Gas Phase Catalytic Hydrogenation of C4 Alkynols over Pd/Al2O3


Abstract
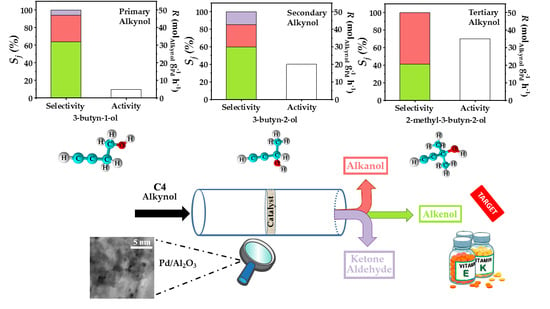
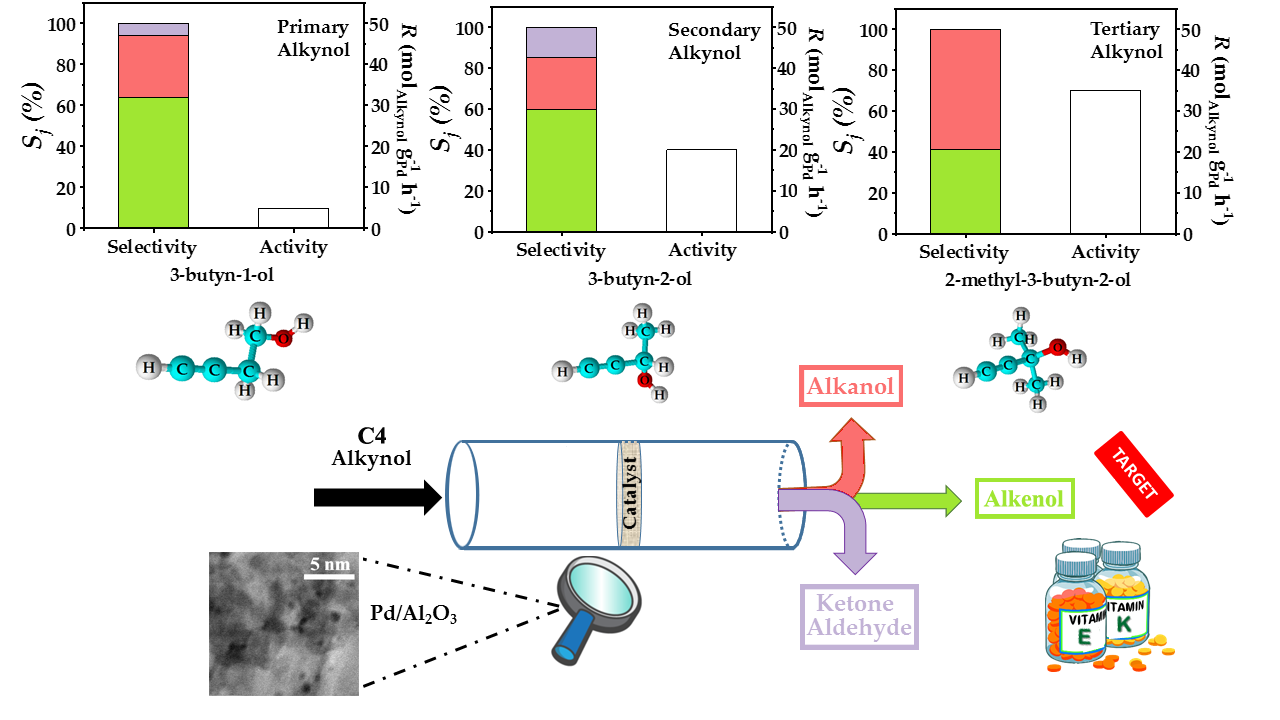{kind=link}
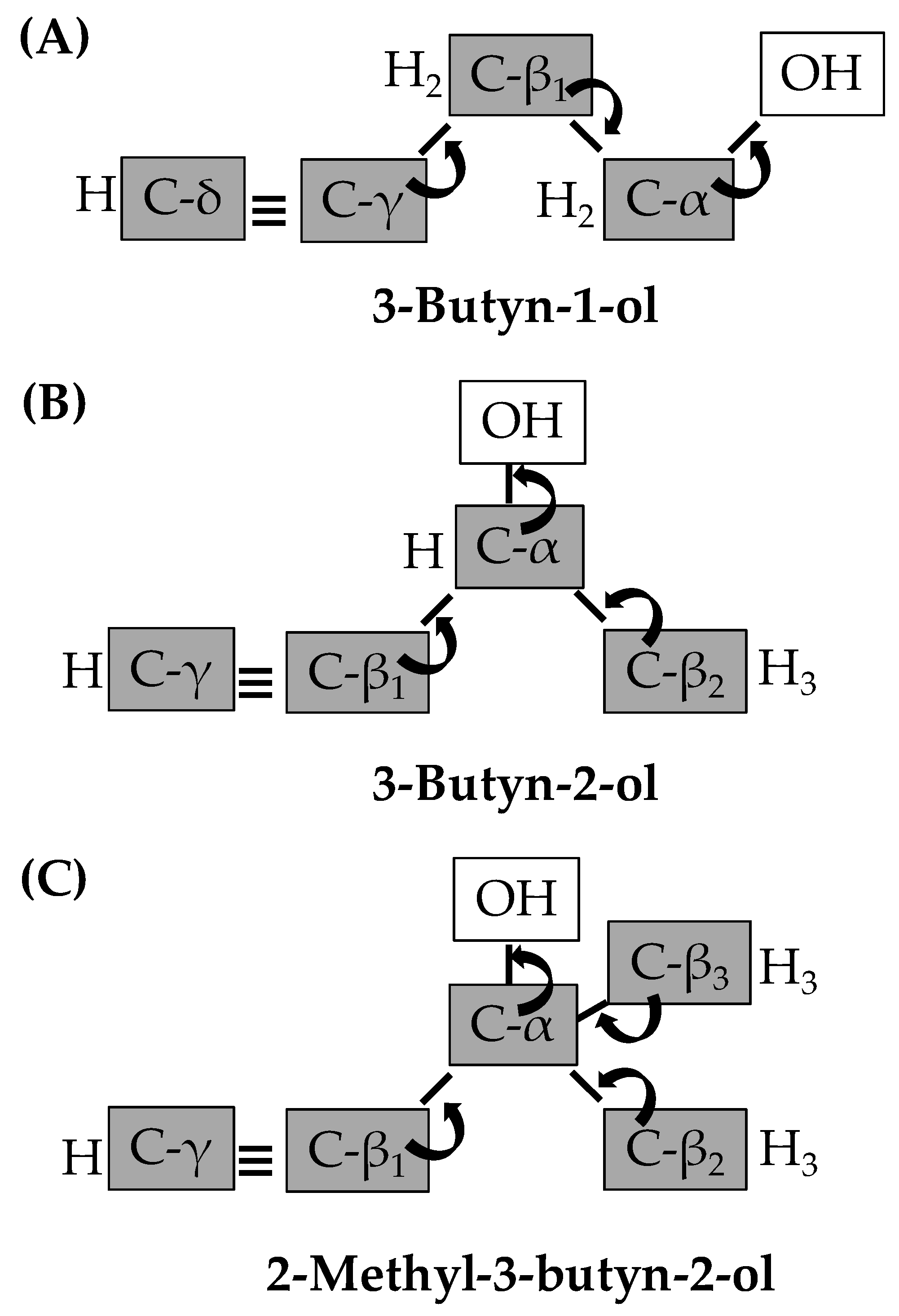{kind=link}
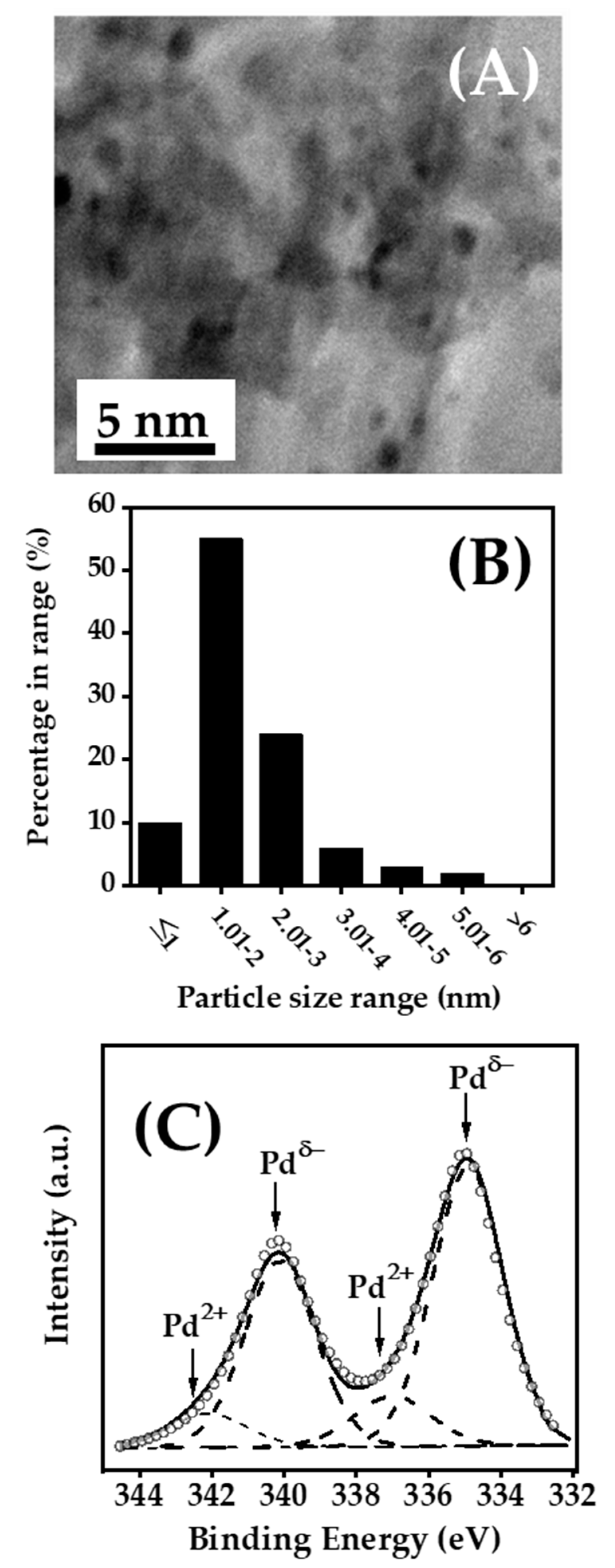{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Catalyst Characterisation
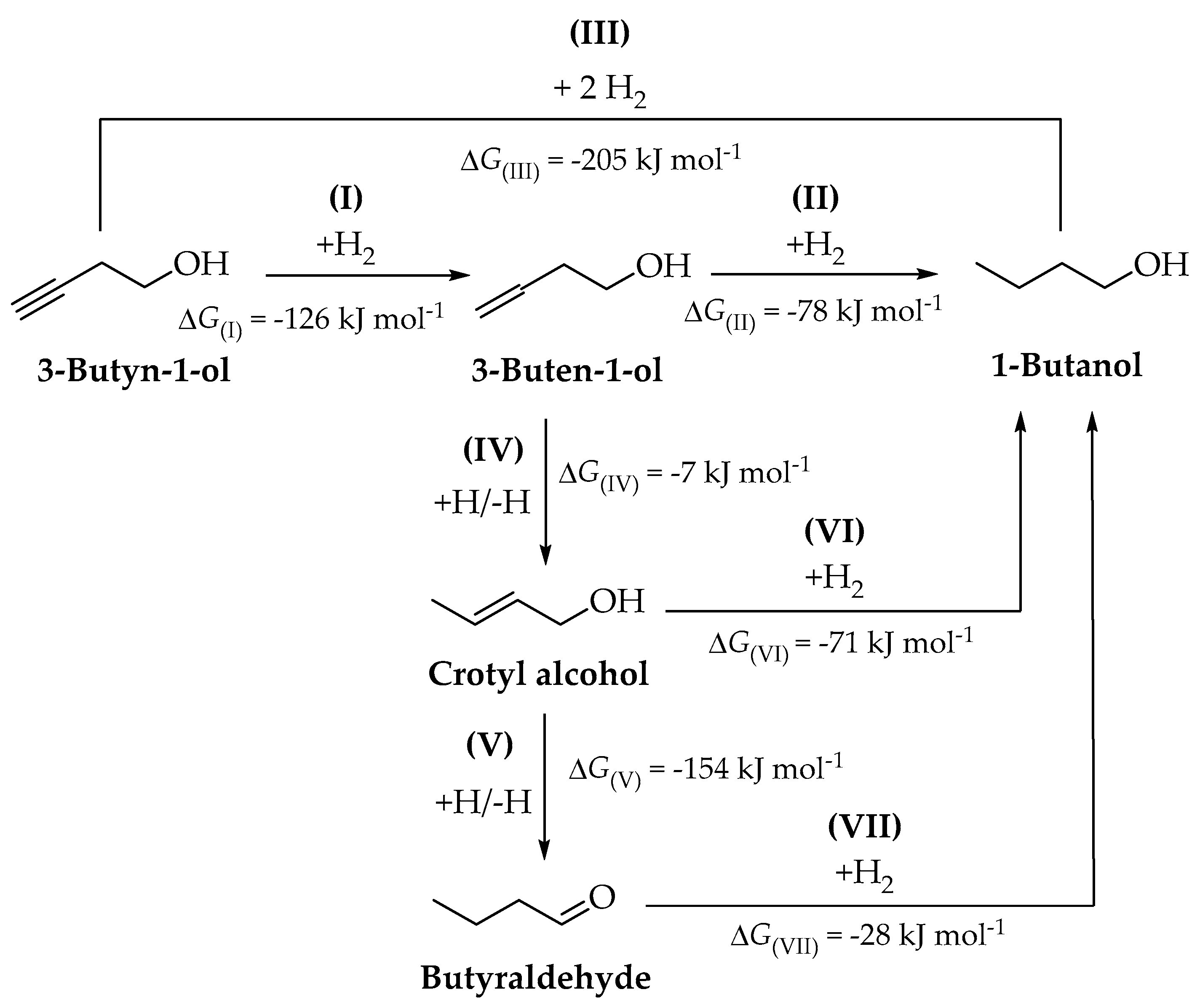
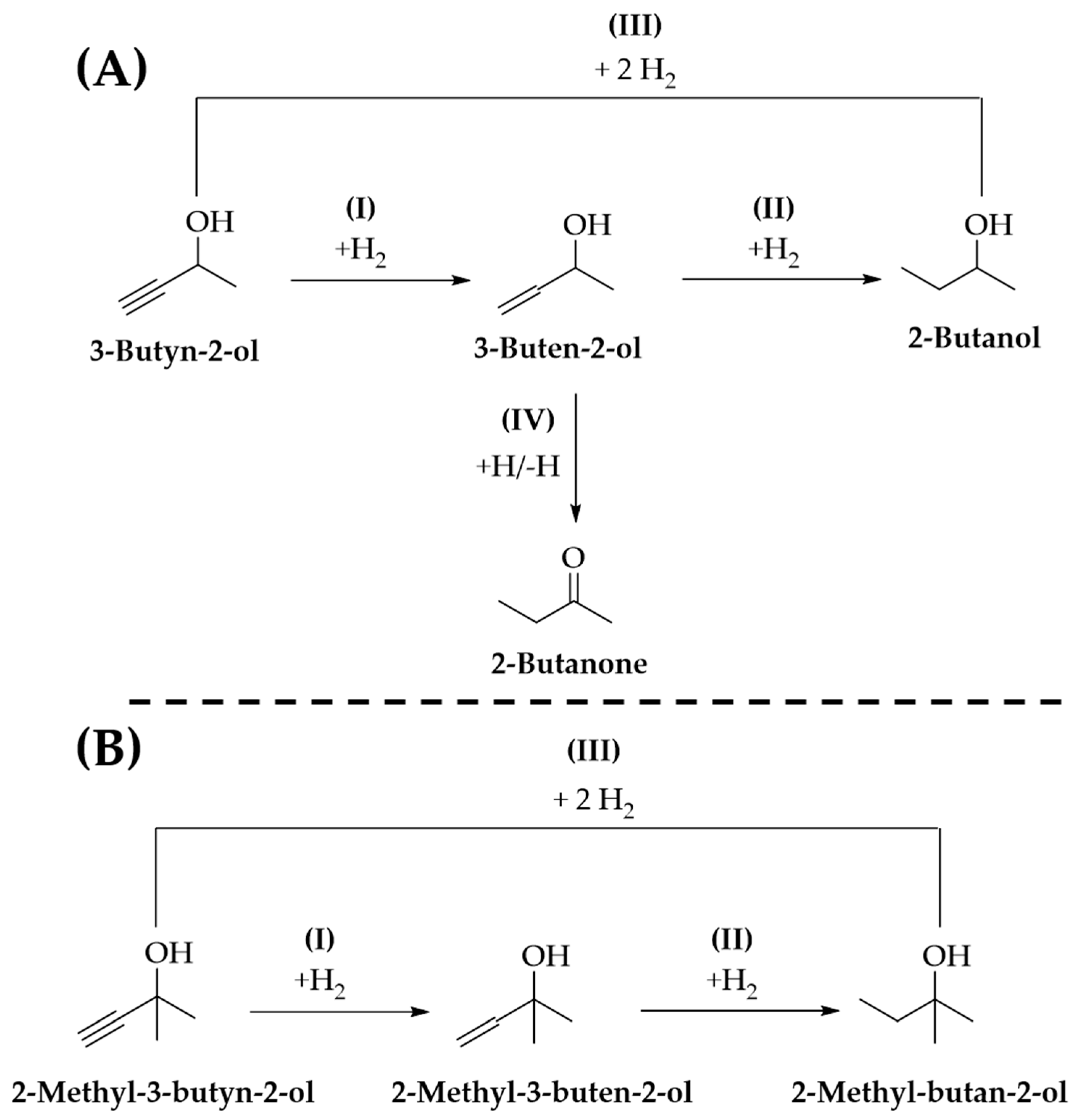
2.2. Reaction Thermodynamics
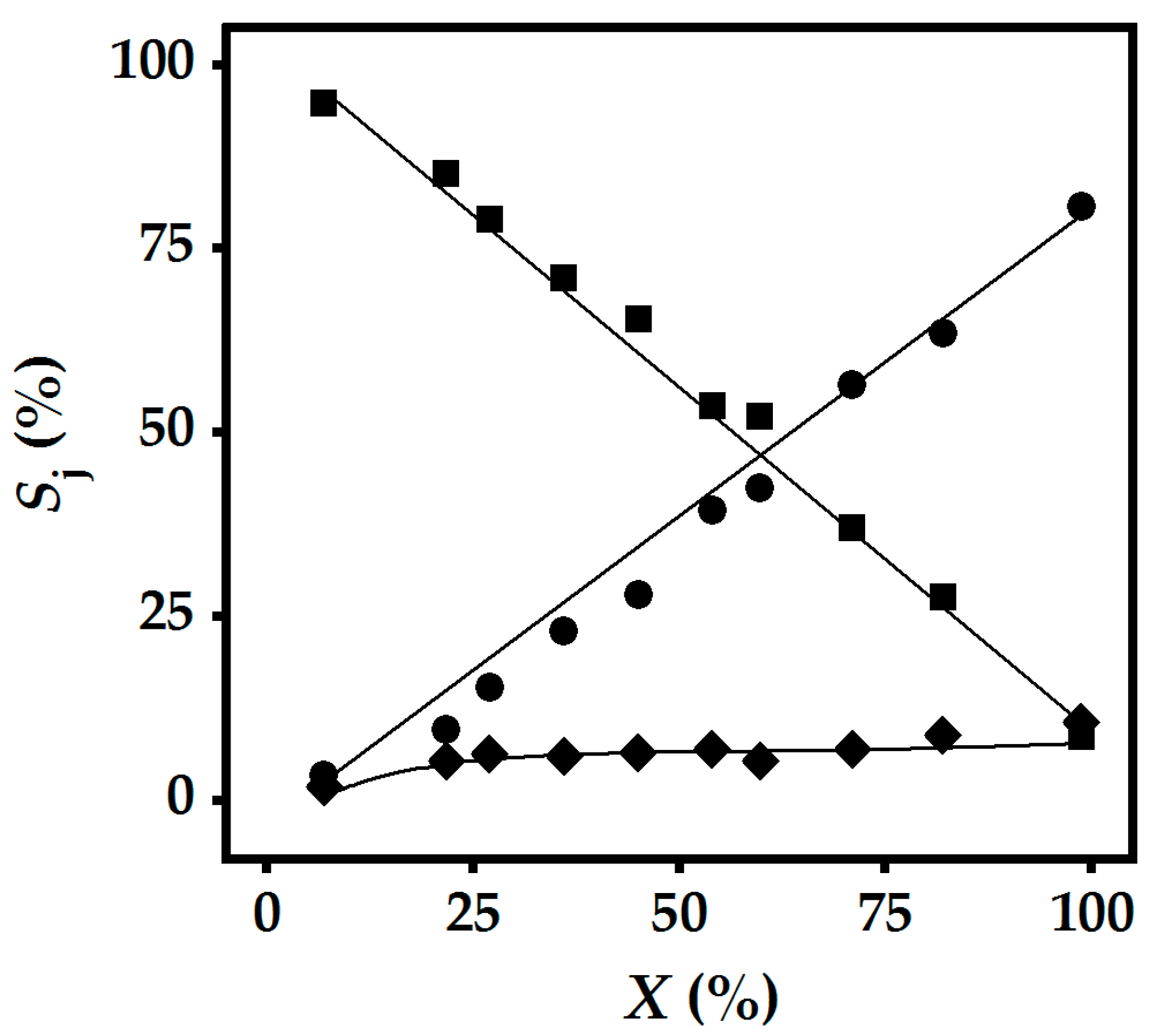
2.3. Gas Phase Hydrogenation of 3-Butyn-1-ol
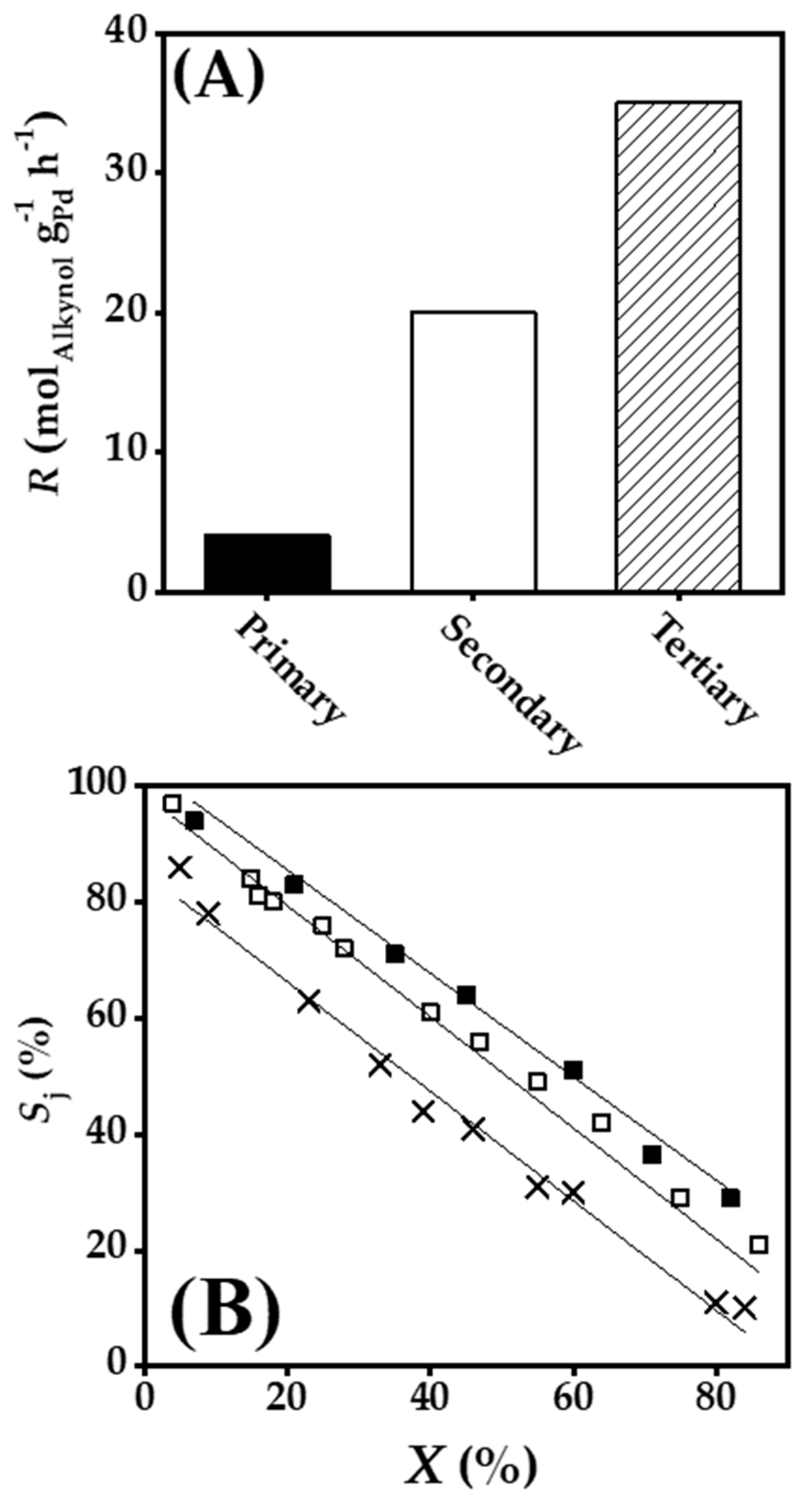
2.4. Gas Phase Hydrogenation of 3-Butyn-2-ol and 2-Methyl-3-butyn-2-ol
3. Materials and Methods
3.1. Catalyst Characterisation
3.2. Catalytic Procedure
3.3. Thermodynamic Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delgado, J.A.; Benkirane, O.; Claver, C.; Curulla-Ferré, D.; Godard, C. Advances in the Preparation of Highly Selective Nanocatalysts for the Semi-Hydrogenation of Alkynes Using Colloidal Approaches. Dalton Trans. 2017, 46, 12381–12403. [Google Scholar] [CrossRef]
- Rajaram, J.; Narula, A.P.S.; Chawla, H.P.S.; Dev, S. Semihydrogenation of Acetylenes. Tetrahedron 1983, 39, 2315–2322. [Google Scholar] [CrossRef]
- Molnár, Á.; Sárkány, A.; Varga, M. Hydrogenation of Carbon-Carbon Multiple Bonds: Chemo-, Regio- and Stereo-Selectivity. J. Mol. Catal. A Chem. 2001, 173, 185–221. [Google Scholar] [CrossRef]
- Mei, D.; Sheth, P.; Neurock, M.; Smith, C. First-Principles-Based Kinetic Monte Carlo Simulation of the Selective Hydrogenation of Acetylene over Pd(111). J. Catal. 2006, 242, 1–15. [Google Scholar] [CrossRef]
- Karpiński, Z. Catalysis by Supported, Unsupported, and Electron-Deficient Palladium. In Advances in Catalysis; Academic Press: Warsaw, Poland, 1990; Volume 37, pp. 45–100. [Google Scholar]
- Semagina, N.; Kiwi-Minsker, L. Recent Advances in the Liquid-Phase Synthesis of Metal Nanostructures with Controlled Shape and Size for Catalysis. Catal. Rev. 2009, 51, 147–217. [Google Scholar] [CrossRef]
- Sárkány, A.; Weiss, A.H.; Guczi, L. Structure Sensitivity of Acetylene-Ethylene Hydrogenation over Pd Catalysts. J. Catal. 1986, 98, 550–553. [Google Scholar] [CrossRef]
- Hub, S.; Touroude, R. Mechanism of Catalytic Hydrogenation of But-1-yne on Palladium. J. Catal. 1988, 114, 411–421. [Google Scholar] [CrossRef]
- Semagina, N.; Renken, A.; Kiwi-Minsker, L. Palladium Nanoparticle Size Effect in 1-Hexyne Selective Hydrogenation. J. Phys. Chem. C 2007, 111, 13933–13937. [Google Scholar] [CrossRef]
- Boitiaux, J.P.; Cosyns, J.; Vasudevan, S. Hydrogenation of Highly Unsaturated Hydrocarbons over Highly Dispersed Pd Catalyst. Part II: Ligand Effect of Piperidine. Appl. Catal. 1985, 15, 317–326. [Google Scholar] [CrossRef]
- Terasawa, M.; Yamamoto, H.; Kaneda, K.; Imanaka, T.; Teranishi, S. Selective Hydrogenation of Acetylenes to Olefins Catalyzed by Polymer-Bound Palladium(II) Complexes. J. Catal. 1979, 57, 315–325. [Google Scholar] [CrossRef]
- Karavanov, A.N.; Gryaznov, V.M. Effect of the Structure of Substituted Propargyl and Allyl Alcohols on the Rate of their Liquid Phase Hydrogenation on a Pd-Ru Alloy Membrane Catalyst. Bull. Acad. Sci. USSR Division Chem. Sci. 1989, 38, 1593–1596. [Google Scholar] [CrossRef]
- Bonrath, W.; Netscher, T. Catalytic Processes in Vitamins Synthesis and Production. Appl. Catal. A Gen. 2005, 280, 55–73. [Google Scholar] [CrossRef]
- Bonrath, W.; Medlock, J.; Schütz, J.; Wüstenberg, B.; Netscher, T. Hydrogenation in the Vitamins and Fine Chemicals Industry—An Overview. In Hydrogenation; InTech: Rijeka, Croatia, 2012; pp. 69–90. [Google Scholar]
- Bonrath, W.; Eggersdorfer, M.; Netscher, T. Catalysis in the Industrial Preparation of Vitamins and Nutraceuticals. Catal. Today 2007, 121, 45–57. [Google Scholar] [CrossRef]
- Crespo-Quesada, M.; Cárdenas-Lizana, F.; Dessimoz, A.L.; Kiwi-Minsker, L. Modern Trends in Catalyst and Process Design for Alkyne Hydrogenations. ACS Catal. 2012, 2, 1773–1786. [Google Scholar] [CrossRef]
- Izumi, Y.; Tanaka, Y.; Urabe, K. Selective Catalytic Hydrogenation of Propargyl Alcohol with Heteropoly Acid-Modified Palladium. Chem. Lett. 1982, 11, 679–682. [Google Scholar] [CrossRef]
- Uberman, P.M.; Costa, N.J.S.; Philippot, K.; Carmona, R.; dos Santos, A.A.; Rossi, L.M. A Recoverable Pd Nanocatalyst for Selective Semi-Hydrogenation of Alkynes: Hydrogenation of Benzyl-Propargylamines as a Challenging Model. Green Chem. 2014, 16, 4566–4574. [Google Scholar] [CrossRef]
- Navarro-Fuentes, F.; Keane, M.; Ni, X.-W. A Comparative Evaluation of Hydrogenation of 3-Butyn-2-ol over Pd/Al2O3 in an Oscillatory Baffled Reactor and a Commercial Parr Reactor. Org. Process Res. Dev. 2019, 23, 38–44. [Google Scholar] [CrossRef]
- Hou, R.; Wang, T.; Lan, X. Enhanced Selectivity in the Hydrogenation of Acetylene due to the Addition of a Liquid Phase as a Selective Solvent. Ind. Eng. Chem. Res. 2013, 52, 13305–13312. [Google Scholar] [CrossRef]
- Pérez, D.; Olivera-Fuentes, C.; Curbelo, S.; Rodríguez, M.J.; Zeppieri, S. Study of the Selective Hydrogenation of 1,3-Butadiene in Three Types of Industrial Reactors. Fuel 2015, 149, 34–45. [Google Scholar] [CrossRef]
- Prestianni, A.; Crespo-Quesada, M.; Cortese, R.; Ferrante, F.; Kiwi-Minsker, L.; Duca, D. Structure Sensitivity of 2-Methyl-3-Butyn-2-ol Hydrogenation on Pd: Computational and Experimental Modeling. J. Phys. Chem. C 2014, 118, 3119–3128. [Google Scholar] [CrossRef]
- Yarulin, A.; Yuranov, I.; Cárdenas-Lizana, F.; Abdulkin, P.; Kiwi-Minsker, L. Size-Effect of Pd-(Poly(N -vinyl-2-pyrrolidone)) Nanocatalysts on Selective Hydrogenation of Alkynols with Different Alkyl Chains. J. Phys. Chem. C 2013, 117, 13424–13434. [Google Scholar] [CrossRef]
- Smirnov, M.Y.; Klembovskii, I.O.; Kalinkin, A.V.; Bukhtiyarov, V.I. An XPS Study of the Interaction of a Palladium Foil with NO2. Kinet. Catal. 2018, 59, 786–791. [Google Scholar] [CrossRef]
- Juan-Juan, J.; Román-Martínez, M.C.; Illán-Gómez, M.J. Catalytic Activity and Characterization of Ni/Al2O3 and NiK/Al2O3 Catalysts for CO2 Methane Reforming. Appl. Catal. A Gen. 2004, 264, 169–174. [Google Scholar] [CrossRef]
- Nag, N.K. A Study on the Dispersion and Catalytic Activity of Gamma Alumina-Supported Palladium Catalysts. Catal. Lett. 1994, 24, 37–46. [Google Scholar] [CrossRef]
- McCarty, J.G. Kinetics of PdO Combustion Catalysis. Catal. Today 1995, 26, 283–293. [Google Scholar] [CrossRef]
- Weissman, D.L.; Shek, M.L.; Spicer, W.E. Photoemission Spectra and Thermal Desorption Characteristics of Two States of Oxygen on Pd. Surf. Sci. 1980, 92, L59–L66. [Google Scholar] [CrossRef]
- Papp, A.; Molnár, Á.; Mastalir, Á. Catalytic Investigation of Pd Particles Supported on MCM-41 for the Selective Hydrogenations of Terminal and Internal Alkynes. Appl. Catal. A Gen. 2005, 289, 256–266. [Google Scholar] [CrossRef]
- Da Silva, F.P.; Rossi, L.M. Palladium on Magnetite: Magnetically Recoverable Catalyst for Selective Hydrogenation of Acetylenic to Olefinic Compounds. Tetrahedron 2014, 70, 3314–3318. [Google Scholar] [CrossRef]
- Derrien, M.L. Selective Hydrogenation Applied to the Refining of Petrochemical Raw Materials Produced by Steams Cracking. Stud. Surf. Sci. Catal. 1986, 27, 613–666. [Google Scholar]
- Nikolaev, S.A.; Zanaveskin, L.N.; Smirnov, V.V.; Averyanov, V.A.; Zanaveskin, K.L. Catalytic Hydrogenation of Alkyne and Alkadiene Impurities from Alkenes. Practical and Theoretical Aspects. Russ. Chem. Rev. 2009, 78, 231–247. [Google Scholar] [CrossRef]
- Morrill, C.; Grubbs, R.H. Highly Selective 1,3-Isomerization of Allylic Alcohols via Rhenium Oxo Catalysis. J. Am. Chem. Soc. 2005, 127, 2842–2843. [Google Scholar] [CrossRef]
- Jewell, L.; Davis, B. Review of Absorption and Adsorption in the Hydrogen–Palladium System. Appl. Catal. A Gen. 2006, 310, 1–15. [Google Scholar] [CrossRef]
- Nikoshvili, L.; Shimanskaya, E.; Bykov, A.; Yuranov, I.; Kiwi-Minsker, L.; Sulman, E. Selective Hydrogenation of 2-Methyl-3-Butyn-2-ol over Pd-Nanoparticles Stabilized in Hypercrosslinked Polystyrene: Solvent Effect. Catal. Today 2014, 241, 179–188. [Google Scholar] [CrossRef]
- Sulman, E.M. Selective Hydrogenation of Unsaturated Ketones and Acetylene Alcohols. Russ. Chem. Rev. 1994, 63, 923–936. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Semagina, N.; Renken, A.; Laub, D.; Kiwi-Minsker, L. Synthesis of Monodispersed Palladium Nanoparticles to Study Structure Sensitivity of Solvent-Free Selective Hydrogenation of 2-Methyl-3-butyn-2-ol. J. Catal. 2007, 246, 308–314. [Google Scholar] [CrossRef]
- Boitiaux, J.P.; Cosyns, J.; Robert, E. Liquid Phase Hydrogenation of Unsaturated Hydrocarbons on Palladium, Platinum and Rhodium Catalysts. Part II: Kinetic Study of 1-Butene, 1,3-Butadiene and 1-Butyne Hydrogenation on Rhodium; Comparison with Platinum and Palladium Part II. Appl. Catal. 1987, 32, 169–183. [Google Scholar] [CrossRef]
- Morrill, T.C.; D’Souza, C.A. Efficient Hydride-Assisted Isomerization of Alkenes via Rhodium Catalysis. Organometallics 2003, 22, 1626–1629. [Google Scholar] [CrossRef]
- Karlsson, E.A.; Bäckvall, J.-E. Mechanism of the Palladium-Catalyzed Carbohydroxylation of Allene-Substituted Conjugated Dienes: Rationalization of the Recently Observed Nucleophilic Attack by Water on a (π-Allyl)palladium Intermediate. Chem. A Eur. J. 2008, 14, 9175–9180. [Google Scholar] [CrossRef]
- Behm, R.J.; Penka, V.; Cattania, M.-G.; Christmann, K.; Ertl, G. Evidence for ‘“Subsurface”’ Hydrogen on Pd(110): An Intermediate Between Chemisorbed and Dissolved Species. J. Chem. Phys. 1983, 78, 7486–7490. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W. Isomerization of Allylic Alcohols to Carbonyl Compounds by Aqueous-Biphase Rhodium Catalysis. New J. Chem. 2001, 25, 11–12. [Google Scholar] [CrossRef]
- Ide, M.S.; Hao, B.; Neurock, M.; Davis, R.J. Mechanistic Insights on the Hydrogenation of α,β-Unsaturated Ketones and Aldehydes to Unsaturated Alcohols over Metal Catalysts. ACS Catal. 2012, 2, 671–683. [Google Scholar] [CrossRef]
- Ponec, V. On the Role of Promoters in Hydrogenations on Metals; α,β-Unsaturated Aldehydes and Ketones. Appl. Catal. A Gen. 1997, 149, 27–48. [Google Scholar] [CrossRef]
- Amorim, C.; Keane, M.A. Palladium Supported on Structured and Nonstructured Carbon: A Consideration of Pd Particle Size and the Nature of Reactive Hydrogen. J. Colloid Interface Sci. 2008, 322, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Lizana, F.; Wang, X.; Lamey, D.; Li, M.; Keane, M.A.; Kiwi-Minsker, L. An Examination of Catalyst Deactivation in p-Chloronitrobenzene Hydrogenation over Supported Gold. Chem. Eng. J. 2014, 255, 695–704. [Google Scholar] [CrossRef]
- Venezia, A.M.; Liotta, L.F.; Deganello, G.; Schay, Z.; Guczi, L. Characterization of Pumice-Supported Ag–Pd and Cu–Pd Bimetallic Catalysts by X-Ray Photoelectron Spectroscopy and X-Ray Diffraction. J. Catal. 1999, 182, 449–455. [Google Scholar] [CrossRef]
- Fu, Z.; Hu, J.; Hu, W.; Yang, S.; Luo, Y. Quantitative analysis of Ni2+/Ni3+ in Li[NixMnyCoz]O2 Cathode Materials: Non-linear Least-squares Fitting of XPS Spectra. Appl. Surf. Sci. 2018, 441, 1048–1056. [Google Scholar] [CrossRef]
- Cárdenas-Lizana, F.; Lamey, D.; Perret, N.; Gómez-Quero, S.; Kiwi-Minsker, L.; Keane, M.A. Au/Mo2N as a New Catalyst Formulation for the Hydrogenation of p-Chloronitrobenzene in Both Liquid and Gas Phases. Catal. Commun. 2012, 21, 46–51. [Google Scholar] [CrossRef]
- Ye, G.; Xie, D.; Qiao, W.; Grace, J.R.; Lim, C.J. Modelling of Fluidized Bed Membrane Reactors for Hydrogen Production from Steam Methane Reforming with Aspen Plus. Int. J. Hydrogen Energy 2009, 34, 4755–4762. [Google Scholar] [CrossRef]
- Joback, K.G.; Reid, R.C. Estimation of Pure-Component Properties from Group-Contributions. Chem. Eng. Commun. 1987, 57, 233–243. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Fernández, A.; Pischetola, C.; Cárdenas-Lizana, F. Gas Phase Catalytic Hydrogenation of C4 Alkynols over Pd/Al2O3. Catalysts 2019, 9, 924. https://doi.org/10.3390/catal9110924
González-Fernández A, Pischetola C, Cárdenas-Lizana F. Gas Phase Catalytic Hydrogenation of C4 Alkynols over Pd/Al2O3. Catalysts. 2019; 9(11):924. https://doi.org/10.3390/catal9110924
Chicago/Turabian StyleGonzález-Fernández, Alberto, Chiara Pischetola, and Fernando Cárdenas-Lizana. 2019. "Gas Phase Catalytic Hydrogenation of C4 Alkynols over Pd/Al2O3" Catalysts 9, no. 11: 924. https://doi.org/10.3390/catal9110924
APA StyleGonzález-Fernández, A., Pischetola, C., & Cárdenas-Lizana, F. (2019). Gas Phase Catalytic Hydrogenation of C4 Alkynols over Pd/Al2O3. Catalysts, 9(11), 924. https://doi.org/10.3390/catal9110924