Progress in Synthesizing Analogues of Nitrogenase Metalloclusters for Catalytic Reduction of Nitrogen to Ammonia

Abstract
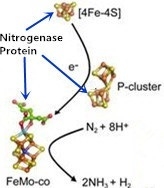
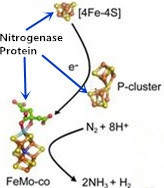
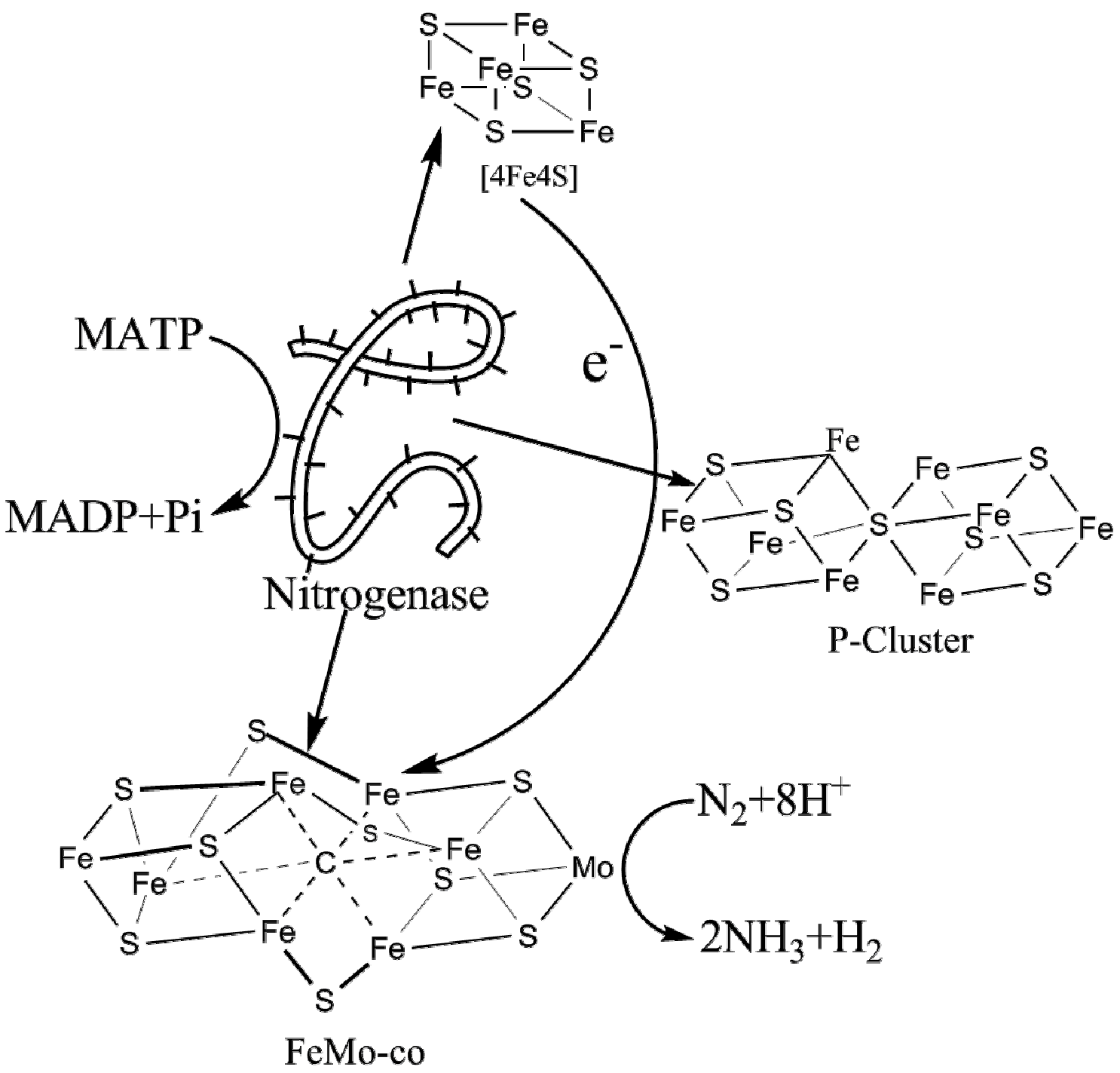
1. Introduction
2. Nitrogenase Metalloclusters
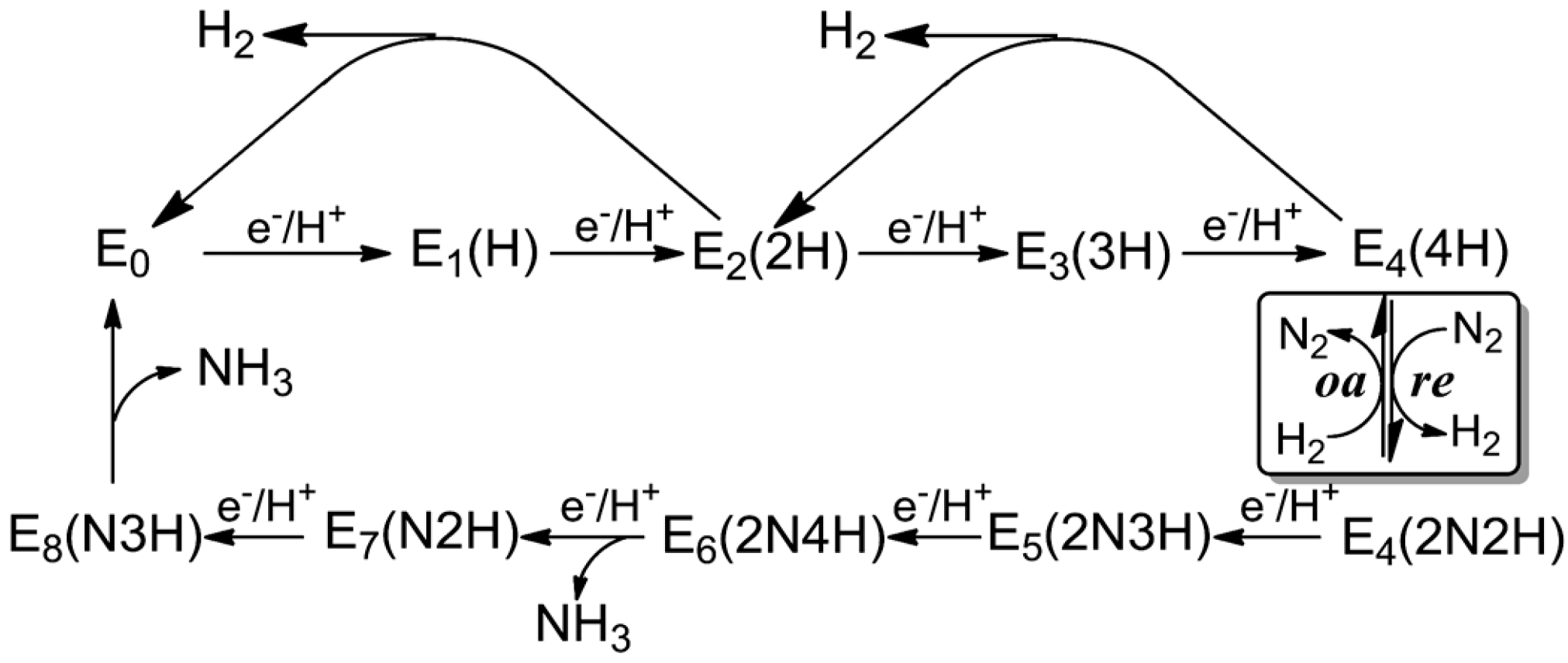
3. Reduction Mechanism of N2 to NH3
4. Synthesizing Analogues of Nitrogenase Metalloclusters
4.1. Synthesizing Analogues of [4Fe4S] Clusters
4.1.1. Structure of [4Fe4S] Clusters
4.1.2. Preparation of [4Fe4S] Clusters
4.2. Synthesizing Analogues of P-Clusters
4.2.1. Structure of P-Clusters
4.2.2. Preparation of P-Clusters
4.3. Synthesizing Analogues of FeMo-co
4.3.1. Structure of FeMo-co
4.3.2. Preparation of FeMo-co Analogues
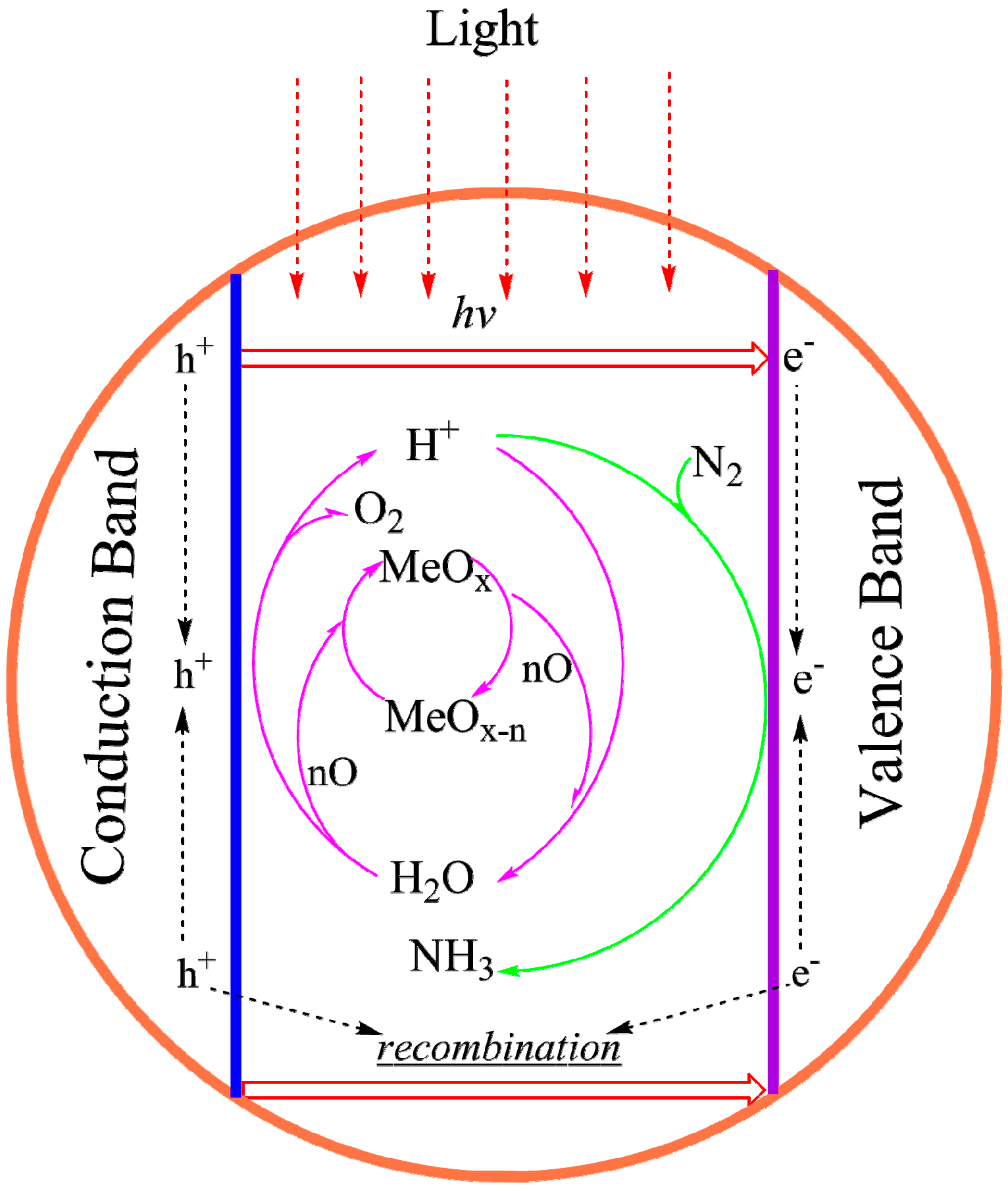
4.3.3. Synthesizing FeMo-co Analogues as Photo-Catalysts
4.3.4. Incorporating Carbon Atoms into FeMo-co Analogues
5. Summary and Outlook
Funding
Conflicts of Interest
References
- Kandemir, T.; Schuster, M.E.; Senyshyn, A.; Behrens, M.; Schlögl, R. The Haber-Bosch Process Revisited: On the Real Structure and Stability of “Ammonia Ferritin” under Working Conditions. Angew. Chem. Int. Ed. 2013, 52, 12723. [Google Scholar] [CrossRef] [PubMed]
- Shipman, M.A.; Symes, M.D. Recent progress towards the electro-synthesis of ammonia from sustainable resources. Cata. Today 2017, 286, 57–68. [Google Scholar] [CrossRef]
- Haber, F.; Leiser, R. Method and Apparatus for Testing Gases. Patent US1269599A, 18 June 1918. [Google Scholar]
- Bosch, C. The development of the chemical high pressure method during the establishment of the new ammonia industry. Nobel Lect. 1932, 1941, 197–235. [Google Scholar]
- Tamaru, K. Catalytic Ammonia Synthesis: Fundamentals and Practice; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Galloway, J.N.; Cowling, E.B. Reactive N2 and The World: 200 Years of Change, AMBIO. J. Hum. Environ. 2002, 31, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.N. N2 Cycles: Past, Present and Future. Biogeochemistry 2004, 70, 153–226. [Google Scholar] [CrossRef]
- Erisman, J.W.; Bleeker, A.; Galloway, J.; Sutton, M.S. Reduced N2 in ecology and the environment. Environ. Pollut. 2007, 150, 140–149. [Google Scholar] [CrossRef]
- Giddey, S.; Badwal, P.S.; Kulkarni, A. Review of electrochemical ammonia production technologies and materials. Inter. J. Hydro. Energy 2013, 38, 14576–14594. [Google Scholar] [CrossRef]
- Schlögl, R. Catalytic Synthesis of Ammonia-A “Never-Ending Story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008. [Google Scholar] [CrossRef]
- Spatzal, T. Reactive Nitrogen-Containing Species: Generation, Characterization and Functionalization. J. Inorg. Gen. Chem. 2015, 641, 1–135. [Google Scholar]
- Garagounis, I.; Kyriakou, V.; Skodra, A.; Vasileiou, E.; Stoukides, M. Electrochemical synthesis of ammonia in solid electrolyte cells. Front. Energy Res. 2014, 2, 1–10. [Google Scholar] [CrossRef]
- Amar, I.A.; Lan, R.; Petit, C.T.G.; Tao, S.W. Solid-state electrochemical synthesis of ammonia: A review. J. Solid State Electrochem. 2011, 15, 1845–1860. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Lukoyanov, D.; Yang, Z.Y.; Dean, D.R.; Seefeldt, L.C. Mechanism of N2 fixation by Nitrogenase: The next stage. Chem. Rev. 2014, 114, 4041–4062. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.K.; Lowe, D.J. Mechanism of Molybdenum Nitrogenase. Chem. Rev. 1996, 96, 2983–3011. [Google Scholar] [CrossRef] [PubMed]
- Modak, J.M. Haber process for ammonia synthesis. Resonance 2002, 7, 69–77. [Google Scholar] [CrossRef]
- Khoenkhoen, N.; de Bruin, B.; Reek, J.N.; Dzik, W.I. Reactivity of DiN2 Bound to Mid—and Late-Transition-Metal Centres. Eur. J. Inorg. Chem. 2015, 4, 567–598. [Google Scholar] [CrossRef]
- Georgiadis, M.M.; Komiya, H.; Chakrabarti, P.; Woo, D.; Kornuc, J.J.; Rees, D.C. Crystallographic structure of the nitrogenase ferritin protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. [Google Scholar] [CrossRef]
- Wiig, J.A.; Hu, Y.; Lee, C.C.; Ribbe, M.W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase FeMo-co. Science 2012, 337, 1672–1675. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Deanb, D.R.; Hoffman, B.M. Nitrogenase Mechanism: Electron and Proton Accumulation and N2 Reduction. In Molybdenum and Tungsten Enzymes: Biochemistry; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Simpson, F.B.; Burris, R.H. A nitrogen pressure of 50 atmospheres does not prevent evolution of hydrogen by nitrogenase. Science 1984, 224, 1095–1097. [Google Scholar] [CrossRef]
- Bjornsson, R.; Delgado-Jaime, M.U.; Lima, F.A.; Sippel, D.; Schlesier, J.; Weyhermüller, T.; Einsle, O.; Neese, F.; DeBeer, S. Molybdenum L-Edge XAS Spectra of MoFe Nitrogenase. Z. Anorg. Allg. Chem. 2015, 641, 65–71. [Google Scholar] [CrossRef]
- Grossman, J.G.; Hasnain, S.S.; Yousafzai, F.K.; Smith, B.E.; Eady, R.R. Comparing crystallographic and solution structures of nitrogenase complexes. Acta. Crystallogr. 1999, 55, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.J.; Peters, J.W.; Lanzilotta, W.N.; Ryle, M.J.; Seefeldt, L.C. MgATP-bound and nucleotide-free structures of a nitrogenase protein complex between the Leu127-Fe-protein and the MoFe-protein. Biogeochemistry 2001, 40, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Danyal, K.; Seefeldt, L.C. Nitrogen Fixation: Mechanism of Mo-Dependent Nitrogenase; Springer: Berlin, Germany, 2011. [Google Scholar]
- Tezcan, F.A.; Kaiser, J.T.; Mustafi, D.; Walton, M.Y.; Howard, J.B.; Rees, D.C. Nitrogenase Complexes: Multiple Docking Sites for a Nucleotide Switch Protein. Science 2005, 309, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, F.A.; Kaiser, J.T.; Howard, J.B.; Rees, D.C. Structural Evidence for Asymmetrical Nucleotide Interactions in Nitrogenase. J. Am. Chem. Soc. 2015, 137, 146–149. [Google Scholar] [CrossRef]
- Lukoyanov, D.; Khadka, N.; Dean, D.R.; Raugei, S.; Seefeldt, L.C.; Hoffman, B.M. Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride (Janus) State Involves a FeMo-cofactor-H2 Intermediate. Inorg. Chem. 2017, 56, 2233–2240. [Google Scholar] [CrossRef]
- Thorneley, R.N.; Lowe, D.J. The mechanism of Klebsiella pneumoniae nitrogenase action. J. Biochem. 1984, 224, 887–894. [Google Scholar] [CrossRef]
- Watt, G.D.; Reddy, K.R.N. Formation of an all ferrous Fe4S4 cluster in the ferritin protein component of Azotobacter vinelandii Nitrogenase. J. Inorg. Biochem. 1994, 53, 281–294. [Google Scholar] [CrossRef]
- Siegbahn, P. The mechanism for nitrogenase including all steps. Phys. Chem. Chem. Phys. 2019, 21, 15747–15759. [Google Scholar] [CrossRef]
- Kaczmarek, M.A.; Malhotra, A.; Balan, G.A.; Timmins, A.; de Visser, S.P. Nitrogen Reduction to Ammonia on a Biomimetic Mononuclear Iron Centre: Insights into the Nitrogenase Enzyme. Chem. Eur. J. 2018, 24, 5293–5302. [Google Scholar] [CrossRef]
- Benedek, Z.; Papp, M.; Oláh, J.; Szilvási, T. Identifying the Rate-Limiting Elementary Steps of Nitrogen Fixation with Single-Site Fe Model Complexes. Inorg. Chem. 2018, 57, 8499–8508. [Google Scholar] [CrossRef]
- Sickerman, N.S.; Tanifuji, K.; Hu, Y.L.; Ribbe, M.W. Synthetic Analogues of Nitrogenase Metallocofactors: Challenges and Developments. Eur. J. Chem. 2017, 23, 12425–12432. [Google Scholar] [CrossRef] [PubMed]
- Ribbe, M.W.; Hu, Y.; Hodgson, K.O. Hedman. Chem. Rev. 2014, 114, 4063–4080. [Google Scholar] [CrossRef] [PubMed]
- Bazhenova, T.; Shilov, A. Nitrogen fixation in solution coordination. Chem. Rev. 1995, 144, 69–145. [Google Scholar]
- Lanzilotta, W. Nucleotide hydrolysis and protein conformational changes in azotobacter. Biochemistry 1995, 34, 10713–10723. [Google Scholar] [CrossRef] [PubMed]
- Nyborg, A.; Johnson, J.; Gunn, A.; Watt, G. Evidence for a two-electron transfer using the all-ferrous Fe-protein during nitrogenase catalysis. J. Biol. Chem. 2000, 275, 39307–39312. [Google Scholar] [CrossRef]
- Schindelin, H.; Kisker, C.; Schlessman, J.L.; Howard, J.B.; Rees, D.C. Structure of ADP·AIF4−-stabilized nitrogenase complex and its implications for signal transduction. Nature 1997, 387, 370–376. [Google Scholar] [CrossRef]
- Herskovitz, T.; Averill, B.A.; Holm, R.H.; Ibers, J.A.; Phillips, W.D.; Weiher, J.F. Structure and Properties of a Synthetic Analogue of Bacterial Ferritin-Sulfur Proteins. Proc. Natl. Acad. Sci. USA 1972, 69, 2437–2441. [Google Scholar] [CrossRef]
- Rao, P.V.; Holm, R.H. Synthetic Analogues of the Active Sites of Ferritin-Sulfur Proteins. Chem. Rev. 2004, 104, 527–559. [Google Scholar]
- Hagen, K.S.; Watson, A.D.; Holm, R.H. Synthetic Routes to Fe2S2, Fe3S4, Fe4S4, and Fe& Clusters from the Common Precursor [Fe(SC2H5)4]2−: Structures and Properties of [Fe3S4(SR),I3− and [Fe&(SC2H5)2]4−, Examples of the Newest Types of Fe-S-SR Clusters. J. Am. Chem. Soc. 1983, 105, 3905–3913. [Google Scholar]
- Cambray, J.; Lane, R.W.; Wedd, A.G.; Johnson, R.W.; Holm, R.H. Chemical and Electrochemical Interrelationships of the 1-Fe, 2-Fe, and 4-Fe Analogues of the Active Sites of Ferritin-Sulfur Proteins. Inorg. Chem. 1977, 16, 2565–2571. [Google Scholar] [CrossRef]
- McSkimming, A.; Daniel, L.M. Suess, Selective Synthesis of Site-Differentiated Fe4S4 and Fe6S6 Clusters. Inorg. Chem. 2018, 57, 14904–14912. [Google Scholar] [CrossRef] [PubMed]
- Ohki, Y.; Tanifuji, K.; Yamada, N.; Imada, M.; Tajima, T.; Tatsumi, K. Synthetic analogues of [Fe4S4(Cys)3(His)] in hydrogenases and [Fe4S4(Cys)4] in HiPIP derived from all-ferric [Fe4S4{N(SiMe3)2}4]. Proc. Natl. Acad. Sci. USA 2011, 108, 12635–12640. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.B.; Bobrik, M.A.; Holm, R.H. Inorganic Derivatives of Ferritin Sulfide Thiolate Dimers and Tetramers: Synthesis and Properties of the Halide Series [Fe2S2X4]2− and [Fe4S4X4]2− (X = Cl, Br, I). Inorg. Chem. 1978, 17, 578. [Google Scholar] [CrossRef]
- Tyson, M.A.; Demadis, K.D.; Coucouvanis, D. Uncharged Mixed-Ligand Clusters with the [Fe4S4]+ and [Fe4S4]2+ Cores. Synthesis, Structural Characterization, and Properties of the Fe4S4X (tBu3P)3 (X = Cl, Br, I) and Fe4S4(SPh)2(tBu3P)2 Cubanes. Inorg. Chem. 1995, 34, 4519. [Google Scholar] [CrossRef]
- Zhou, H.C.; Holm, R.H. Synthesis and reactions of cubane-type ferritin-sulfurphosphine clusters, including soluble clusters of nuclearities 8 and 16. Inorg. Chem. 2003, 42, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.; Segal, B.M.; Huang, J.S.; Long, J.R.; Holm, R.H. Polycubane Clusters: Synthesis of [Fe4S4 (PR3)4]1+,0 (R = But, Cy, Pri) and [Fe4S4]0 Core Aggregation upon Loss of Phosphine. J. Am. Chem. Soc. 1996, 118, 11844. [Google Scholar] [CrossRef]
- Tanifuji, K.; Yamada, N.; Tajima, T.; Sasamori, T.; Tokitoh, N.; Matsuo, T.; Tamao, K.; Ohki, Y.; Tatsumi, K. A convenient route to synthetic analogues of the oxidized form of high-potential iron–sulfur proteins. Inorg. Chem. 2014, 53, 4000–4009. [Google Scholar] [CrossRef]
- Ohta, S.; Ohki, Y. Impact of ligands and media on the structure and properties of biological and biomimetic iron-sulfur clusters. Coord. Chem. Rev. 2017, 338, 207–225. [Google Scholar] [CrossRef]
- Cleland, W.E.; Holtman, D.A.; Sabat, M.; Ibers, J.A.; DeFotis, G.C.; Averill, B.A. Effects of Phenoxide Ligation on Iron-Sulfur Clusters: Preparation and Properties of [Fe4S4(OAr)4]2− Ions and the Structure of [(C2H5)4N]2[Fe4S4(OC6H5)4]. J. Am. Chem. Soc. 1983, 105, 6021. [Google Scholar] [CrossRef]
- Gaillard, J.; Gloux, J.; Gloux, P.; Lamotte, B.; Rius, G. Elucidation of the microscopic mechanism of ferroelectricity and of the associated phase transition from high resolution nmr of 31p in a new hydrogen-bonded ferroelectric: Ammonium phosphotellurate. Ferroelectrics 1984, 54, 6. [Google Scholar] [CrossRef]
- Gloux, J.; Gloux, P.; Lamotte, B.; Mouesca, J.M.; Rius, G. The Different [Fe4S4]3+ and [Fe4S4]+ Species Created by. Gamma. Irradiation in Single Crystals of the (Et4N)2[Fe4S4(SBenz)4] Model Compound: Their EPR Description and Their Biological Significance. J. Am. Chem. Soc. 1994, 116, 1953–1961. [Google Scholar] [CrossRef]
- O’Sullivan, T.; Millar, M.M. Synthesis and study of an analog for the [Fe4S4]3+ center of oxidized high potential iron-sulfur proteins. J. Am. Chem. Soc. 1985, 107, 4096–4097. [Google Scholar] [CrossRef]
- Adman, E.; Watenpaugh, K.D.; Jensen, L.H. NH—S hydrogen bonds in Peptococcus aerogenes ferredoxin, Clostridium pasteurianum rubredoxin and Chromatium high potential iron protein. Proc. Natl. Acad. Sci. USA 1975, 72, 4854–4858. [Google Scholar] [CrossRef] [PubMed]
- Hoveyda, H.R.; Holm, R.H. Characterization of the Self-Condensation Equilibrium of [Fe4S4(SH)4]2−: Spectroscopic Identification of a Unique Sulfido-Bridged Acyclic Tricubane Cluster. Inorg. Chem. 1997, 36, 4571. [Google Scholar] [CrossRef] [PubMed]
- Holm, R.H.; Phillips, W.D.; Averill, B.A.; Mayerle, J.J.; Herskovitz, T. Synthetic analogs of the active sites of iron-sulfur proteins. V. Proton resonance properties of the tetranuclear clusters [tetra-mu-sulfido-tetrakis(alkyl or aryl thiolato)tetraferrate]2−. J. Am. Chem. Soc. 1974, 96, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Que, L.J.; Bobrik, M.A.; Ibers, J.A.; Holm, R.H. Synthetic analogs of the active sites of iron-sulfur proteins. VII. Ligand substitution reactions of the tetranuclear clusters (Fe4S4(SR)4)2− and the structure of ((CH3)4N)2 (Fe4S4(SC6H5)4). J. Am. Chem. Soc. 1974, 96, 4168–4178. [Google Scholar] [CrossRef]
- Sharma, S.; Sivalingam, K.; Garnet, F.N.; Chan, K.L. Low-energy spectrum of iron-sulfur clusters directly from many-particle quantum mechanics. Nat. Chem. 2014, 6, 927–933. [Google Scholar] [CrossRef]
- Barclay, J.E.; Davies, S.C.; Evans, D.J.; Hughes, D.L.; Longhurst, S. Latticeeffects in the Mössbauer spectra of salts of [Fe4S4{S(CH2)nOH4]2−. Crystal structures of [PPh4]2[Fe4S4{S(CH2)nOH4] (n = 2,3 and 4). Inorg. Chim. Acta. 1999, 291, 101. [Google Scholar] [CrossRef]
- Kim, Y.; Han, J. Synthesis, Structure, and Reactivity of the [Fe4S4(SR)4]2− (R = 2-,3-, 4-Pyridinemethane) Clusters. Bul. Korean Chem. Soc. 2012, 33, 48–54. [Google Scholar] [CrossRef]
- Ueyama, N.; Yamada, Y.; Okamura, T.; Kimura, S.; Nakamura, A. Structure and Properties of [Fe4S4 {2,6-bis(acylamino) benzenethiolato-S4]2− and [Fe2S2{2,6-bis(acylamino)benzenethiolato-S4]2−: Protection of the Fe-S Bond by Double NH.S Hydrogen Bonds. Inorg. Chem. 1996, 35, 6473–6484. [Google Scholar] [CrossRef]
- Davies, S.C.; Evans, D.J.; Henderson, R.A.; Hughes, D.L.; Longhurst, S. Mononuclear, binuclear, trinuclear and tetranuclear iron complexes of the N(CH2CH2S)33− (NS3) ligand with nitrosyl co-ligands. J. Chem. Soc. Dalton Trans. 2002, 43, 2473–2482. [Google Scholar] [CrossRef]
- Lo, W.; Huang, S.; Zheng, S.L.; Holm, R.H. Cubane-type Fe4S4 Clusters with Chiral Thiolate Ligation: Formation by Ligand Substitution, Detection of Intermediates by 1H NMR, and Solid State Structures Including Spontaneous Resolution Upon Cristallization. Inorg. Chem. 2011, 50, 11082–11090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carney, M.J.; Papaefthymiou, G.C.; Spartalian, K.; Frankel, R.B.; Holm, R.H. Ground spin state variability in [Fe4S4(SR)4]3− synthetic analogs of the reduced clusters in ferredoxins and other iron-sulfur proteins: Cases of extreme sensitivity of electronic state and structure to extrinsic factors. J. Am. Chem. Soc. 1988, 110, 6084–6095. [Google Scholar] [CrossRef] [PubMed]
- Münck, E.; Kent, T.A. Structure and magnetism of iron-sulfur clusters in proteins. Hyperfine Interact. 1986, 27, 161–172. [Google Scholar] [CrossRef]
- Stephan, D.W.; Papaefthymiou, G.C.; Frankel, R.B.; Holm, R.H. Analogues of the (4Fe-4S)+ sites of reduced ferredoxins: Structural and spectroscopic properties of ((C2H5)4N)3(Fe4S4(S-P-C6H4Br)4) in crystalline and solution phases. Inorg. Chem. 1983, 22, 1550. [Google Scholar] [CrossRef]
- Lee, C.C.; Fay, A.W.; Weng, T.-C.; Krest, C.M.; Hedman, B.; Hodgson, K.O.; Hu, Y.; Ribbe, M.W. Uncoupling binding of substrate CO from turnover by vanadium nitrogenase. Proc. Natl. Acad. Sci. USA 2015, 112, 13845–13849. [Google Scholar] [CrossRef]
- Henderson, R.A. N2 Fixation at the Millenium: Advances Towards the Mechanism of Nitrogenases; Elsevier: Amsterdam, The Netherlands, 2002; pp. 223–261. [Google Scholar]
- Hu, Y.; Fay, A.W.; Lee, C.C.; Yoshizawa, J.; Ribbe, M.W. Assembly of Nitrogenase MoFe-protein. Biogeochemistry 2008, 47, 3973–3981. [Google Scholar] [CrossRef]
- Zhang, Y.; Holm, R.H. Structural conversions of molybdenum-ferritin-sulfur edge-bridged double cubanes and PN-type clusters topologically related to the nitrogenase P-cluster. Inorg. Chem. 2004, 43, 674–682. [Google Scholar] [CrossRef]
- Hlavinka, M.L.; Miyaji, T.; Staples, R.J.; Holm, R.H. Hydroxide-promoted core conversions of molybdenum-ferritin-sulfur edge-bridged double cubanes: Oxygen-ligated topological PN clusters. Inorg. Chem. 2007, 46, 9192–9200. [Google Scholar] [CrossRef][Green Version]
- Corbett, M.C. Comparison of ferritin-molybdenum cofactor-deficient nitrogenase MoFe-proteins by X-ray absorption spectroscopy: Implications for P-cluster biosynthesis. J. Biol. Chem. 2004, 279, 28276–28282. [Google Scholar] [CrossRef]
- Broach, R.B. Variable-temperature, variable-field magnetic circular dichroism spectroscopic study of the metal clusters in the nifB and nifH MoFe-proteins of nitrogenase from Azotobacter vinelandii. Biogeochemistry 2006, 45, 15039–15048. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y. Nitrogenase reactivity with P-cluster variants. Proc. Natl. Acad. Sci. USA 2005, 102, 13825–13830. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, L.C.; Hoffman, B.M.; Dean, D.R. Mechanism of Mo-Dependent Nitrogenase. Annu. Rev. Biochem. 2009, 78, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Danyal, K.; Dean, D.R.; Hoffman, B.M.; Seefeldt, L.C. Electron transfer within nitrogenase: Evidence for a deficit-spending mechanism. Biochem. 2011, 50, 9255–9263. [Google Scholar] [CrossRef]
- Musgrave, K.B.; Liu, H.I.; Ma, L.; Burgess, B.K.; Watt, G.; Hedman, B.; Hodgson, K.O. EXAFS studies on the PN and POX states of the P-clusters in nitrogenase. J. Biol. Inorg. Chem. 1988, 3, 344–352. [Google Scholar] [CrossRef]
- Chan, M.K.; Kim, J.; Rees, D.C. The Nitrogenase FeMo-coand P-cluster pair: A resolution structures. Science 1993, 260, 792–794. [Google Scholar] [CrossRef]
- Challen, P.R.; Koo, S.K.; Dunham, W.R.; Coucouvanis, D. New P2-S2—Coupled, Singly Bridged Double Cubane with the [(Fe4S4C13)2S]4− Core. J. Am. Chem. Soc. 1990, 112, 2455–2456. [Google Scholar] [CrossRef]
- Pierik, A.J.; Wassink, H.; Haaker, H.; Hagen, W.R. Redox properties and EPR spectroscopy of the P clusters of Azotobacter vinelandii MoFe-protein. Eur. J. Biochem. 1993, 212, 51–61. [Google Scholar] [CrossRef]
- Kurts, D.M.; McMillan, R.S.; Burgess, B.K.; Mortenson, L.E.; Holm, R.H. Identification of ferritin-sulfur centers in the ferritin-molybdenum proteins of nitrogenase. Proc. Natl. Acad. Sci. USA 1979, 76, 4986–4989. [Google Scholar] [CrossRef]
- Osterloh, F.; Sanakis, Y.; Staples, R.J.; Münck, E.; Holm, R.H. A Molybdenum-Ferritin-Sulfur Cluster Containing Structural Elements Relevant to the P-Cluster of Nitrogenase. Angew. Chem. Int. Ed. 1999, 38, 2066–2070. [Google Scholar] [CrossRef]
- Osterloh, F.; Achim, C.; Holm, R.H. Molybdenum-Ferritin-Sulfur Clusters of Nuclearities Eight and Sixteen, Including a Topological Analogue of the P-Cluster of Nitrogenase. Inorg. Chem. 2001, 40, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Ohki, Y.; Tatsumi, K. New Synthetic Routes to Metal-Sulfur Clusters Relevant to the Nitrogenase Metallo-Clusters, Z. Anorg. Allg. Chem. 2013, 639, 1340–1349. [Google Scholar] [CrossRef]
- Eady, R.R. Structure−Function Relationships of Alternative Nitrogenases. Chem. Rev. 1996, 96, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Rees, D.C. Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257, 1677–1682. [Google Scholar] [CrossRef]
- Spatzal, T.; Aksoyoglu, M.; Zhang, L.M.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef]
- Rittle, J.; Peters, J.C. Fe–N2/CO complexes that model a possible role for the interstitial C atom of FeMo-co(FeMoco). Proc. Natl. Acad. Sci. USA 2013, 110, 15898–15903. [Google Scholar] [CrossRef]
- Christiansen, J.; Cash, V.L.; Seefeldt, L.C.; Dean, D.R. Isolation and Characterisation of Acetylene-resistant Nitrogenase. J. Bio. Chem. 2000, 275, 11459–11464. [Google Scholar] [CrossRef]
- Djurdjevic, I.; Einsle, O.; Decamps, L. Nitrogenase Cofactor: Inspiration for Model Chemistry. Chem. Asian J. 2017, 12, 1447–1455. [Google Scholar] [CrossRef]
- Holm, R.H.; Lo, W. Structural Conversions of Synthetic and Protein-Bound Ferritin-Sulfur Clusters. Chem. Rev. 2016, 116, 13685–13713. [Google Scholar] [CrossRef]
- Shah, V.K.; Brill, W.J. Isolation of an ferritin-molybdenum cofactor from nitrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3249–3253. [Google Scholar] [CrossRef]
- Wolff, T.E.; Power, P.P.; Frankel, R.B.; Holm, R.H. Synthesis and Electronic and Redox Properties of “Double-Cubane” Cluster Complexes Containing MoFe3S4 and WFe3S4 Cores. J. Am. Chem. Soc. 1980, 102, 4694–4703. [Google Scholar] [CrossRef]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray Emission Spectroscopy Evidences a Central Carbon in the nitrogenase Ferritin-Molybdenum Cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Spatzal, T.; Perez, K.A.; Einsle, O.; Howard, J.B.; Rees, D.C. Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620. [Google Scholar] [CrossRef] [PubMed]
- Wolff, T.E.; Berg, J.M.; Hodgson, K.O.; Frankel, R.B.; Holm, R.H. Synthetic Approaches to the Molybdenum Site in Nitrogenase. Preparation and Structural Properties of the Molybdenum-Ferritin-Sulfur “Double-Cubane” Cluster Complexes [MO&& (SC2H&J3- and [Mo2Fe6S9(Sc2H5)8]3−. J. Am. Chem. Soc. 1979, 101, 4140–4150. [Google Scholar]
- Demadis, K.D.; Campana, C.F.; Coucouvanis, D. Synthesis Structural Characterization of the New Mo2Fe6S8(PR3)6(CL4-cat)2 Clusters. J. Am. Chem. Soc. 1995, 117, 7832–7833. [Google Scholar] [CrossRef]
- Zhang, Y.; Holm, R.H. Synthesis of a Molecular Mo2Fe6S9 Cluster with the Topology of the PN Cluster of Nitrogenase by Rearrangement of an Edge-Bridged Mo2Fe6S8 Double Cubane. J. Am. Chem. Soc. 2003, 125, 3910–3920. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, Y.; Yu, H.; Zhao, J.F.; Chen, Y.H.; Hou, Z.M.; Qu, J.P. DFT Studies on the Reduction of Dinitrogen to Ammonia by a Thiolate-Bridged Diferritin Complex as a Nitrogenase Mimic. Organometallics 2012, 31, 335–344. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Wang, B.M.; Luo, Y.; Yang, D.W.; Tong, P.; Zhao, J.F.; Luo, L.; Zhou, Y.H.; Chen, S.; et al. Ammonia formation by a thiolate-bridged diferritin amide complex as a nitrogenase mimic. Nat. Chem. 2013, 5, 320–326. [Google Scholar] [CrossRef]
- Dance, I. Mimicking Nitrogenase. Dalton Trans. 2010, 39, 2972–2983. [Google Scholar] [CrossRef]
- Leigh, G.J. Nitrogen Fixation at the Millennium; Leigh, G.J., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 299–322. [Google Scholar]
- Demadis, K.D.; Malinak, S.M.; Coucouvanis, D. Catalytic Reduction of Hydrazine to Ammonia with MoFe3S4−Polycarboxylate Clusters. Possible Relevance Regarding the Function of the Molybdenum-Coord—inated Homocitrate in Nitrogenase. Inorg. Chem. 1996, 35, 4038–4046. [Google Scholar] [CrossRef]
- Malinak, S.M.; Demadis, K.D.; Coucouvanis, D. Catalytic Reduction of Hydrazine to Ammonia by the VFe3S4 Cubanes. Further Evidence for the Direct Involvement of the Heterometal in the Reduction of Nitrogenase Substrates and Possible Relevance to the Vanadium Nitrogenases. J. Am. Chem. Soc. 1995, 117, 3126–3133. [Google Scholar] [CrossRef]
- McEvoy, J.P.; Brudvig, G.W. Water-splitting chemistry of photo system II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, J.; Xiao, H.; Li, J. Surface cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 2018, 140, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 1–10. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Ai, Z.H.; Jia, F.L.; Zhang, L.Z. Oxygen Vacancy—Mediated Photocatalysis of BiOCl: Reactivity, Selectivity, and Perspectives. Angew. Chem. 2018, 57, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, J. Progress of metal oxide(sulfide)-based photocatalytic materials for reducing nitrogen to ammonia. J. Chem. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Thacker, C.M.; Folkins, H.O.; Miller, E.L. Free Energies of Formation of Gaseous Hydrocarbons and Related Substances. Ind. Eng. Chem. 1941, 33, 584–590. [Google Scholar] [CrossRef]
- Brown, K.A.; Harris, D.F.; Wilker, M.B. Light-driven dinitrogen reduction catalyzed by a CdS: Nitrogenase MoFe-protein biohybrid. Science 2016, 352, 448–450. [Google Scholar] [CrossRef]
- Liu, J.; Kelley, M.S.; Wu, W.Q.; Banerjee, A.; Douvalis, A.P.; Wu, J.S.; Zhang, Y.B.; Schatz, G.C.; Kanatzidis, M.G. Nitrogenase-mimic ferritin-containing chalcogels for photochemical reduction of dinitrogen to ammonia. Proc. Natl. Acad. Sci. USA 2016, 113, 5530–5535. [Google Scholar] [CrossRef]
- Banerjee, A.; Yuhas, B.D.; Margulies, E.A.; Zhang, Y.; Shim, Y.; Wasielewski, M.R.; Kanatzidis, M.G. Photochemical nitrogen conversion to ammonia in ambient conditions with FeMoS-chalcogels. J. Am. Chem. Soc. 2015, 137, 2030–2034. [Google Scholar] [CrossRef]
- Xu, G.; Wang, Z.; Ling, R.; Zhou, J.; Chen, X.D.; Holm, R.H. Ligand metathesis as rational strategy for the synthesis of cubane-type heteroleptic ferritin–sulfur clusters relevant to the FeMo cofactor. Proc. Natl. Acad. Sci. USA 2018, 115, 5089–5092. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Chen, X.D.; Zheng, S.L.; Holm, R.H. Selenium as a structural surrogate of sulfur: Template-assisted assembly of five types of tungsten-ferritin-sulfur/selenium clusters and the structural fate of chalcogenide reactants. J. Am. Chem. Soc. 2012, 134, 6479–6490. [Google Scholar] [CrossRef] [PubMed]
- DeRosha, D.E.; Holland, P.L. Incorporating light atoms into synthetic analogues of FeMoco. Proc. Natl. Acad. Sci. USA 2018, 115, 5054–5056. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analogues | [Fe4S4(SR)4]0 | [Fe4S4(SR)4]1− | [Fe4S4(SR)4]2− | [Fe4S4(SR)4]3− | [Fe4S4(SR)4]4− |
|---|---|---|---|---|---|
| Core oxidation state | [Fe4S4]4+ | [Fe4S4]3+ | [Fe4S4]2+ | [Fe4S4]1+ | [Fe4S4]0 |
| Composition of oxidation state | 4Fe(III) | 3Fe(III) + Fe(II) | 2Fe(III) + 2Fe(II) | Fe(III) + 3Fe(III) | 4Fe (II) |
| Analogues | R | Year | Refs |
|---|---|---|---|
| [Fe4S4(SR)4]1− | Ph | 1984 | [54] |
| CH2Ph | 1994 | [55] | |
| 2,4,6-triisopropylphenyl | 1985 | [56] | |
| 2,6-di(mesityl)phenyl | 1975 | [57] | |
| [Fe4S4(SR)4]2− | H | 1997 | [58] |
| Me | 1974 | [59] | |
| Bn | 1972 | [41] | |
| Ph | 1974 | [60] | |
| Et | 2003 | [50] | |
| CH3 | 2014 | [61] | |
| CH2CH2OH | 1999 | [62] | |
| 2,3,4-pyridinemethane | 2012 | [63] | |
| 2,6-bis(acylamino)benzenethiolato | 1996 | [64] | |
| CH2CH(OH)Me | 2002 | [65] | |
| CH(Me)Ph CH2CH(Me)Et CH2CH(OH)CH2OH | 2011 | [66] | |
| [Fe4S4(SR)4]3− | H | 1997 | [58] |
| Me | 1988 | [67] | |
| C6H11 | 1986 | [68] | |
| 4-BrC6H4 | 1983 | [69] | |
| CH2Ph | 2015 | [70] |
| No. | Reaction | E0 (V) |
|---|---|---|
| 1 | 1.33 b | |
| 2 | −0.42 a | |
| 3 | −4.16 b | |
| 4 | −3.2 b | |
| 5 | 0.55 b |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J. Progress in Synthesizing Analogues of Nitrogenase Metalloclusters for Catalytic Reduction of Nitrogen to Ammonia. Catalysts 2019, 9, 939. https://doi.org/10.3390/catal9110939
Yang J. Progress in Synthesizing Analogues of Nitrogenase Metalloclusters for Catalytic Reduction of Nitrogen to Ammonia. Catalysts. 2019; 9(11):939. https://doi.org/10.3390/catal9110939
Chicago/Turabian StyleYang, Jianjun. 2019. "Progress in Synthesizing Analogues of Nitrogenase Metalloclusters for Catalytic Reduction of Nitrogen to Ammonia" Catalysts 9, no. 11: 939. https://doi.org/10.3390/catal9110939
APA StyleYang, J. (2019). Progress in Synthesizing Analogues of Nitrogenase Metalloclusters for Catalytic Reduction of Nitrogen to Ammonia. Catalysts, 9(11), 939. https://doi.org/10.3390/catal9110939