Non-Monotonic Trends of Hydrogen Adsorption on Single Atom Doped g-C3N4




Abstract
1. Introduction
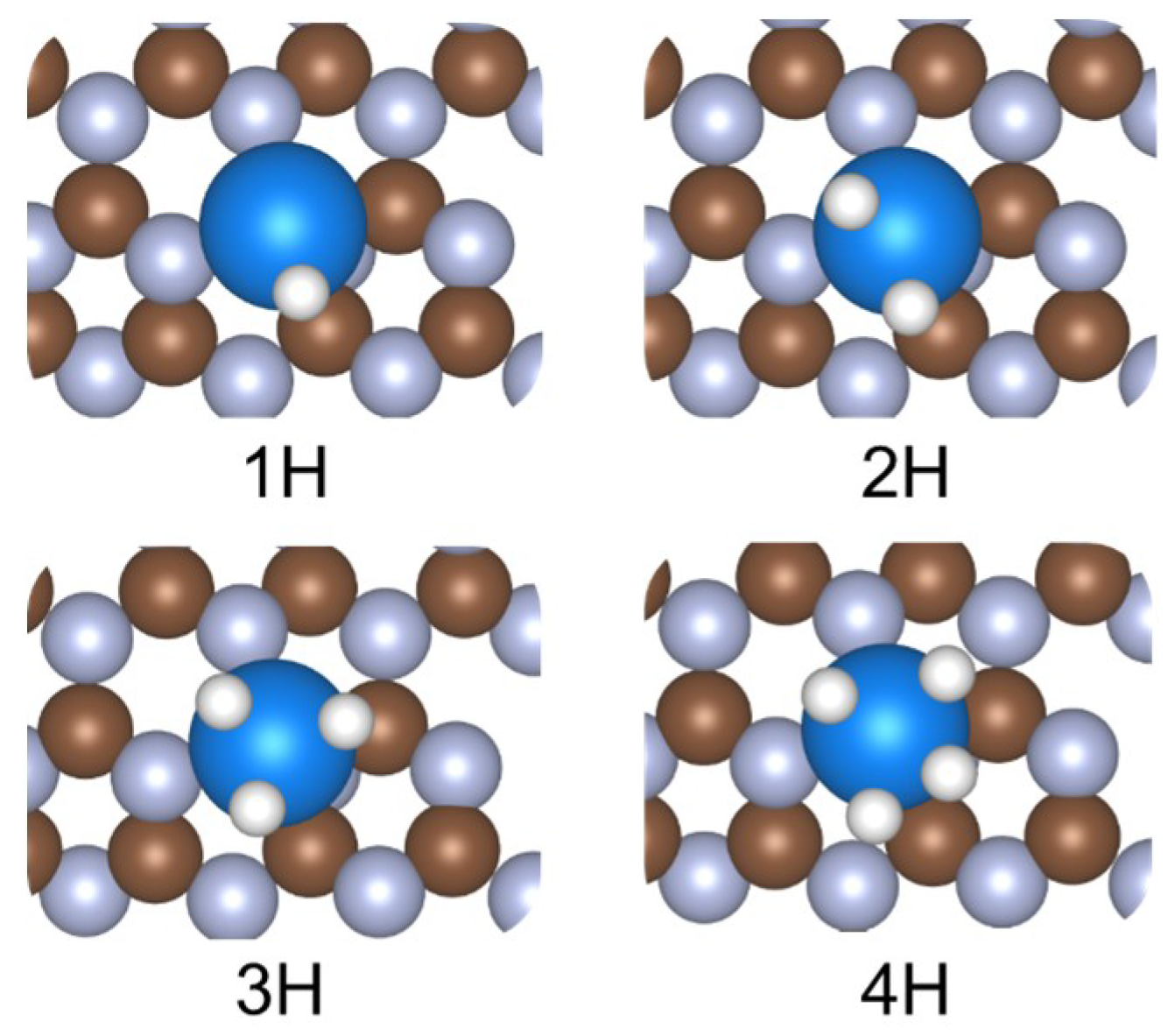
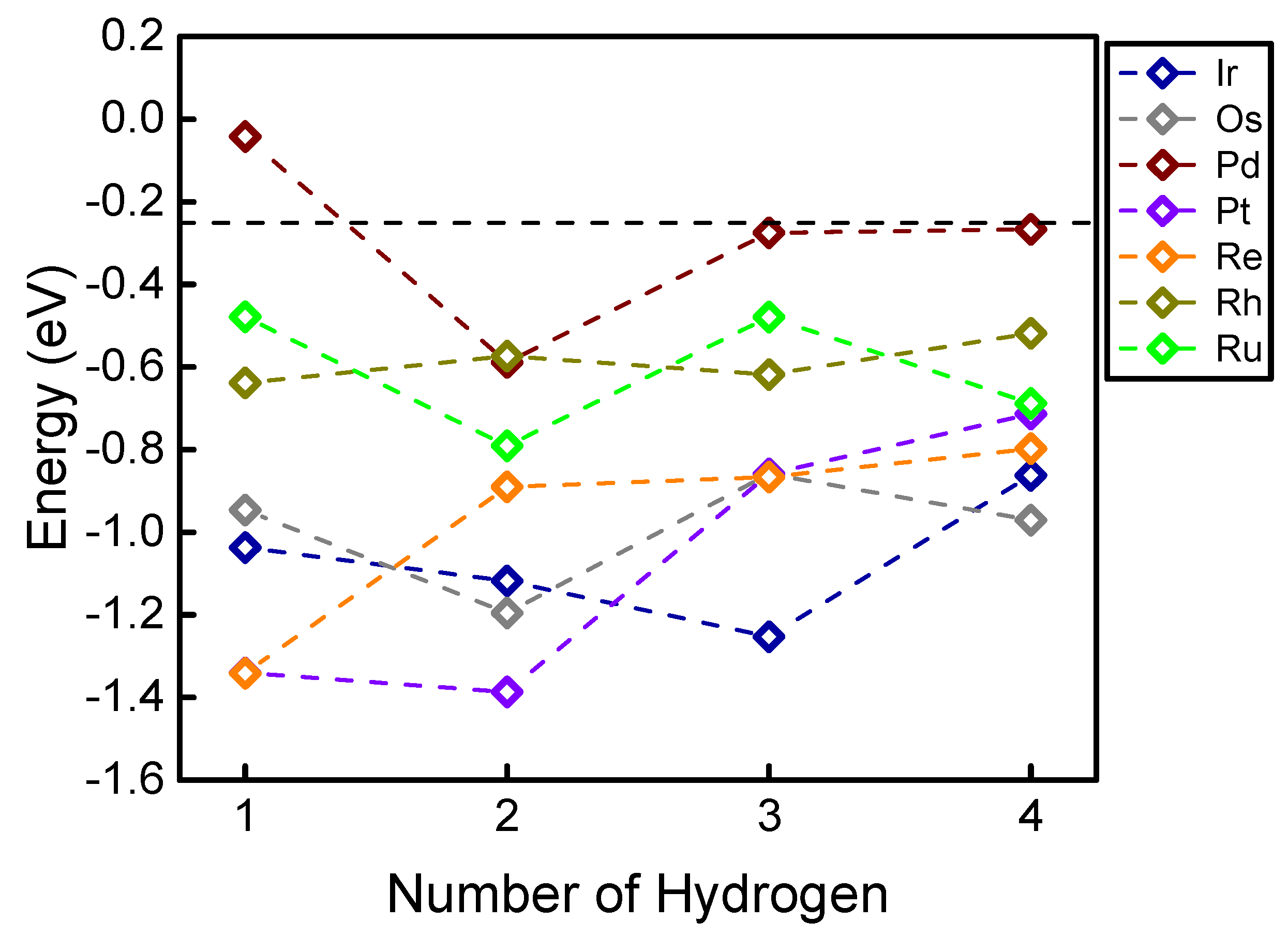
2. Results and Discussion
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| g-C3N4 | graphitic carbon nitrite |
| HER | hydrogen evolution reaction |
| H | Hydrogen |
| C | Carbon |
| O | Oxygen |
| Ir | Iridium |
| Os | Osmium |
| Pd | Palladium |
| Pt | Platinum |
| Re | Rhenium |
| Rh | Rhodium |
| Ru | Ruthenium |
| adsorption energy, eV | |
| total energy of the adsorption system, eV | |
| total energy of the bare system, eV | |
| total energy of a hydrogen molecule in vacuum, eV | |
| number of adsorbed hydrogens | |
| reaction free energy of hydrogen evolution reaction, eV |
References
- Wang, Y.; Arandiyan, H.; Scott, J.; Amal, R. Single-atom and Nano-clustered Pt Catalysts for Selective CO2 Reduction Single-atom and Nano-clustered Pt Catalysts for Selective CO2 Reduction. ACS Appl. Energy Mater. 2018, 1, 6781–6789. [Google Scholar] [CrossRef]
- Back, S.; Lim, J.; Kim, N.Y.; Kim, Y.H.; Jung, Y. Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, Z. Single Mo Atom Supported on Defective Boron Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study. J. Am. Chem. Soc. 2017, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Jung, Y. On the mechanism of electrochemical ammonia synthesis on the Ru catalyst. Phys. Chem. Chem. Phys. 2016. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.H.; Tsai, C.; Vojvodic, A.; Nørskov, J.K. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 2015. [Google Scholar] [CrossRef]
- Back, S.; Yeom, M.S.; Jung, Y. Active Sites of Au and Ag Nanoparticle Catalysts for CO2 Electroreduction to CO. ACS Catal. 2015, 5, 5089–5096. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Liu, Z. Application of Artificial Neural Networks for Catalysis: A Review. Catalysts 2017, 7, 306. [Google Scholar] [CrossRef]
- Li, H.; Shin, K.; Henkelman, G. Effects of Ensembles, Ligand, and Strain on Adsorbate Binding to Alloy Surfaces. J. Chem. Phys. 2018, 149, 174705. [Google Scholar] [CrossRef]
- Li, H.; Evans, E.J.; Mullins, C.B.; Henkelman, G. Ethanol Decomposition on Pd–Au Alloy Catalysts. J. Phys. Chem. C 2018, 122, 22024–22032. [Google Scholar] [CrossRef]
- Li, H.; Luo, L.; Kunal, P.; Bonifacio, C.S.; Duan, Z.; Yang, J.C.; Humphrey, S.M.; Crooks, R.M.; Henkelman, G. Oxygen Reduction Reaction on Classically Immiscible Bimetallics: A Case Study of RhAu. J. Phys. Chem. C 2018, 122, 2712–2716. [Google Scholar] [CrossRef]
- Li, H.; Henkelman, G. Dehydrogenation Selectivity of Ethanol on Close-Packed Transition Metal Surfaces: A Computational Study of Monometallic, Pd/Au, and Rh/Au Catalysts. J. Phys. Chem. C 2017, 121, 27504–27510. [Google Scholar] [CrossRef]
- Wu, K.; Du, K.; Hu, G. Red-blood-cell-like (NH4)[Fe2(OH)(PO4)2]·2H2O particles: Fabrication and application in high-performance LiFePO4 cathode materials. J. Mater. Chem. A 2018, 6, 1057–1066. [Google Scholar] [CrossRef]
- Wu, K.; Du, K.; Hu, G. A novel design concept for fabricating 3D graphene with the assistant of anti-solvent precipitated sulphates and its Li-ion storage properties. J. Mater. Chem. A 2018. [Google Scholar] [CrossRef]
- Wu, K.; Liu, D.; Tang, Y. In-situ single-step chemical synthesis of graphene-decorated CoFe2O4composite with enhanced Li ion storage behaviors. Electrochim. Acta 2018. [Google Scholar] [CrossRef]
- Luo, Y.; Dolder, C.K.; Walker, J.M.; Mishra, R.; Dean, D.; Becker, M.L. Synthesis and Biological Evaluation of Well-Defined Poly(propylene fumarate) Oligomers and Their Use in 3D Printed Scaffolds. Biomacromolecules 2016. [Google Scholar] [CrossRef]
- Walker, J.M.; Bodamer, E.; Kleinfehn, A.; Luo, Y.; Becker, M.; Dean, D. Design and mechanical characterization of solid and highly porous 3D printed poly(propylene fumarate) scaffolds. Prog. Addit. Manuf. 2017. [Google Scholar] [CrossRef]
- Walker, J.M.; Bodamer, E.; Krebs, O.; Luo, Y.; Kleinfehn, A.; Becker, M.L.; Dean, D. Effect of Chemical and Physical Properties on the In Vitro Degradation of 3D Printed High Resolution Poly(propylene fumarate) Scaffolds. Biomacromolecules 2017. [Google Scholar] [CrossRef] [PubMed]
- Cabán-Acevedo, M.; Stone, M.L.; Schmidt, J.R.; Thomas, J.G.; Ding, Q.; Chang, H.C.; Tsai, M.L.; He, H.; Jin, S. Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide. Nat. Mater. 2015. [Google Scholar] [CrossRef]
- Li, J.-S.; Wang, Y.; Liu, C.-H.; Li, S.-L.; Wang, Y.-G.; Dong, L.-Z.; Dai, Z.-H.; Li, Y.-F.; Lan, Y.-Q. Coupled molybdenum carbide and reduced graphene oxide electrocatalysts for efficient hydrogen evolution. Nat. Commun. 2016, 7, 11204. [Google Scholar] [CrossRef]
- Vesborg, P.C.K.; Seger, B.; Chorkendorff, I. Recent development in hydrogen evolution reaction catalysts and their practical implementation. J. Phys. Chem. Lett. 2015. [Google Scholar] [CrossRef]
- Ito, Y.; Cong, W.; Fujita, T.; Tang, Z.; Chen, M. High catalytic activity of nitrogen and sulfur co-doped nanoporous graphene in the hydrogen evolution reaction. Angew. Chem. Int. Ed. 2015. [Google Scholar] [CrossRef]
- Morales-Guio, C.G.; Stern, L.A.; Hu, X. Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution. Chem. Soc. Rev. 2014, 43, 6555–6569. [Google Scholar] [CrossRef] [PubMed]
- Kai, W.; Liwei, L.; Wen, X.; Shengzhe, Z.; Yong, L.; Hongwei, Z.; Zongqiang, S. Electrodeposition synthesis of PANI/MnO2/graphene composite materials and its electrochemical performance. Int. J. Electrochem. Sci. 2017, 12, 8306–8314. [Google Scholar] [CrossRef]
- Wang, K.; Pang, J.; Li, L.; Zhou, S.; Li, Y.; Zhang, T. Synthesis of hydrophobic carbon nanotubes/reduced graphene oxide composite films by flash light irradiation. Front. Chem. Sci. Eng. 2018, 12, 376–382. [Google Scholar] [CrossRef]
- Kai, W.; Shengzhe, Z.; Yanting, Z.; Jun, R.; Li, L.; Yong, Y. Synthesis of Porous Carbon by Activation Method and its Electrochemical Performance. Int. J. Electrochem. Sci. 2018, 13, 10766–10773. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Tan, S.H.; Chen, K.Q. Enhance the stability of α-graphyne nanoribbons by dihydrogenation. Org. Electron. Phys. Mater. Appl. 2014. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Zeng, J.; Chen, K.Q. Spin filtering effect and magnetoresistance in zigzag 6, 6, 12-graphyne nanoribbon system. Carbon 2014. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Chen, C.Y.; Li, B.L.; Chen, K.Q. Characteristics of classical Kirchhoff’s superposition law in carbon atomic wires connected in parallel. Carbon 2015. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; López, N.; Pérez-Ramírez, J. A Stable Single-Site Palladium Catalyst for Hydrogenations. Angew. Chemie Int. Ed. 2015. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Supported metal catalysts at the single-atom—A viewpoint. Cuihua Xuebao/Chin. J. Catal. 2017. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Liu, Y.; Cen, W.; Luo, X. Functional Group Effects on the HOMO—LUMO Gap of g-C3N4. Nanomaterials 2018, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Qiu, Y.; Peng, S.; Chen, C.; Zeng, J.; Zhang, S.; Xiao, R. Enhanced hydrogen production performance through controllable redox exsolution within CoFeAlO: Xspinel oxygen carrier materials. J. Mater. Chem. A 2018. [Google Scholar] [CrossRef]
- Zeng, D.; Xiao, R.; Zhang, S.; Zeng, J.; Huang, Z. Bio-oil heavy fraction as a feedstock for hydrogen generation via chemical looping process: Reactor design and hydrodynamic analysis. Int. J. Chem. React. Eng. 2017. [Google Scholar] [CrossRef]
- Zeng, D.W.; Xiao, R.; Huang, Z.C.; Zeng, J.M.; Zhang, H.Y. Continuous hydrogen production from non-aqueous phase bio-oil via chemical looping redox cycles. Int. J. Hydrogen Energy 2016, 41, 6676–6684. [Google Scholar] [CrossRef]
- Duan, C.; Cao, Y.; Hu, L.; Fu, D.; Ma, J. Synergistic effect of TiF3 on the dehydriding property of α-AlH3 nano-composite. Mater. Lett. 2019, 238, 254–257. [Google Scholar] [CrossRef]
- Duan, C.W.; Hu, L.X.; Ma, J.L. Ionic liquids as an efficient medium for the mechanochemical synthesis of α-AlH3 nano-composites. J. Mater. Chem. A 2018, 6, 6309–6318. [Google Scholar] [CrossRef]
- Xin, H.; Linic, S. Communications: Exceptions to the d -band model of chemisorption on metal surfaces: The dominant role of repulsion between adsorbate states and metal d-states. J. Chem. Phys. 2010, 132, 10–14. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.; Jiang, D.E. Stronger-than-Pt hydrogen adsorption in a Au22 nanocluster for the hydrogen evolution reaction. J. Mater. Chem. A 2018, 6, 7532–7537. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Tang, H.; Van Der Ven, A.; Trout, B.L. Phase diagram of oxygen adsorbed on platinum (111) by first-principles investigation. Phys. Rev. B Condens. Matter Mater. Phys. 2004. [Google Scholar] [CrossRef]
- Reuter, K.; Scheffler, M. First-Principles Atomistic Thermodynamics for Oxidation Catalysis: Surface Phase Diagrams and Catalytically Interesting Regions. Phys. Rev. Lett. 2003. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; He, Y.; Ding, M.; Wang, Y.; Zhong, J. Nanoimaging of food proteins by atomic force microscopy. Part I: Components, imaging modes, observation ways, and research types. Trends Food Sci. Technol. 2018. [Google Scholar] [CrossRef]
- Shi, C.; He, Y.; Ding, M.; Wang, Y.; Zhong, J. Nanoimaging of food proteins by atomic force microscopy. Part II: Components, imaging modes, observation ways, and research types. Trends Food Sci. Technol. 2018. [Google Scholar] [CrossRef]
- Melissen, S.T.A.G.; Steinmann, S.N.; Le Bahers, T.; Sautet, P. DFT Perspective on the Thermochemistry of Carbon Nitride Synthesis. J. Phys. Chem. C 2016. [Google Scholar] [CrossRef]
- Li, X.; Melissen, S.T.A.G.; Le Bahers, T.; Sautet, P.; Masters, A.F.; Steinmann, S.N.; Maschmeyer, T. Shining Light on Carbon Nitrides: Leveraging Temperature to Understand Optical Gap Variations. Chem. Mater. 2018. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140. [Google Scholar] [CrossRef]
- Monkhorst, H.; Pack, J. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z. Mining the intrinsic trends of CO2 solubility in blended solutions. J. CO2 Util. 2018, 26, 496–502. [Google Scholar] [CrossRef]
- Liu, S.; White, M.G.; Liu, P. Mechanism of Oxygen Reduction Reaction on Pt(111) in Alkaline Solution: Importance of Chemisorbed Water on Surface. J. Phys. Chem. C 2016. [Google Scholar] [CrossRef]
- Liu, S.; Liu, P. Optimized Pt-Based Catalysts for Oxygen Reduction Reaction in Alkaline Solution: A First Principle Study. J. Electrochem. Soc. 2018. [Google Scholar] [CrossRef]
- Liu, S.; White, M.G.; Liu, P. Oxygen Reduction Reaction on Ag(111) in Alkaline Solution: A Combined Density Functional Theory and Kinetic Monte Carlo Study. ChemCatChem 2018. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Li, Y.; Xia, H.; Engelhard, M.H.; Fu, S.; Du, D.; Lin, Y. A Facile Method for Synthesizing Dendritic Core-Shell Structured Ternary Metallic Aerogels and Their Enhanced Electrochemical Performances. Chem. Mater. 2016. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Zhong, H.; Su, D.; Li, N.; Engelhard, M.H.; Xia, H.; Zhang, Q.; Feng, S.; Beckman, S.P.; et al. Nanovoid Incorporated IrxCu Metallic Aerogels for Oxygen Evolution Reaction Catalysis. ACS Energy Lett. 2018. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Tian, M.; Su, D.; Fu, M.; Engelhard, M.H.; Chowdhury, I.; Feng, S.; Du, D.; Lin, Y. Ultrafine Pd ensembles anchored-Au2Cu aerogels boost ethanol electrooxidation. Nano Energy 2018. [Google Scholar] [CrossRef]
- Shao, P.; Tian, J.; Duan, X.; Yang, Y.; Shi, W.; Luo, X.; Cui, F.; Luo, S.; Wang, S. Cobalt silicate hydroxide nanosheets in hierarchical hollow architecture with maximized cobalt active site for catalytic oxidation. Chem. Eng. J. 2019. [Google Scholar] [CrossRef]
- Shao, P.; Tian, J.; Yang, F.; Duan, X.; Gao, S.; Shi, W.; Luo, X.; Cui, F.; Luo, S.; Wang, S. Identification and Regulation of Active Sites on Nanodiamonds: Establishing a Highly Efficient Catalytic System for Oxidation of Organic Contaminants. Adv. Funct. Mater. 2018. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Z.; Cui, D.; Luo, X.; Liang, B.; Yang, L.; Liu, T.; Wang, A.; Luo, S. Ultrafine palladium nanoparticles supported on 3D self-supported Ni foam for cathodic dechlorination of florfenicol. Chem. Eng. J. 2018. [Google Scholar] [CrossRef]
- Duan, C.; Huo, J.; Li, F.; Yang, M.; Xi, H. Ultrafast room-temperature synthesis of hierarchically porous metal–organic frameworks by a versatile cooperative template strategy. J. Mater. Sci. 2018. [Google Scholar] [CrossRef]
- Duan, C.; Li, F.; Xiao, J.; Liu, Z.; Li, C.; Xi, H. Rapid room-temperature synthesis of hierarchical porous zeolitic imidazolate frameworks with high space-time yield. Sci. China Mater. 2017. [Google Scholar] [CrossRef]
- Lu, F.; Liu, S.; Zeng, G.; Li, J.; Yang, Y. Probing the Electrochemical Reaction Mechanism and Crystallinity Effect of RuO2 for Sodium Storage. J. Electrochem. Soc. 2018. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Q.; Huang, M.; Zhang, X.; Ouyang, X. Chemisorption of metallic radionuclides on a monolayer MoS2 nanosheet. Nanoscale Adv. 2019, 20–22. [Google Scholar] [CrossRef]
- Min, X.; Wu, X.; Shao, P.; Ren, Z.; Ding, L.; Luo, X. Ultra-high capacity of lanthanum-doped UiO-66 for phosphate capture: Unusual doping of lanthanum by the reduction of coordination number. Chem. Eng. J. 2019. [Google Scholar] [CrossRef]
- Zhou, Y.; Cao, S.; Wang, J.; Zhu, H.; Yang, S.; Wang, X.; Kong, D. Bright Stretchable Electroluminescent Devices based on Silver Nanowire Electrodes and High-k Thermoplastic Elastomers. ACS Appl. Mater. Interfaces 2018, 10, 44760–44767. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Q.; Huang, M.; Ouyang, X. Adsorption of hazardous gases in nuclear islands on monolayer MoS2 sheet. Adsorption 2019. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, H.; Liang, J.; Feng, D.; Tang, Q.; Yu, K.; Shang, Z. Effect of tourmaline addition on the structure of silica hollow microspheres prepared by a novel template method. J. Alloys Compd. 2017, 693, 1323–1327. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, H.; Liang, J.; Tang, Q.; Li, Y.; Shang, Z. High emission reduction performance of a novel organic-inorganic composite filters containing sepiolite mineral nanofibers. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Element | H-Metal Bond Length (with 1H) | H-Metal Bond Length (with 2H) a | H-Metal Bond Length (with 3H) a | H-Metal Bond Length (with 4H) a | Number of Favorable Adsorbed H |
|---|---|---|---|---|---|
| Ir | 1.597 | 1.604 | 1.591 | 1.589 | 3 |
| Os | 1.660 | 1.648 | 1.636 | 1.627 | 2 |
| Pd | 1.571 | 1.552 | 1.653 | 1.689 | 2 |
| Pt | 1.575 | 1.563 | 1.595 | 1.596 | 2 |
| Re | 1.736 | 1.700 | 1.641 | 1.670 | 1 |
| Rh | 1.585 | 1.579 | 1.631 | 1.613 | 1 |
| Ru | 1.631 | 1.620 | 1.615 | 1.641 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zhang, Z.; Liu, Z. Non-Monotonic Trends of Hydrogen Adsorption on Single Atom Doped g-C3N4. Catalysts 2019, 9, 84. https://doi.org/10.3390/catal9010084
Li H, Zhang Z, Liu Z. Non-Monotonic Trends of Hydrogen Adsorption on Single Atom Doped g-C3N4. Catalysts. 2019; 9(1):84. https://doi.org/10.3390/catal9010084
Chicago/Turabian StyleLi, Hao, Zhien Zhang, and Zhijian Liu. 2019. "Non-Monotonic Trends of Hydrogen Adsorption on Single Atom Doped g-C3N4" Catalysts 9, no. 1: 84. https://doi.org/10.3390/catal9010084
APA StyleLi, H., Zhang, Z., & Liu, Z. (2019). Non-Monotonic Trends of Hydrogen Adsorption on Single Atom Doped g-C3N4. Catalysts, 9(1), 84. https://doi.org/10.3390/catal9010084