | Me | 1d | 4d | 84 | 75 |
| 5 d | 4-CF3Ph | Me | 1e | 4e | 95 | 50 |
| 6 | 4-MePh | Me | 1f | 4f | 25 | 75 |
| 7 | 4-MeOPh | Me | 1g | 4g | 10 | 65 |
| 8 | Me | Me | 1h | 4h | 92 | 39 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kon, K.; Takai, H.; Kohari, Y.; Murata, M. Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions. Catalysts 2019, 9, 514. https://doi.org/10.3390/catal9060514
Kon K, Takai H, Kohari Y, Murata M. Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions. Catalysts. 2019; 9(6):514. https://doi.org/10.3390/catal9060514
Chicago/Turabian StyleKon, Kazumasa, Hiromu Takai, Yoshihito Kohari, and Miki Murata. 2019. "Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions" Catalysts 9, no. 6: 514. https://doi.org/10.3390/catal9060514
APA StyleKon, K., Takai, H., Kohari, Y., & Murata, M. (2019). Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions. Catalysts, 9(6), 514. https://doi.org/10.3390/catal9060514