Recent In Situ/Operando Spectroscopy Studies of Heterogeneous Catalysis with Reducible Metal Oxides as Supports

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Intimate Metal-Support Interaction
2.1. Strong Metal-Support Interaction
2.2. Adsorbate-Mediated SMSI
2.3. Wet-Chemistry SMSI Effect
2.4. Electronic Metal-Support Interactions
3. The Active Site at the Metal-Support Interface
3.1. CO Oxidation
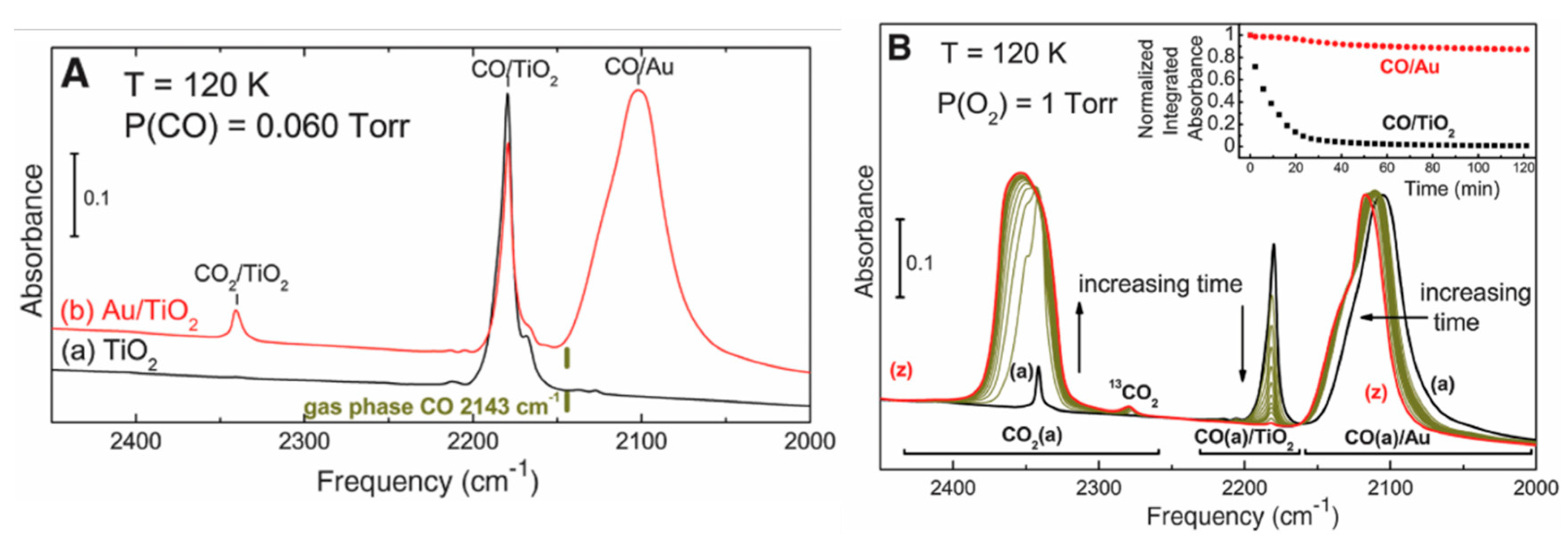
3.1.1. The Adsorption of CO on Au and TiO2 at the Metal-Support Interface
3.1.2. The Adsorption of CO on Au Nanoparticle with Bulk Reduced TiO2 Support
3.1.3. The Participation of Ce3+ Species in the Reaction at the Pt/CeO2 Interface
3.1.4. The Adsorbate-Driven Metal-Metal Oxide Interface Formation on the Surface of Bimetal Catalysts
3.2. Methane Oxidation
3.3. Water–Gas Shift Reaction (WGSR)
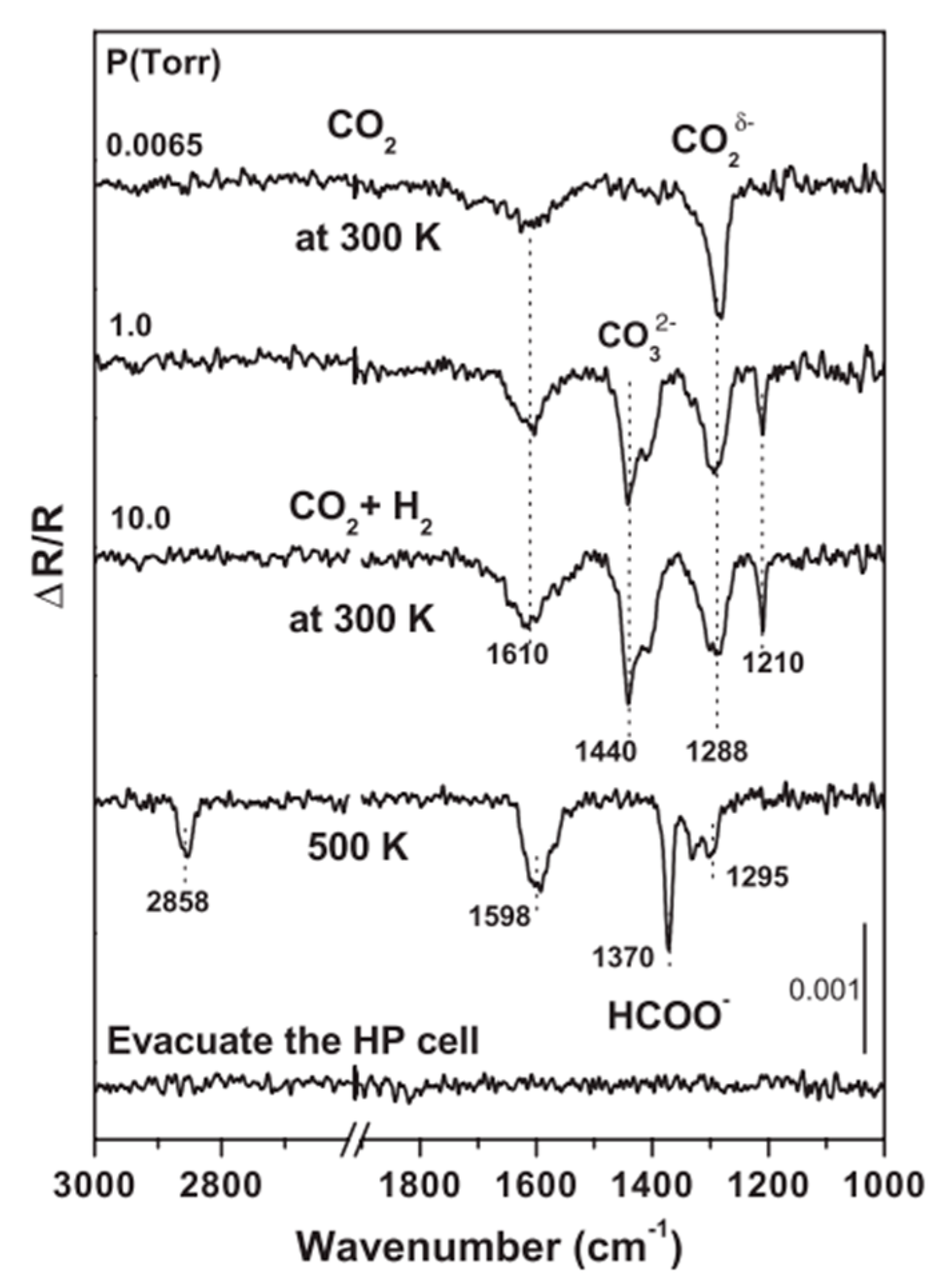
3.3.1. The EMSI at Niδ−–Ov–Ti3+ Interfacial Site
3.3.2. H2O Dissociation at Ni/TiO2 Interface
3.4. CO2 Hydrogenation
3.4.1. The CO2 Hydrogenation to CH3OH at Cu/CeO2 Interface
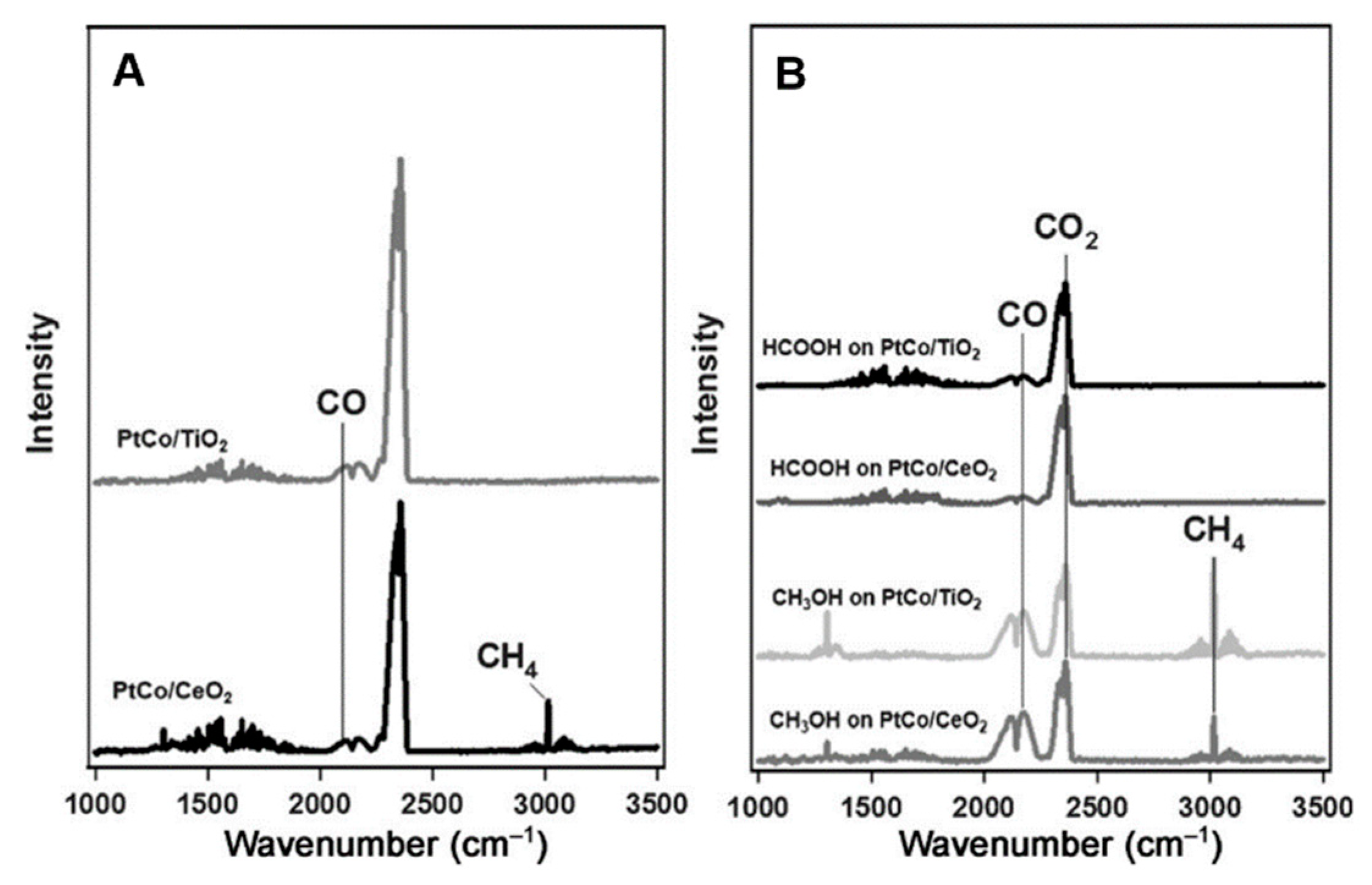
3.4.2. CO2 Hydrogenation to CO/CH4 at PtCo/CeO2 and PtCo/TiO2 Interface
3.4.3. CO2 Hydrogenation to CH3OH/CO at the Cu/TiO2 Interface
3.4.4. CO2 Hydrogenation to CH4 at the Ru/CeO2 Interface
3.5. Unsaturated Aldehyde Hydrogenation
4. Conclusions
Funding
Conflicts of Interest
References
- Ryczkowski, J. IR spectroscopy in catalysis. Catal. Today 2001, 68, 263–381. [Google Scholar] [CrossRef]
- Venezia, A.M. X-ray photoelectron spectroscopy (XPS) for catalysts characterization. Catal. Today 2003, 77, 359–370. [Google Scholar] [CrossRef]
- Stacchiola, D.J. Tuning the properties of copper-based catalysts based on molecular in situ studies of model systems. Acc. Chem. Res. 2015, 48, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Bañares, M.A. Operando methodology: Combination of in situ spectroscopy and simultaneous activity measurements under catalytic reaction conditions. Catal. Today 2005, 100, 71–77. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Ford, M.E.; Gregory, D.; Hu, R.; Keturakis, C.J.; Lwin, S.; Tang, Y.; Yang, Z.; Zhu, M.; Bañares, M.A.; et al. A decade plus of operando spectroscopy studies. Catal. Today 2017, 283, 27–53. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Shin, C.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]
- Zhu, H.; Qin, Z.; Shan, W.; Shen, W.; Wang, J. Pd/CeO2-TiO2 catalyst for CO oxidation at low temperature: A TPR study with H2 and CO as reducing agents. J. Catal. 2004, 225, 267–277. [Google Scholar] [CrossRef]
- Carrettin, S.; Concepciόn, P.; Corma, A.; Lόpez Nieto, J.M.; Puntes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Overbury, S.H. On the structure dependence of CO oxidation over CeO2 nanocrystals with well-defined surface planes. J. Catal. 2012, 285, 61–73. [Google Scholar] [CrossRef]
- Luengnaruemitchai, A.; Osuwan, S.; Gulari, E. Comparative studies of low-temperature water–gas shift reaction over Pt/CeO2, Au/CeO2, and Au/Fe2O3 catalysts. Catal. Commun. 2003, 4, 215–221. [Google Scholar] [CrossRef]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au-CeO2 catalysts for low-temperature water–gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I. Effect of morphological characteristics of TiO2-supported noble metal catalysts on their activity for the water–gas shift reaction. J. Catal. 2004, 225, 327–336. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Hrbek, J.; Evans, J.; Pérez, M. Water gas shift reaction on Cu and Au nanoparticles supported on CeO2(111) and ZnO(0001): Intrinsic activity and importance of support interactions. Angew. Chem. Int. Ed. 2007, 46, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Park, J.N.; McFarland, E.W. A highly dispersed Pd-Mg/SiO2 catalyst active for methanation of CO2. J. Catal. 2009, 266, 92–97. [Google Scholar] [CrossRef]
- Corma, A.; Garcia, H. Photocatalytic reduction of CO2 for fuel production: Possibilities and challenges. J. Catal. 2013, 308, 168–175. [Google Scholar] [CrossRef]
- Hakim, S.H.; Sener, C.; Alba-Rubio, A.C.; Gostanian, T.M.; O’Neill, B.J.; Ribeiro, F.H.; Miller, J.T.; Dumesic, J.A. Synthesis of supported bimetallic nanoparticles with controlled size and composition distributions for active site elucidation. J. Catal. 2015, 328, 75–90. [Google Scholar] [CrossRef]
- Centi, G.; Iaquaniello, G.; Perathoner, S. Can we afford to waste carbon dioxide? Carbon dioxide as a valuable source of carbon for the production of light olefins. ChemSusChem 2011, 4, 1265–1273. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.N.; Zhang, X.L.; Zhao, N.; Mao, F.K.; Wei, W.; Sun, Y.H. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Haller, G.L.; Resasco, D.E. Metal support interaction-group—VIII metals and reducible oxides. Adv. Catal. 1989, 36, 173–235. [Google Scholar] [CrossRef]
- Ioannides, T.; Verykios, X.E. Charge transfer in metal catalysts supported on doped TiO2: A theoretical approach based on metal-semiconductor contact theory. J. Catal. 1996, 161, 560–569. [Google Scholar] [CrossRef]
- Farmer, J.A.; Campbell, C.T. Ceria maintains smaller metal catalyst particles by strong metal-support bonding. Science 2010, 329, 933–936. [Google Scholar] [CrossRef]
- Saavedra, J.; Doan, H.A.; Pursell, C.J.; Grabow, L.C.; Chandler, B.D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Garten, R.L. Strong metal-support interactions. Group VIII noble metals supported on Titania. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]
- Datye, A.K.; Kalakkad, D.S.; Yao, M.H.; Smith, D.J. Comparison of metal-support interactions in Pt/TiO2 and Pt/CeO2. J. Catal. 1995, 155, 148–153. [Google Scholar] [CrossRef]
- Freund, H.J.; Pacchioni, G. Oxide ultra-thin films on metals: New materials for the design of supported metal catalysts. Chem. Soc. Rev. 2008, 37, 2224–2242. [Google Scholar] [CrossRef]
- Suzuki, T.; Souda, R. The encapsulation of Pd by the supporting TiO2(110) surface induced by strong metal-support interactions. Surf. Sci. 2000, 448, 33–39. [Google Scholar] [CrossRef]
- Bowker, M.; Stone, P.; Morrall, P.; Smith, R.; Bennett, R.; Perkins, N.; Kvon, R.; Pang, C.; Fourre, E.; Hall, M. Model catalyst studies of the strong metal−support interaction: Surface structure identified by STM on Pd nanoparticles on TiO2(110). J. Catal. 2005, 234, 172–181. [Google Scholar] [CrossRef]
- Bennett, R.A.; Pang, C.L.; Perkins, N.; Smith, R.D.; Morrall, P.; Kvon, R.I.; Bowker, M. Surface structures in the SMSI state; Pd on (1 × 2) reconstructed TiO2(110). J. Phys. Chem. B 2002, 106, 4688–4696. [Google Scholar] [CrossRef]
- Fu, Q.; Wagner, T.; Olliges, S.; Carstanjen, H.-Z. Metal-oxide interfacial reactions: Encapsulation of Pd on TiO2(110). J. Phys. Chem. B 2005, 109, 944–951. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Park, J.Y. Frontiers of surface science. Phys. Today 2007, 60, 48–53. [Google Scholar] [CrossRef]
- Somorjai, G.A.; York, R.L.; Butcher, D.; Park, J.Y. The evolution of model catalytic systems; studies of structure, bonding and dynamics from single crystal metal surfaces to nanoparticles, and from low pressure (˂10 ‒ 3 Torr) to high pressure (˃10 ‒ 3 Torr) to liquid interfaces. Phys. Chem. Chem. Phys. 2007, 9, 3500–3513. [Google Scholar] [CrossRef]
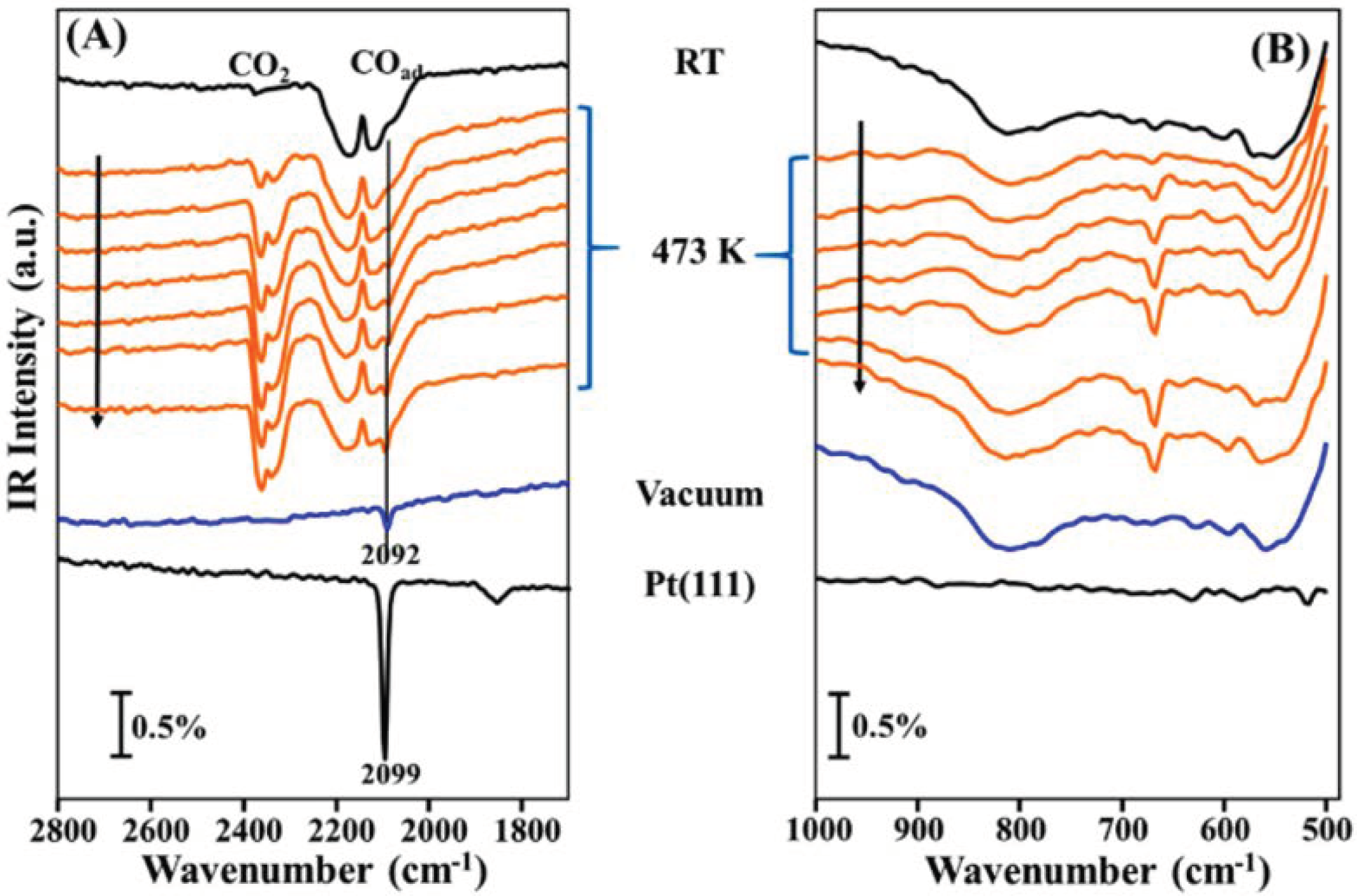
- Li, H.; Weng, X.; Tang, Z.; Zhang, H.; Ding, D.; Chen, M.; Wan, H. Evidence of the encapsulation model for strong metal-support interaction under oxidized conditions: A case study on TiOx/Pt(111) for CO oxidation by in situ wide spectral range infrared reflection adsorption spectroscopy. ACS Catal. 2018, 8, 10156–10163. [Google Scholar] [CrossRef]
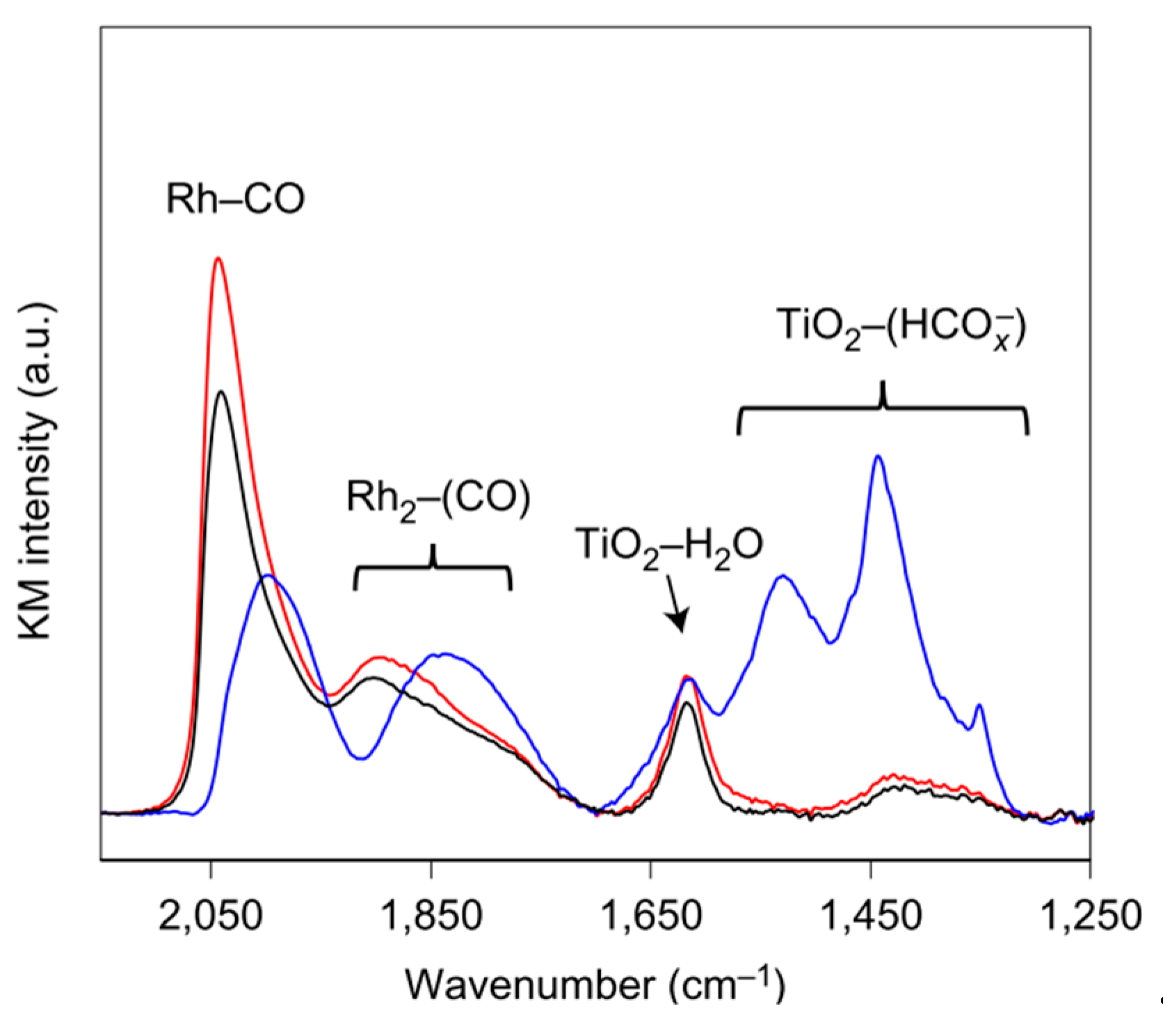
- Matsubu, J.C.; Zhang, S.; DeRita, L.; Marinkovic, N.S.; Chen, J.G.; Graham, G.W.; Pan, X.; Christopher, P. Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 2017, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.A. Complexity in the decomposition of formic acid on the TiO2(110) surface. J. Phys. Chem. B 1997, 101, 221–229. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Morikawa, Y.; Takahashi, I.; Aizawa, M.; Namai, Y.; Sasaki, T.; Iwasawa, Y. First-principles theoretical study and scanning tunneling microscopic observation of dehydration process of formic acid on a TiO2(110) surface. J. Phys. Chem. B 2004, 108, 14446–14451. [Google Scholar] [CrossRef]
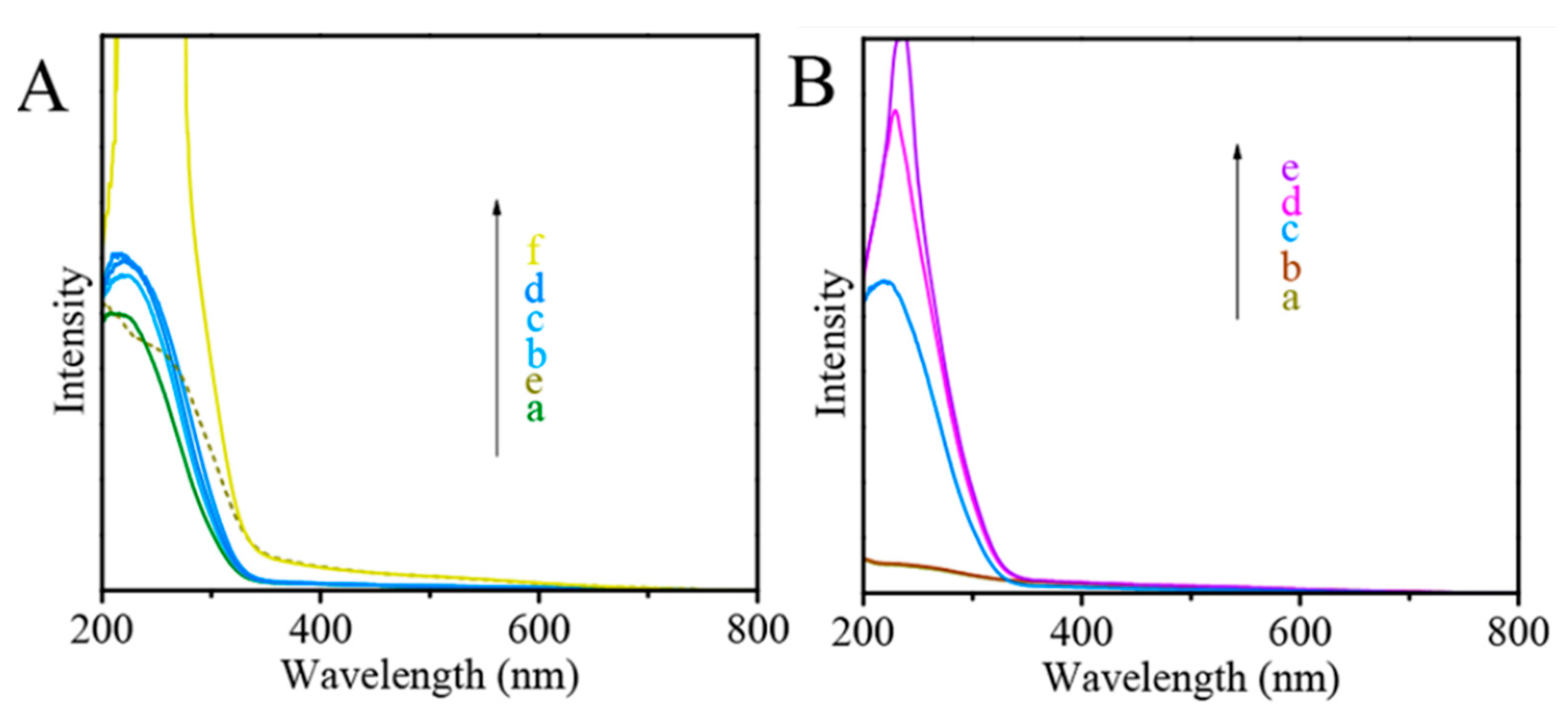
- Zhang, J.; Wang, H.; Wang, L.; Ali, S.; Wang, C.; Wang, L.; Meng, X.; Li, B.; Su, D.S.; Xiao, F. Wet-chemistry strong metal−support interactions in titania supported Au catalysts. J. Am. Chem. Soc. 2019, 141, 2975–2983. [Google Scholar] [CrossRef]
- Bruix, A.; Rodriguez, J.A.; Ramrez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. A new type of strong metal-support interaction and the production of H2 through the transformation of water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) Catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef]
- Campbell, C.T. Catalyst-support interactions electronic perturbations. Nat. Chem. 2012, 4, 597–598. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Alayoglu, S.; Musselwhite, N.; Plamthottam, S.; Melaet, G.; Lindeman, A.E.; Somorjai, G.A. Enhanced CO oxidation rates at the interface of mesoporous oxides and Pt nanoparticles. J. Am. Chem. Soc. 2013, 135, 16689–16696. [Google Scholar] [CrossRef] [PubMed]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T., Jr. Spectroscopic observation of dual catalytic sites during oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Widmann, D.; Behm, R.J. Influence of TiO2 bulk defects on CO adsorption and CO oxidation on Au/TiO2: Electronic metal−support interactions (EMSIs) in supported Au catalysts. ACS Catal. 2017, 7, 2339–2345. [Google Scholar] [CrossRef]
- Wang, Y.; Widmann, D.; Heenemann, M.; Diemant, T.; Biskupek, J.; Schlögl, R.; Behm, R.J. The role of electronic metal-support interactions and its temperature dependence: CO adsorption and CO oxidation on Au/TiO2 catalysts in the presence of TiO2 bulk defects. J. Catal. 2017, 354, 46–60. [Google Scholar] [CrossRef]
- Kopelent, R.; van Bokhoven, J.A.; Szlachetko, J.; Edebeli, J.; Paun, C.; Nachtegaal, M.; Safonova, O.V. Catalytically active and spectator Ce3+ in ceria-supported metal catalysts. Angew. Chem. Int. Ed. 2015, 54, 8728–8731. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, W.H.; Doh, W.H.; Lee, S.W.; Noh, M.C.; Gallet, J.; Bournel, F.; Kondoh, H.; Mase, K.; Jung, Y.; et al. Adsorbate-driven reactive interfacial Pt-NiO1−x nanostructure formation on the Pt3Ni(111) alloy surface. Sci. Adv. 2018, 4, eaat3151. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lim, J.; Lee, C.; Back, S.; An, K.; Shin, J.W.; Ryoo, R.; Jung, Y.; Park, J.Y. Boosting hot electron flux and catalytic activity at metal-oxide interfaces of PtCo bimetallic nanoparticles. Nat. Commun. 2018, 9, 2235. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Laffir, F.; Curtin, T.; Thompson, J.M.; Rooney, D.W. A bimetallic catalyst on a dual component support for low temperature total methane oxidation. Appl. Catal. B 2016, 187, 408–418. [Google Scholar] [CrossRef]
- Osman, A.I.; Meudal, J.; Laffir, F.; Thompson, J.; Rooney, D. Enhanced catalytic activity of Ni on η-Al2O3 and ZSM-5 on addition of ceria zirconia for the partial oxidation of methane. Appl. Catal. B 2017, 212, 68–79. [Google Scholar] [CrossRef]
- Fouladvand, S.; Skoglundh, M.; Carlsson, P. A transient in situ infrared spectroscopy study on methane oxidation over supported Pt catalysts. Catal. Sci. Technol. 2014, 4, 3463–3473. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; McLaren, M.; Laffir, F.; Rooney, D.W. Characterisation of robust combustion catalyst from aluminium foil waste. ChemistrySelect 2018, 3, 1545–1550. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Cherkasov, N.; Fernandez-Garcia, J.; Walker, D.; Walton, R.I.; Rooney, D.W.; Rebrov, E. A highly active and synergistic Pt/Mo2C/Al2O3 catalyst for water–gas shift reaction. Mol. Catal. 2018, 455, 38–47. [Google Scholar] [CrossRef]
- Mudiyanselage, K.; Senanayake, S.D.; Feria, L.; Kundu, S.; Baber, A.E.; Graciani, J.; Vidal, A.B.; Agnoli, S.; Evans, J.; Chang, R.; et al. Importance of the metal-oxide interface in catalysis: In situ studies of the water–gas shift reaction by ambient-pressure X-ray photoelectron spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; He, S.; Chen, H.; Cui, G.; Zheng, L.; Wang, B.; Wei, M. TiO2−x-modified Ni nanocatalyst with tunable metal−support interaction for water−gas shift reaction. ACS Catal. 2017, 7, 7600–7609. [Google Scholar] [CrossRef]
- Xu, M.; Yao, S.; Rao, D.; Niu, Y.; Liu, N.; Peng, M.; Zhai, P.; Man, Y.; Zheng, L.; Wang, B.; et al. Insights into interfacial synergistic catalysis over Ni@TiO2−x catalyst toward water−gas shift reaction. J. Am. Chem. Soc. 2018, 140, 11241–11252. [Google Scholar] [CrossRef] [PubMed]
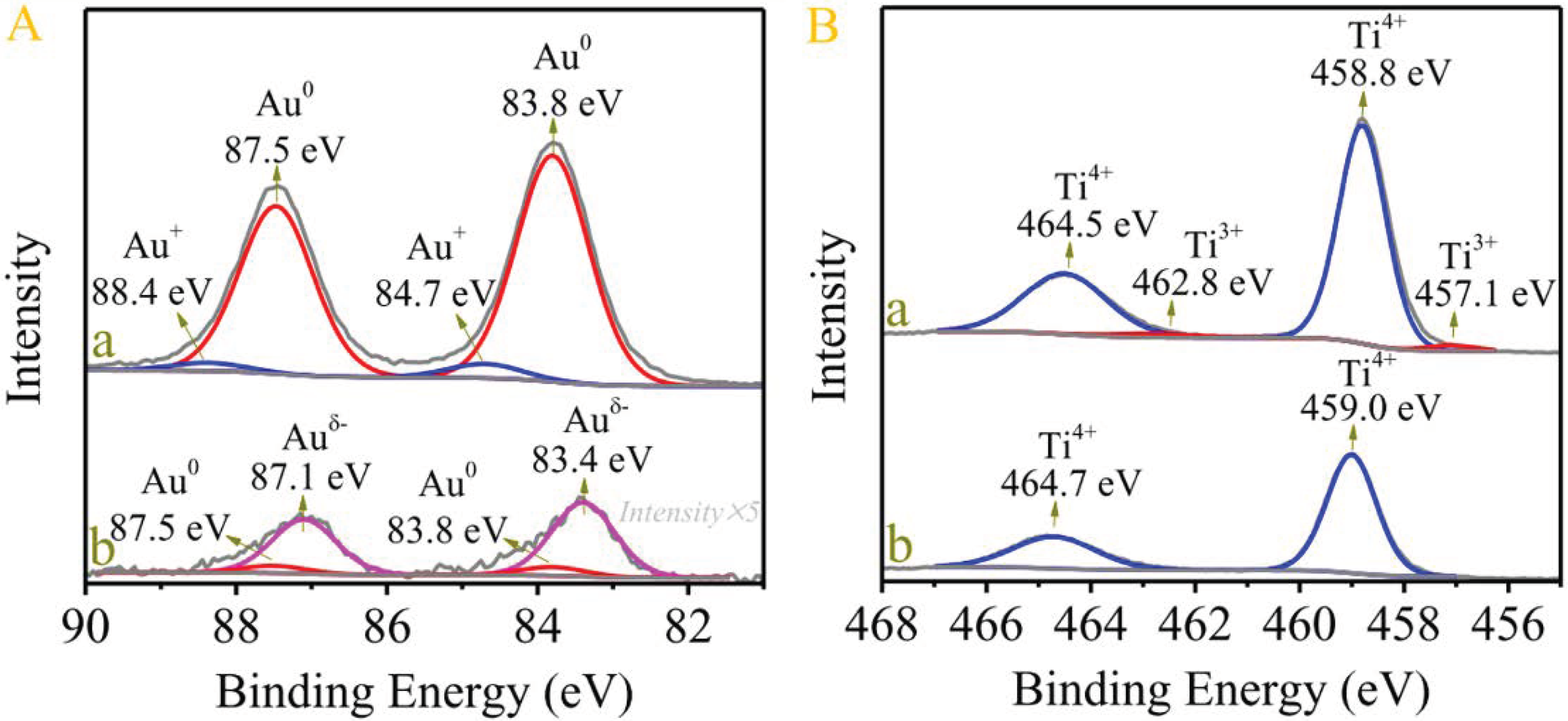
- Liu, N.; Xu, M.; Yang, Y.; Zhang, S.; Zhang, J.; Wang, W.; Zheng, L.; Hong, S.; Wei, M. Auδ−−Ov−Ti3+ interfacial site: Catalytic active center toward low-temperature water gas shift reaction. ACS Catal. 2019, 9, 2707–2717. [Google Scholar] [CrossRef]
- Wang, F.; Wei, M.; Evans, D.G.; Duan, X. CeO2-based heterogeneous catalysts toward catalytic conversion of CO2. J. Mater. Chem. A 2016, 4, 5773–5783. [Google Scholar] [CrossRef]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F.; et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kattel, S.; Senanayake, S.D.; Boscoboinik, J.A.; Nie, X.; Graciani, J.; Rodriguez, J.A.; Liu, P.; Stacchiola, D.J.; Chen, J.G. Low pressure CO2 hydrogenation to methanol over gold nanoparticles activated on a CeOx/TiO2 interface. J. Am. Chem. Soc. 2015, 137, 10104–10107. [Google Scholar] [CrossRef]
- Kattel, S.; Yu, W.; Yang, X.; Yan, B.; Huang, Y.; Wan, W.; Liu, P.; Chen, J.G. CO2 hydrogenation over oxide-supported PtCo catalysts: The role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 2016, 55, 7968–7973. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Yang, Y.; Chen, J.G.; Liu, P. Optimizing binding energies of key intermediates for CO2 hydrogenation to methanol over oxide-supported copper. J. Am. Chem. Soc. 2016, 138, 12440–12450. [Google Scholar] [CrossRef] [PubMed]
- Esch, F.; Fabris, S.; Zhou, L.; Montini, T.; Africh, C.; Fornasiero, P.; Comelli, G.; Roser, R. Electron localization determines defect formation on ceria substrates. Science 2005, 309, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, C.; Zhang, X.; Wei, M.; Evans, D.G.; Duan, X. Catalytic behavior of supported Ru nanoparticles on the {100}, {110}, and {111} facet of CeO2. J. Catal. 2015, 329, 177–186. [Google Scholar] [CrossRef]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.; Wei, M.; Evans, D.G.; Duan, X. Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.R. Surface properties probed by second-harmonic and sum-frequency generation. Nature 1989, 337, 519–525. [Google Scholar] [CrossRef]
- Baker, L.R.; Kennedy, G.; Spronsen, M.V.; Hervier, A.; Cai, X.; Chen, S.; Wang, L.; Somorjai, G.A. Furfuraldehyde hydrogenation on titanium oxide-supported platinum nanoparticles studied by sum frequency generation vibrational spectroscopy: Acid−base catalysis explains the molecular origin of strong metal−support interactions. J. Am. Chem. Soc. 2012, 134, 14208–14216. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.; Baker, L.R.; Somorjai, G.A. Selective amplification of C=O bond hydrogenation on Pt/TiO2: Catalytic reaction and sum-frequency generation vibrational spectroscopy studies of crotonaldehyde hydrogenation. Angew. Chem. Int. Ed. 2014, 53, 3405–3408. [Google Scholar] [CrossRef] [PubMed]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Jiang, J.; Wang, B. Recent In Situ/Operando Spectroscopy Studies of Heterogeneous Catalysis with Reducible Metal Oxides as Supports. Catalysts 2019, 9, 477. https://doi.org/10.3390/catal9050477
Wang F, Jiang J, Wang B. Recent In Situ/Operando Spectroscopy Studies of Heterogeneous Catalysis with Reducible Metal Oxides as Supports. Catalysts. 2019; 9(5):477. https://doi.org/10.3390/catal9050477
Chicago/Turabian StyleWang, Fei, Jianzhun Jiang, and Bin Wang. 2019. "Recent In Situ/Operando Spectroscopy Studies of Heterogeneous Catalysis with Reducible Metal Oxides as Supports" Catalysts 9, no. 5: 477. https://doi.org/10.3390/catal9050477
APA StyleWang, F., Jiang, J., & Wang, B. (2019). Recent In Situ/Operando Spectroscopy Studies of Heterogeneous Catalysis with Reducible Metal Oxides as Supports. Catalysts, 9(5), 477. https://doi.org/10.3390/catal9050477