Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals


Abstract

1. Introduction
2. Lignin Chemistry
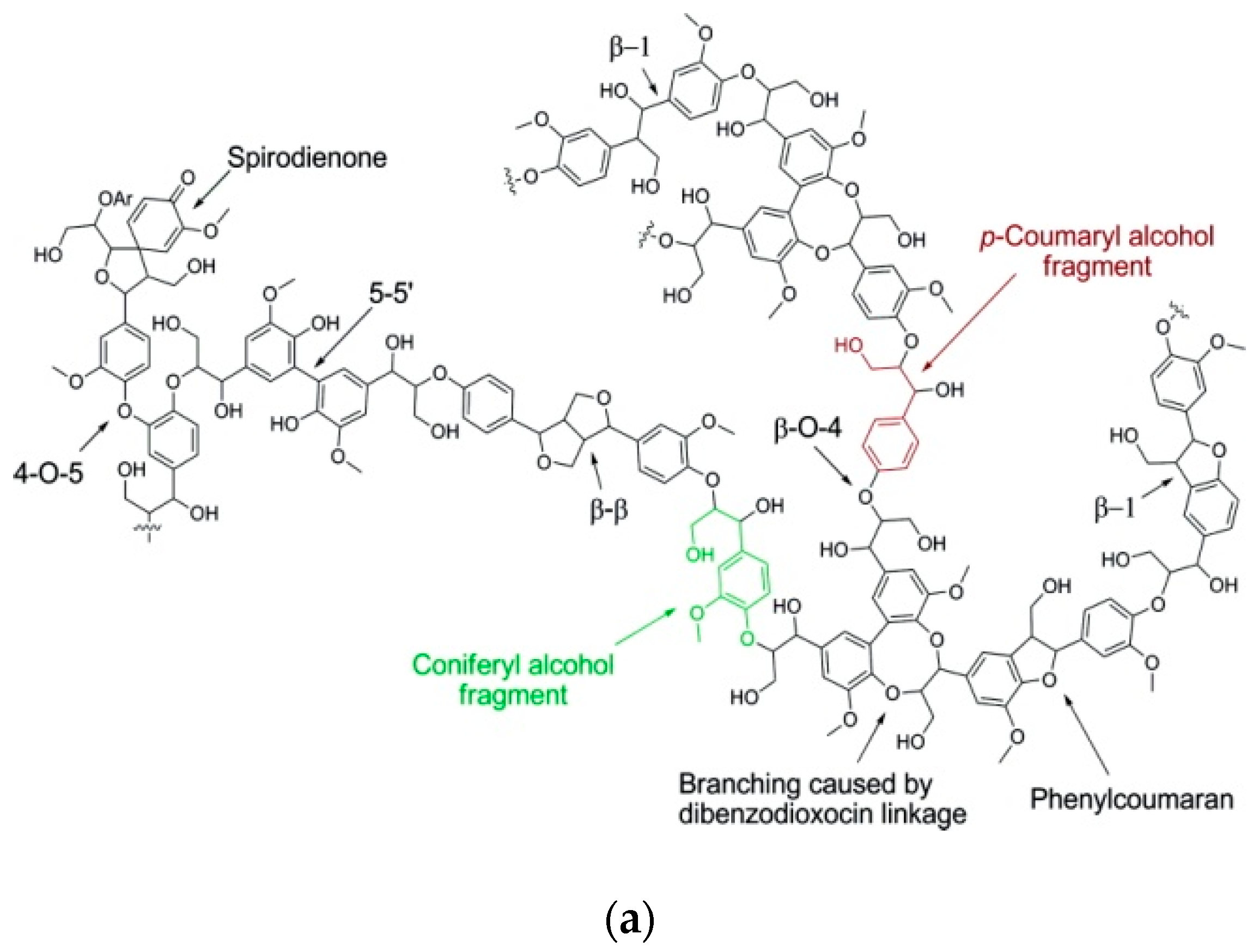
2.1. Lignin Structure and Isolation
2.2. Lignin Valorization
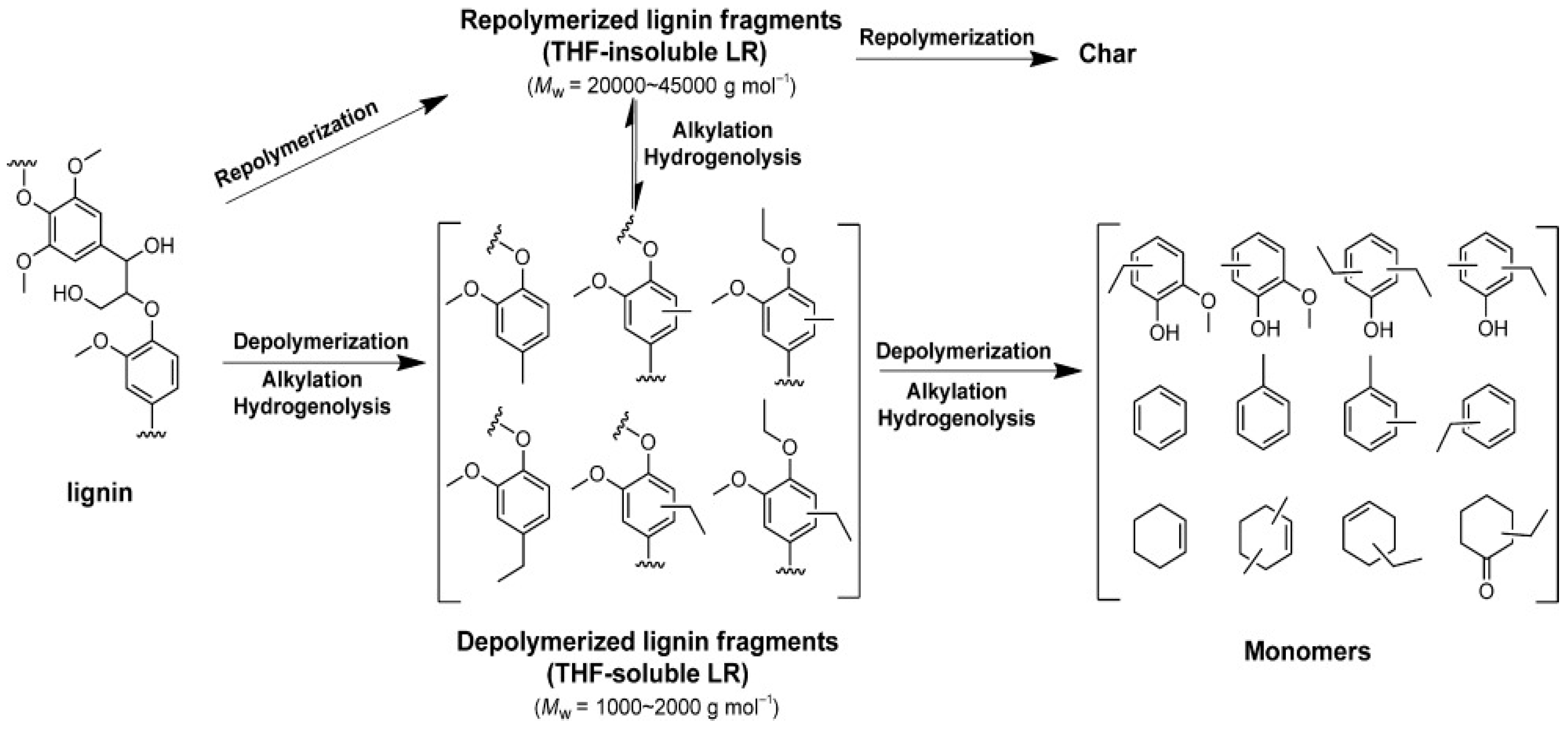
3. Reductive Depolymerization
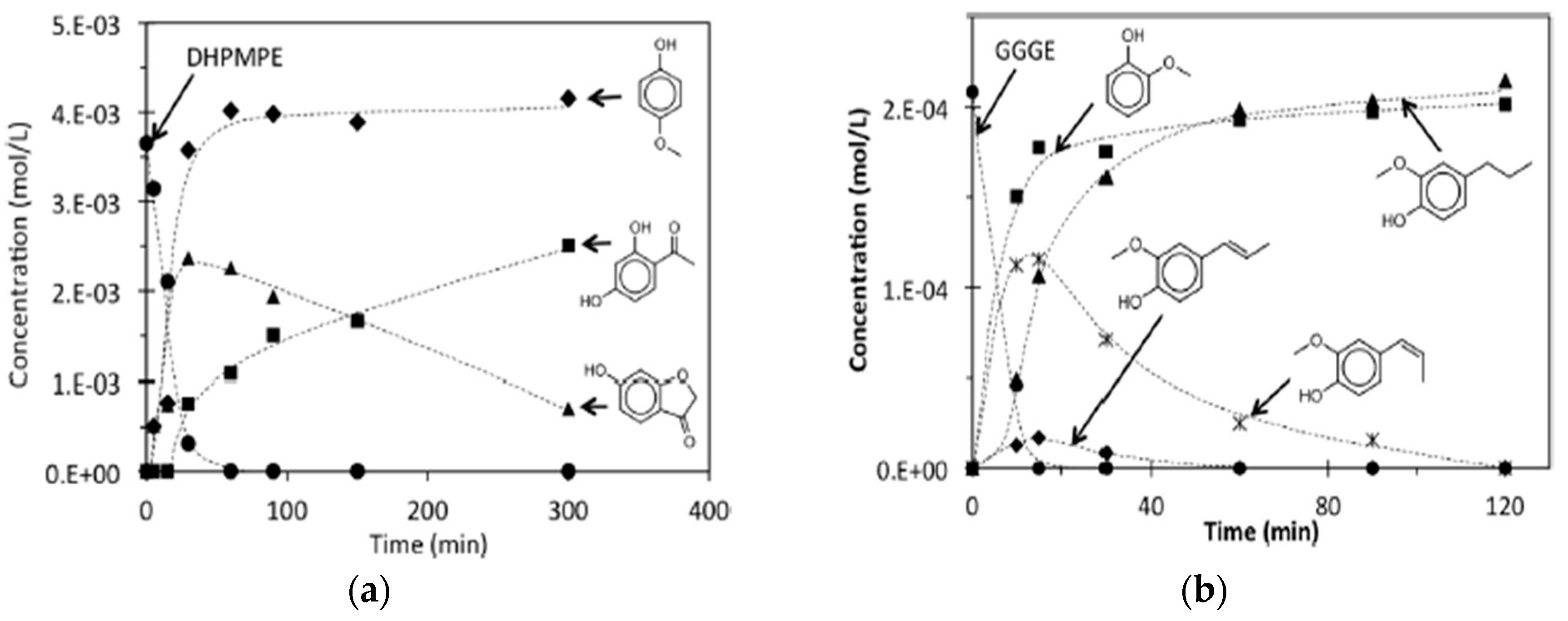
3.1. Reductive Depolymerization of Lignin Model Compounds
3.2. Lignin-First Strategy
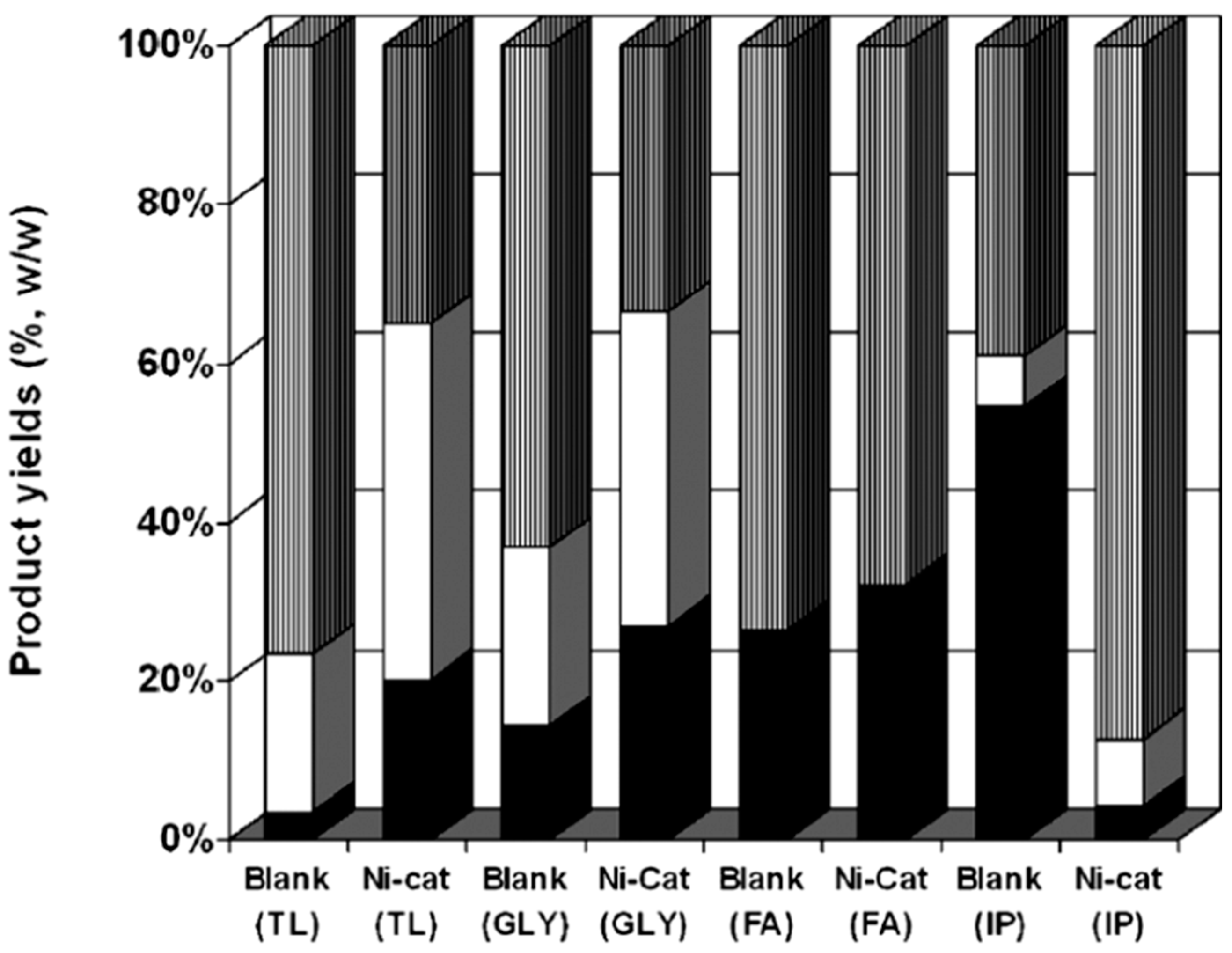
3.3. Depolymerization with External Hydrogen Source
4. Catalytic Hydrogenolysis of Lignins Using Hydrogen Donors
4.1. Kraft Lignins
4.2. Soda Lignins
4.3. Alkali Lignins
4.4. Organosolv Lignins
4.5. Lignosulfonate
4.6. Enzymatic and Acid Hydrolysis Lignins
4.7. Lignins Extracted by Deep Eutectic Solvents
4.8. Selection and Design Criteria for an Effective Catalyst in Reductive Depolymerization of Lignin Using Hydrogen Donors
5. Conclusions and Outlook
Acknowledgements
Conflicts of Interest
References
- European Parliament and the Council, Directive 2009/28/EC. 2009. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex%3A32009L0028 (accessed on 21 December 2018).
- Perlack, R.D.; Wright, L.L.; Turhollow, A.F.; Graham, R.L.; Stokes, B.J.; Erbach, D.C. Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion-Ton Annual Supply; U.S. Department of Energy: Oak Ridge National Laboratory, Oak Ridge, TN, USA, 2005.
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Samec, J.S. Lignin Valorization through Catalytic Lignocellulose Fractionation: A Fundamental Platform for the Future Biorefinery. ChemSusChem 2016, 9, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.S.; Inoue, H.; Endo, T.; Yano, S.; Bon, E.P. Milling pretreatment of sugarcane bagasse and straw for enzymatic hydrolysis and ethanol fermentation. Bioresour. Technol. 2010, 101, 7402–7409. [Google Scholar] [CrossRef]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Mayer-Laigle, C.; Solhy, A.; Arancon, R.A.D.; de Vries, H.; Luque, R. Mechanical pretreatments of lignocellulosic biomass: Towards facile and environmentally sound technologies for biofuels production. RSC Adv. 2014, 4, 48109–48127. [Google Scholar] [CrossRef]
- Gonzales, R.R.; Sivagurunathan, P.; Kim, S.H. Effect of severity on dilute acid pretreatment of lignocellulosic biomass and the following hydrogen fermentation. Int. J. Hydrogen Energy 2016, 41, 21678–21684. [Google Scholar] [CrossRef]
- Xu, J.K.; Sun, R.C. Chapter 19—Recent Advances in Alkaline Pretreatment of Lignocellulosic Biomass. In Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery; Mussatto, S.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 431–459. [Google Scholar]
- Garcia-Cubero, M.A.; Gonzalez-Benito, G.; Indacoechea, I.; Coca, M.; Bolado, S. Effect of ozonolysis pretreatment on enzymatic digestibility of wheat and rye straw. Bioresour. Technol. 2009, 100, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Nitsos, C.; Stoklosa, R.; Karnaouri, A.; Vörös, D.; Lange, H.; Hodge, D.; Crestini, C.; Rova, U.; Christakopoulos, P. Isolation and Characterization of Organosolv and Alkaline Lignins from Hardwood and Softwood Biomass. ACS Sustain. Chem. Eng. 2016, 4, 5181–5193. [Google Scholar] [CrossRef]
- Dale, B.E.; Leong, C.K.; Pham, T.K.; Esquivel, V.M.; Rios, I.; Latimer, V.M. Hydrolysis of lignocellulosics at low enzyme levels: Application of the AFEX process. Bioresour. Technol. 1996, 56, 111–116. [Google Scholar] [CrossRef]
- Duque, A.; Manzanares, P.; Ballesteros, I.; Ballesteros, M. Chapter 15—Steam Explosion as Lignocellulosic Biomass Pretreatment. In Biomass Fractionation Technologies for a Lignocellulosic Feedstock Based Biorefinery; Mussatto, S.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 349–368. [Google Scholar]
- Tang, Y.; Chandra, R.P.; Sokhansanj, S.; Saddler, J.N. Influence of steam explosion processes on the durability and enzymatic digestibility of wood pellets. Fuel 2018, 211, 87–94. [Google Scholar] [CrossRef]
- Schmidt, A.S.; Thomsen, A.B. Optimization of wet oxidation pretreatment of wheat straw. Bioresour. Technol. 1998, 64, 139–151. [Google Scholar] [CrossRef]
- Nitsos, C.K.; Matis, K.A.; Triantafyllidis, K.S. Optimization of hydrothermal pretreatment of lignocellulosic biomass in the bioethanol production process. ChemSusChem 2013, 6, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Foston, M.; Ragauskas, A.J. Comparative studies on hydrothermal pretreatment and enzymatic saccharification of leaves and internodes of alamo switchgrass. Bioresour. Technol. 2011, 102, 7224–7228. [Google Scholar] [CrossRef] [PubMed]
- Oksman, K.; Aitomäki, Y.; Mathew, A.P.; Siqueira, G.; Zhou, Q.; Butylina, S.; Tanpichai, S.; Zhou, X.; Hooshmand, S. Review of the recent developments in cellulose nanocomposite processing. Compos. Part A Appl. Sci. Manuf. 2016, 83, 2–18. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Liu, X.; Lin, Q.; Yan, Y.; Peng, F.; Sun, R.; Ren, J. Hemicellulose from Plant Biomass in Medical and Pharmaceutical Application: A Critical Review. Curr. Med. Chem. 2017, 24, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Espro, C.; Gumina, B.; Szumelda, T.; Paone, E.; Mauriello, F. Catalytic Transfer Hydrogenolysis as an Effective Tool for the Reductive Upgrading of Cellulose, Hemicellulose, Lignin, and Their Derived Molecules. Catalysts 2018, 8, 313. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef]
- Zhou, C.H.; Xia, X.; Lin, C.X.; Tong, D.S.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588–5617. [Google Scholar] [CrossRef]
- Lappas, A.A.; Kalogiannis, K.G.; Iliopoulou, E.F.; Triantafyllidis, K.S.; Stefanidis, S.D. Catalytic pyrolysis of biomass for transportation fuels. Wires Energy Environ. 2012, 1, 285–297. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Pineda, A.; Balu, A.M.; Luque, R.; Campelo, J.M.; Romero, A.A.; Ramos-Fernandez, J.M. Catalytic transformations of biomass-derived acids into advanced biofuels. Catal. Today 2012, 195, 162–168. [Google Scholar] [CrossRef]
- Espro, C.; Gumina, B.; Paone, E.; Mauriello, F. Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts. Catalysts 2017, 7, 78. [Google Scholar] [CrossRef]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. 2016, 55, 8164–8215. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crop. Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Dorrestijn, E.; Laarhoven, L.J.J.; Arends, I.W.C.E.; Mulder, P. The occurrence and reactivity of phenoxyl linkages in lignin and low rank coal. J. Anal. Appl. Pyrol. 2000, 54, 153–192. [Google Scholar] [CrossRef]
- Gillet, S.; Aguedo, M.; Petitjean, L.; Morais, A.R.C.; Lopes, A.M.D.; Lukasik, R.M.; Anastas, P.T. Lignin transformations for high value applications: Towards targeted modifications using green chemistry. Green Chem. 2017, 19, 4200–4233. [Google Scholar] [CrossRef]
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Lora, J. Chapter 10—Industrial Commercial Lignins: Sources, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 225–241. [Google Scholar] [CrossRef]
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lei, F.; Li, P.; Jiang, J. Lignocellulosic biomass to biofuels and biochemicals: A comprehensive review with a focus on ethanol organosolv pretreatment technology. Biotechnol. Bioeng. 2018, 115, 2683–2702. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Harrison, M.D.; Rackemann, D.W.; Doherty, W.O.S.; O’Hara, I.M. Organosolv pretreatment of plant biomass for enhanced enzymatic saccharification. Green Chem. 2016, 18, 360–381. [Google Scholar] [CrossRef]
- Villaverde, J.; Ligero, P.; Vega, A. Formic and acetic acid as agents for a cleaner fractionation of Miscanthus x giganteus. J. Clean. Prod. 2010, 18, 395–401. [Google Scholar] [CrossRef]
- Katahira, R.; Mittal, A.; McKinney, K.; Ciesielski, P.N.; Donohoe, B.S.; Black, S.K.; Johnson, D.K.; Biddy, M.J.; Beckham, G.T. Evaluation of Clean Fractionation Pretreatment for the Production of Renewable Fuels and Chemicals from Corn Stover. ACS Sustain. Chem. Eng. 2014, 2, 1364–1376. [Google Scholar] [CrossRef]
- Smit, A.; Huijgen, W. Effective fractionation of lignocellulose in herbaceous biomass and hardwood using a mild acetone organosolv process. Green Chem. 2017, 19, 5505–5514. [Google Scholar] [CrossRef]
- Pan, X.; Gilkes, N.; Kadla, J.; Pye, K.; Saka, S.; Gregg, D.; Ehara, K.; Xie, D.; Lam, D.; Saddler, J. Bioconversion of hybrid poplar to ethanol and co-products using an organosolv fractionation process: Optimization of process yields. Biotechnol. Bioeng. 2006, 94, 851–861. [Google Scholar] [CrossRef]
- Shuai, L.; Amiri, M.T.; Questell-Santiago, Y.M.; Heroguel, F.; Li, Y.; Kim, H.; Meilan, R.; Chapple, C.; Ralph, J.; Luterbacher, J.S. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 2016, 354, 329–333. [Google Scholar] [CrossRef]
- Raghavendran, V.; Nitsos, C.; Matsakas, L.; Rova, U.; Christakopoulos, P.; Olsson, L. A comparative study of the enzymatic hydrolysis of batch organosolv-pretreated birch and spruce biomass. AMB Express 2018, 8, 114. [Google Scholar] [CrossRef]
- Matsakas, L.; Nitsos, C.; Raghavendran, V.; Yakimenko, O.; Persson, G.; Olsson, E.; Rova, U.; Olsson, L.; Christakopoulos, P. A novel hybrid organosolv: Steam explosion method for the efficient fractionation and pretreatment of birch biomass. Biotechnol. Biofuels 2018, 11, 160. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Jiang, H.; Yu, H.Q. Thermochemical conversion of lignin to functional materials: A review and future directions. Green Chem. 2015, 17, 4888–4907. [Google Scholar] [CrossRef]
- Bjørsvik, H.-R.; Liguori, L. Organic Processes to Pharmaceutical Chemicals Based on Fine Chemicals from Lignosulfonates. Org. Process. Res. Dev. 2002, 6, 279–290. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Koelewijn, S.F.; Renders, T.; Van den Bossche, G.; Vangeel, T.; Schutyser, W.; Sels, B.F. Catalytic Strategies Towards Lignin-Derived Chemicals. Top. Curr. Chem. 2018, 376, 36. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, P.A.; Fotopoulos, A.P.; Karakoulia, S.A.; Triantafyllidis, K.S. Catalytic Fast Pyrolysis of Kraft Lignin With Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. [Google Scholar] [CrossRef] [PubMed]
- Custodis, V.B.F.; Karakoulia, S.A.; Triantafyllidis, K.S.; van Bokhoven, J.A. Catalytic Fast Pyrolysis of Lignin over High-Surface-Area Mesoporous Aluminosilicates: Effect of Porosity and Acidity. ChemSusChem 2016, 9, 1134–1145. [Google Scholar] [CrossRef]
- Guvenatam, B.; Heeres, E.H.J.; Pidko, E.A.; Hensen, E.J.M. Lewis-acid catalyzed depolymerization of Protobind lignin in supercritical water and ethanol. Catal. Today 2016, 259, 460–466. [Google Scholar] [CrossRef]
- Wang, H.; Tucker, M.; Ji, Y. Recent Development in Chemical Depolymerization of Lignin: A Review. J. Appl. Chem. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Hydrolytic depolymerization of hydrolysis lignin: Effects of catalysts and solvents. Bioresour. Technol. 2015, 190, 416–419. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Labidi, J. Organosolv lignin depolymerization with different base catalysts. J. Chem. Technol. Biotechol. 2012, 87, 1593–1599. [Google Scholar] [CrossRef]
- Abdelaziz, O.Y.; Li, K.; Tunå, P.; Hulteberg, C.P. Continuous catalytic depolymerisation and conversion of industrial kraft lignin into low-molecular-weight aromatics. Biomass Convers. Biorefinery 2017, 8, 455–470. [Google Scholar] [CrossRef]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.; Ahmad, M.; Hardiman, E.M.; Rahmanpour, R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011, 28, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Sena-Martins, G.; Almeida-Vara, E.; Duarte, J.C. Eco-friendly new products from enzymatically modified industrial lignins. Ind. Crops Prod. 2008, 27, 189–195. [Google Scholar] [CrossRef]
- Kumar, C.R.; Anand, N.; Kloekhorst, A.; Cannilla, C.; Bonura, G.; Frusteri, F.; Barta, K.; Heeres, H.J. Solvent free depolymerization of Kraft lignin to alkyl-phenolics using supported NiMo and CoMo catalysts. Green Chem. 2015, 17, 4921–4930. [Google Scholar] [CrossRef]
- Kloekhorst, A.; Heeres, H.J. Catalytic Hydrotreatment of Alcell Lignin Using Supported Ru, Pd, and Cu Catalysts. ACS Sustain. Chem. Eng. 2015, 3, 1905–1914. [Google Scholar] [CrossRef]
- Zhu, G.D.; Ouyang, X.P.; Jiang, L.F.; Zhu, Y.; Jin, D.X.; Pang, Y.X.; Qiu, X.Q. Effect of functional groups on hydrogenolysis of lignin model compounds. Fuel Process. Technol. 2016, 154, 132–138. [Google Scholar] [CrossRef]
- Zhang, J.G.; Asakura, H.; van Rijn, J.; Yang, J.; Duchesne, P.; Zhang, B.; Chen, X.; Zhang, P.; Saeys, M.; Yan, N. Highly efficient, NiAu-catalyzed hydrogenolysis of lignin into phenolic chemicals. Green Chem. 2014, 16, 2432–2437. [Google Scholar] [CrossRef]
- Zhang, J.; Teo, J.; Chen, X.; Asakura, H.; Tanaka, T.; Teramura, K.; Yan, N. A Series of NiM (M = Ru, Rh, and Pd) Bimetallic Catalysts for Effective Lignin Hydrogenolysis in Water. ACS Catal. 2014, 4, 1574–1583. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Cai, Y.; Lu, G.-P.; Cai, C. Facile and selective hydrogenolysis of β-O-4 linkages in lignin catalyzed by Pd–Ni bimetallic nanoparticles supported on ZrO2. Green Chem. 2016, 18, 6229–6235. [Google Scholar] [CrossRef]
- Mauriello, F.; Paone, E.; Pietropaolo, R.; Balu, A.M.; Luque, R. Catalytic Transfer Hydrogenolysis of Lignin-Derived Aromatic Ethers Promoted by Bimetallic Pd/Ni Systems. ACS Sustain. Chem. Eng. 2018, 6, 9269–9276. [Google Scholar] [CrossRef]
- Galkin, M.V.; Sawadjoon, S.; Rohde, V.; Dawange, M.; Samec, J.S.M. Mild Heterogeneous Palladium-Catalyzed Cleavage of β-O-4′-Ether Linkages of Lignin Model Compounds and Native Lignin in Air. ChemCatChem 2014, 6, 179–184. [Google Scholar] [CrossRef]
- Galkin, M.V.; Dahlstrand, C.; Samec, J.S. Mild and Robust Redox-Neutral Pd/C-Catalyzed Lignol beta-O-4’ Bond Cleavage Through a Low-Energy-Barrier Pathway. ChemSusChem 2015, 8, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Besse, X.; Schuurman, Y.; Guilhaume, N. Reactivity of lignin model compounds through hydrogen transfer catalysis in ethanol/water mixtures. Appl. Catal. B Environ. 2017, 209, 265–272. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. Solvent effects on the hydrogenolysis of diphenyl ether with Raney nickel and their implications for the conversion of lignin. ChemSusChem 2012, 5, 1455–1466. [Google Scholar] [CrossRef]
- Macala, G.S.; Matson, T.D.; Johnson, C.L.; Lewis, R.S.; Iretskii, A.V.; Ford, P.C. Hydrogen transfer from supercritical methanol over a solid base catalyst: A model for lignin depolymerization. ChemSusChem 2009, 2, 215–217. [Google Scholar] [CrossRef]
- Subbotina, E.; Galkin, M.V.; Samec, J.S.M. Pd/C-Catalyzed Hydrogenolysis of Dibenzodioxocin Lignin Model Compounds Using Silanes and Water as Hydrogen Source. ACS Sustain. Chem. Eng. 2017, 5, 3726–3731. [Google Scholar] [CrossRef]
- Klein, I.; Saha, B.; Abu-Omar, M.M. Lignin depolymerization over Ni/C catalyst in methanol, a continuation: Effect of substrate and catalyst loading. Catal. Sci. Technol. 2015, 5, 3242–3245. [Google Scholar] [CrossRef]
- Galkin, M.V.; Smit, A.T.; Subbotina, E.; Artemenko, K.A.; Bergquist, J.; Huijgen, W.J.; Samec, J.S. Hydrogen-free catalytic fractionation of woody biomass. ChemSusChem 2016, 9, 3280–3287. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, S.; Schutyser, W.; Vanholme, R.; Driessen, T.; Koelewijn, S.F.; Renders, T.; De Meester, B.; Huijgen, W.J.J.; Dehaen, W.; Courtin, C.M.; et al. Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ. Sci. 2015, 8, 1748–1763. [Google Scholar] [CrossRef]
- Song, Q.; Wang, F.; Cai, J.; Wang, Y.; Zhang, J.; Yu, W.; Xu, J. Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process. Energy Environ. Sci. 2013, 6, 994. [Google Scholar] [CrossRef]
- Godard, H.P.; McCarthy, J.L.; Hibbert, H. Hydrogenation of wood. J. Am. Chem. Soc. 1940, 62, 988. [Google Scholar] [CrossRef]
- Yan, N.; Zhao, C.; Dyson, P.J.; Wang, C.; Liu, L.T.; Kou, Y. Selective degradation of wood lignin over noble-metal catalysts in a two-step process. ChemSusChem 2008, 1, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Torr, K.M.; van de Pas, D.J.; Cazeils, E.; Suckling, I.D. Mild hydrogenolysis of in-situ and isolated Pinus radiata lignins. Bioresour. Technol. 2011, 102, 7608–7611. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Samec, J.S. Selective route to 2-propenyl aryls directly from wood by a tandem organosolv and palladium-catalysed transfer hydrogenolysis. ChemSusChem 2014, 7, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Parsell, T.; Yohe, S.; Degenstein, J.; Jarrell, T.; Klein, I.; Gencer, E.; Hewetson, B.; Hurt, M.; Kim, J.I.; Choudhari, H.; et al. A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass. Green Chem. 2015, 17, 1492–1499. [Google Scholar] [CrossRef]
- Luo, H.; Klein, I.M.; Jiang, Y.; Zhu, H.Y.; Liu, B.Y.; Kenttamaa, H.I.; Abu-Omar, M.M. Total Utilization of Miscanthus Biomass, Lignin and Carbohydrates, Using Earth Abundant Nickel Catalyst. ACS Sustain. Chem. Eng. 2016, 4, 2316–2322. [Google Scholar] [CrossRef]
- Ferrini, P.; Rinaldi, R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem. 2014, 53, 8634–8639. [Google Scholar] [CrossRef]
- Van den Bosch, S.; Schutyser, W.; Koelewijn, S.F.; Renders, T.; Courtin, C.M.; Sels, B.F. Tuning the lignin oil OH-content with Ru and Pd catalysts during lignin hydrogenolysis on birch wood. Chem. Commun. 2015, 51, 13158–13161. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, S.; Renders, T.; Kennis, S.; Koelewijn, S.F.; Van den Bossche, G.; Vangeel, T.; Deneyer, A.; Depuydt, D.; Courtin, C.M.; Thevelein, J.M.; et al. Integrating lignin valorization and bio-ethanol production: On the role of Ni-Al2O3 catalyst pellets during lignin-first fractionation. Green Chem. 2017, 19, 3313–3326. [Google Scholar] [CrossRef]
- Schutyser, W.; Van den Bosch, S.; Renders, T.; De Boe, T.; Koelewijn, S.F.; Dewaele, A.; Ennaert, T.; Verkinderen, O.; Goderis, B.; Courtin, C.M.; et al. Influence of bio-based solvents on the catalytic reductive fractionation of birch wood. Green Chem. 2015, 17, 5035–5045. [Google Scholar] [CrossRef]
- Huang, X.; Ouyang, X.; Hendriks, B.M.S.; Gonzalez, O.M.M.; Zhu, J.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Selective production of mono-aromatics from lignocellulose over Pd/C catalyst: The influence of acid co-catalysts. Faraday Discuss. 2017, 202, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Renders, T.; Schutyser, W.; Van den Bosch, S.; Koelewijn, S.F.; Vangeel, T.; Courtin, C.M.; Sels, B.F. Influence of Acidic (H3PO4) and Alkaline (NaOH) Additives on the Catalytic Reductive Fractionation of Lignocellulose. ACS Catal. 2016, 6, 2055–2066. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Schutyser, W.; Sels, B.F. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energy Environ. Sci. 2017, 10, 1551–1557. [Google Scholar] [CrossRef]
- Shu, R.; Long, J.; Xu, Y.; Ma, L.; Zhang, Q.; Wang, T.; Wang, C.; Yuan, Z.; Wu, Q. Investigation on the structural effect of lignin during the hydrogenolysis process. Bioresour. Technol. 2016, 200, 14–22. [Google Scholar] [CrossRef]
- Shu, R.Y.; Xu, Y.; Ma, L.L.; Zhang, Q.; Wang, T.J.; Chen, P.R.; Wu, Q.Y. Hydrogenolysis process for lignosulfonate depolymerization using synergistic catalysts of noble metal and metal chloride. RSC Adv. 2016, 6, 88788–88796. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.; Hwang, H.; Kim, J.K.; Song, I.K.; Choi, J.W. Catalytic depolymerization of lignin macromolecule to alkylated phenols over various metal catalysts in supercritical tert-butanol. J. Anal. Appl. Pyrol. 2015, 113, 99–106. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.; Kim, U.J.; Choi, J.W. Conversion of Lignin to Phenol-Rich Oil Fraction under Supercritical Alcohols in the Presence of Metal Catalysts. Energy Fuels 2015, 29, 5154–5163. [Google Scholar] [CrossRef]
- Yuan, Z.S.; Tymchyshyn, M.; Xu, C.B. Reductive Depolymerization of Kraft and Organosolv Lignin in Supercritical Acetone for Chemicals and Materials. ChemCatChem 2016, 8, 1968–1976. [Google Scholar] [CrossRef]
- Lama, S.M.G.; Pampel, J.; Fellinger, T.P.; Beskoski, V.P.; Slavkovic-Beskoski, L.; Antonietti, M.; Molinari, V. Efficiency of Ni Nanoparticles Supported on Hierarchical Porous Nitrogen-Doped Carbon for Hydrogenolysis of Kraft Lignin in Flow and Batch Systems. ACS Sustain. Chem. Eng. 2017, 5, 2415–2420. [Google Scholar] [CrossRef]
- Park, J.; Oh, S.; Kim, J.Y.; Park, S.Y.; Song, I.K.; Choi, J.W. Comparison of degradation features of lignin to phenols over Pt catalysts prepared with various forms of carbon supports. RSC Adv. 2016, 6, 16917–16924. [Google Scholar] [CrossRef]
- Bouxin, F.P.; McVeigh, A.; Tran, F.; Westwood, N.J.; Jarvis, M.C.; Jackson, S.D. Catalytic depolymerisation of isolated lignins to fine chemicals using a Pt/alumina catalyst: Part 1—Impact of the lignin structure. Green Chem. 2015, 17, 1235–1242. [Google Scholar] [CrossRef]
- Barta, K.; Warner, G.R.; Beach, E.S.; Anastas, P.T. Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides. Green Chem. 2014, 16, 191–196. [Google Scholar] [CrossRef]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640–1647. [Google Scholar] [CrossRef]
- Narani, A.; Chowdari, R.K.; Cannilla, C.; Bonura, G.; Frusteri, F.; Heeres, H.J.; Barta, K. Efficient catalytic hydrotreatment of Kraft lignin to alkylphenolics using supported NiW and NiMo catalysts in supercritical methanol. Green Chem. 2015, 17, 5046–5057. [Google Scholar] [CrossRef]
- Verziu, M.; Tirsoaga, A.; Cojocaru, B.; Bucur, C.; Tudora, B.; Richel, A.; Aguedo, M.; Samikannu, A.; Mikkola, J.P. Hydrogenolysis of lignin over Ru-based catalysts: The role of the ruthenium in a lignin fragmentation process. Mol. Catal. 2018, 450, 65–76. [Google Scholar] [CrossRef]
- Konnerth, H.; Zhang, J.G.; Ma, D.; Prechtl, M.H.G.; Yan, N. Base promoted hydrogenolysis of lignin model compounds and organosolv lignin over metal catalysts in water. Chem. Eng. Sci. 2015, 123, 155–163. [Google Scholar] [CrossRef]
- Ma, X.L.; Tian, Y.; Hao, W.Y.; Ma, R.; Li, Y.D. Production of phenols from catalytic conversion of lignin over a tungsten phosphide catalyst. Appl. Catal. A Gen. 2014, 481, 64–70. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, S.Y.; Choi, I.G.; Choi, J.W. Evaluation of RuxNi1-x/SBA-15 catalysts for depolymerization features of lignin macromolecule into monomeric phenols. Chem. Eng. J. 2018, 336, 640–648. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, Q.; Shu, R.; Xu, Y.; Ma, L.; Wang, T. Catalytic depolymerization of the hydrolyzed lignin over mesoporous catalysts. Bioresour. Technol. 2017, 226, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.P.; Wang, S.Z.; Li, H.L.; Li, Z.W.; Shi, Z.J.; Xiao, L.; Sun, R.G.; Fang, Y.M.; Song, G.Y. Catalytic Hydrogenolysis of Lignins into Phenolic Compounds over Carbon Nanotube Supported Molybdenum Oxide. ACS Catal. 2017, 7, 7535–7542. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Wang, H.; Ma, Q.; Li, S.; Chang, H.M.; Jameel, H. Liquefaction of kraft lignin by hydrocracking with simultaneous use of a novel dual acid-base catalyst and a hydrogenation catalyst. Bioresour. Technol. 2017, 243, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Hao, W.; Ma, X.; Tian, Y.; Li, Y. Catalytic ethanolysis of Kraft lignin into high-value small-molecular chemicals over a nanostructured alpha-molybdenum carbide catalyst. Angew. Chem. Int. Ed. 2014, 53, 7310–7315. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Ma, R.; Hao, W.Y.; Chen, M.M.; Iran, F.; Cui, K.; Tian, Y.; Li, Y.D. Common Pathways in Ethanolysis of Kraft Lignin to Platform Chemicals over Molybdenum-Based Catalysts. ACS Catal. 2015, 5, 4803–4813. [Google Scholar] [CrossRef]
- Ma, X.; Cui, K.; Hao, W.; Ma, R.; Tian, Y.; Li, Y. Alumina supported molybdenum catalyst for lignin valorization: Effect of reduction temperature. Bioresour. Technol. 2015, 192, 17–22. [Google Scholar] [CrossRef]
- Singh, S.K.; Ekhe, J.D. Cu–Mo doped zeolite ZSM-5 catalyzed conversion of lignin to alkyl phenols with high selectivity. Catal. Sci. Technol. 2015, 5, 2117–2124. [Google Scholar] [CrossRef]
- Huang, S.H.; Mahmood, N.; Zhang, Y.S.; Tymchyshyn, M.; Yuan, Z.S.; Xu, C.B. Reductive de-polymerization of kraft lignin with formic acid at low temperatures using inexpensive supported Ni-based catalysts. Fuel 2017, 209, 579–586. [Google Scholar] [CrossRef]
- Huang, S.; Mahmood, N.; Tymchyshyn, M.; Yuan, Z.; Xu, C.C. Reductive de-polymerization of kraft lignin for chemicals and fuels using formic acid as an in-situ hydrogen source. Bioresour. Technol. 2014, 171, 95–102. [Google Scholar] [CrossRef]
- Liguori, L.; Barth, T. Palladium-Nafion SAC-13 catalysed depolymerisation of lignin to phenols in formic acid and water. J. Anal. Appl. Pyrol. 2011, 92, 477–484. [Google Scholar] [CrossRef]
- Limarta, S.O.; Ha, J.M.; Park, Y.K.; Lee, H.; Suh, D.J.; Jae, J. Efficient depolymerization of lignin in supercritical ethanol by a combination of metal and base catalysts. J. Ind. Eng. Chem. 2018, 57, 45–54. [Google Scholar] [CrossRef]
- Molinari, V.; Clavel, G.; Graglia, M.; Antonietti, M.; Esposito, D. Mild Continuous Hydrogenolysis of Kraft Lignin over Titanium Nitride–Nickel Catalyst. ACS Catal. 2016, 6, 1663–1670. [Google Scholar] [CrossRef]
- Luo, L.; Yang, J.; Yao, G.; Jin, F. Controlling the selectivity to chemicals from catalytic depolymerization of kraft lignin with in-situ H2. Bioresour. Technol. 2018, 264, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Nandeshwar, K.; Ekhe, J.D. Thermochemical lignin depolymerization and conversion to aromatics in subcritical methanol: Effects of catalytic conditions. New J. Chem. 2016, 40, 3677–3685. [Google Scholar] [CrossRef]
- Huang, X.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J. Catalytic depolymerization of lignin in supercritical ethanol. ChemSusChem 2014, 7, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atay, C.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Role of Cu–Mg–Al Mixed Oxide Catalysts in Lignin Depolymerization in Supercritical Ethanol. ACS Catal. 2015, 5, 7359–7370. [Google Scholar] [CrossRef]
- Huang, X.M.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef]
- Huang, X.; Atay, C.; Zhu, J.; Palstra, S.W.L.; Koranyi, T.I.; Boot, M.D.; Hensen, E.J.M. Catalytic Depolymerization of Lignin and Woody Biomass in Supercritical Ethanol: Influence of Reaction Temperature and Feedstock. ACS Sustain. Chem. Eng. 2017, 5, 10864–10874. [Google Scholar] [CrossRef]
- Jeong, S.; Yang, S.; Kim, D.H. Depolymerization of Protobind lignin to produce monoaromatic compounds over Cu/ZSM-5 catalyst in supercritical ethanol. Mol. Catal. 2017, 442, 140–146. [Google Scholar] [CrossRef]
- Zhou, M.H.; Sharma, B.K.; Liu, P.; Ye, J.; Xu, J.M.; Jiang, J.C. Catalytic in Situ Hydrogenolysis of Lignin in Supercritical Ethanol: Effect of Phenol, Catalysts, and Reaction Temperature. ACS Sustain. Chem. Eng. 2018, 6, 6867–6875. [Google Scholar] [CrossRef]
- Li, Z.; Bi, Z.; Yan, L. Two-step hydrogen transfer catalysis conversion of lignin to valuable small molecular compounds. Green Process. Synth. 2017, 6, 363–370. [Google Scholar] [CrossRef]
- Onwudili, J.A.; Williams, P.T. Catalytic depolymerization of alkali lignin in subcritical water: Influence of formic acid and Pd/C catalyst on the yields of liquid monomeric aromatic products. Green Chem. 2014, 16, 4740–4748. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Pineda, A.; Romero, A.A.; Luque, R.; Labidi, J. Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: Catalyst screening. Appl. Catal. B Envrion. 2014, 145, 43–55. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Labidi, J.; Pineda, A.; Balu, A.M.; Luque, R. Heterogeneously Catalysed Mild Hydrogenolytic Depolymerisation of Lignin Under Microwave Irradiation with Hydrogen-Donating Solvents. ChemCatChem 2013, 5, 977–985. [Google Scholar] [CrossRef]
- Xu, W.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid. ChemSusChem 2012, 5, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kloekhorst, A.; Shen, Y.; Yie, Y.; Fang, M.; Heeres, H.J. Catalytic hydrodeoxygenation and hydrocracking of Alcell® lignin in alcohol/formic acid mixtures using a Ru/C catalyst. Biomass Bioenergy 2015, 80, 147–161. [Google Scholar] [CrossRef]
- Park, J.; Riaz, A.; Insyani, R.; Kim, J. Understanding the relationship between the structure and depolymerization behavior of lignin. Fuel 2018, 217, 202–210. [Google Scholar] [CrossRef]
- Warner, G.; Hansen, T.S.; Riisager, A.; Beach, E.S.; Barta, K.; Anastas, P.T. Depolymerization of organosolv lignin using doped porous metal oxides in supercritical methanol. Bioresour. Technol. 2014, 161, 78–83. [Google Scholar] [CrossRef]
- Klamrassamee, T.; Laosiripojana, N.; Faungnawakij, K.; Moghaddam, L.; Zhang, Z.Y.; Doherty, W.O.S. Co- and Ca-phosphate-based catalysts for the depolymerization of organosolv eucalyptus lignin. RSC Adv. 2015, 5, 45618–45621. [Google Scholar] [CrossRef]
- Regmi, Y.N.; Mann, J.K.; McBride, J.R.; Tao, J.M.; Barnes, C.E.; Labbe, N.; Chmely, S.C. Catalytic transfer hydrogenolysis of organosolv lignin using B-containing FeNi alloyed catalysts. Catal. Today 2018, 302, 190–195. [Google Scholar] [CrossRef]
- Liu, S.; Lin, Z.; Cai, Z.; Long, J.; Li, Z.; Li, X. Selective depolymerization of lignosulfonate via hydrogen transfer enhanced in an emulsion microreactor. Bioresour. Technol. 2018, 264, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Oregui-Bengoechea, M.; Gandarias, I.; Arias, P.L.; Barth, T. Unraveling the Role of Formic Acid and the Type of Solvent in the Catalytic Conversion of Lignin: A Holistic Approach. ChemSusChem 2017, 10, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Kristianto, I.; Limarta, S.O.; Lee, H.; Ha, J.M.; Suh, D.J.; Jae, J. Effective depolymerization of concentrated acid hydrolysis lignin using a carbon-supported ruthenium catalyst in ethanol/formic acid media. Bioresour. Technol. 2017, 234, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.T.; Li, Z.; Tang, X.; Sun, Y.; Zeng, X.H.; Liu, S.J.; Lin, L. Depolymerization of Cellulolytic Enzyme Lignin for the Production of Monomeric Phenols over Raney Ni and Acidic Zeolite Catalysts. Energy Fuels 2015, 29, 1662–1668. [Google Scholar] [CrossRef]
- Das, L.; Li, M.; Stevens, J.; Li, W.Q.; Pu, Y.Q.; Ragauskas, A.J.; Shi, J. Characterization and Catalytic Transfer Hydrogenolysis of Deep Eutectic Solvent Extracted Sorghum Lignin to Phenolic Compounds. ACS Sustain. Chem. Eng. 2018, 6, 10408–10420. [Google Scholar] [CrossRef]
- Seo, M.; Yoon, D.; Hwang, K.S.; Kang, J.W.; Kim, J. Supercritical alcohols as solvents and reducing agents for the synthesis of reduced graphene oxide. Carbon 2013, 64, 207–218. [Google Scholar] [CrossRef]
- Tudorache, M.; Opris, C.; Cojocaru, B.; Apostol, N.G.; Tirsoaga, A.; Coman, S.M.; Parvulescu, V.I.; Duraki, B.; Krumeich, F.; van Bokhoven, J.A. Highly Efficient, Easily Recoverable, and Recyclable Re–SiO2–Fe3O4 Catalyst for the Fragmentation of Lignin. ACS Sustain. Chem. Eng. 2018, 6, 9606–9618. [Google Scholar] [CrossRef]
- Opris, C.; Cojocaru, B.; Gheorghe, N.; Tudorache, M.; Coman, S.M.; Parvulescu, V.I.; Duraki, B.; Krumeich, F.; van Bokhoven, J.A. Lignin fragmentation over magnetically recyclable composite Co@Nb2O5@Fe3O4 catalysts. J. Catal. 2016, 339, 209–227. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Agent | T (°C) | MW (Da) | Polydispersity | Contamination |
|---|---|---|---|---|---|
| Kraft | NaOH + Na2S | 170 | 1000–3000 | 2.5–3.5 | Sulfur |
| Soda | NaOH + anthraquinone | 140–170 | 1000–3000 | 2.5–3.5 | Sulfur |
| Sulfite | sulfite salts | 140–170 | 1000–50,000 | 4.2–7.0 | Sulfur |
| Organosolv | organic solvents | 180–200 | 500–5000 | 1.5 | Sulfur free |
| Catalyst | Product Yield (wt.%) | Molecular Weight of Bio-Oil (g/mol) | Monomer Yield (wt.%) | ||||
|---|---|---|---|---|---|---|---|
| Bio-Oil | SR | Mn (g/mol) | Mw (g/mol) | Total | Phenolic Monomers | Aliphatic Esters | |
| No catalyst | 36.2 | 47.3 | 347 | 737 | 1.75 | 1.70 | 0.04 |
| MgO/C 10% a | 43.3 | 37.3 | 370 | 834 | 2.10 | 1.83 | 0.26 |
| MgO/Al2O3 10% | 42.1 | 37.5 | 383 | 844 | 1.98 | 1.76 | 0.23 |
| MgO/ZrO2 10% | 47.5 | 32.7 | 365 | 776 | 4.22 | 3.90 | 0.32 |
| Ru/C 10% | 88.1 | 8.8 | 398 | 906 | 5.05 | 4.54 | 0.51 |
| Ru/C 10% + MgO/C 10% | 79.2 | 10.0 | 371 | 944 | 5.73 | 5.15 | 0.58 |
| Ru/C 10% + MgO/Al2O3 10% | 70.9 | 21.0 | 434 | 1064 | 4.52 | 3.76 | 0.77 |
| Ru/C 10% + MgO/ZrO2 10% | 82.7 | 11.3 | 323 | 832 | 6.10 | 5.16 | 0.94 |
| Sample | Hold Time (h) | Liquid (wt.%) | Solid Residue (wt.%) | Gas (wt.%) | % Balance |
|---|---|---|---|---|---|
| Lignin | 1.0 | 58.2 | 30.6 | 5.56 | 94.4 |
| Lignin | 3.0 | 51.6 | 35.9 | 9.12 | 96.6 |
| Lignin | 6.0 | 33.3 | 46.0 | 17.0 | 96.3 |
| Lignin/FA | 1.0 | 61.6 | 0.64 | 36.5 | 98.7 |
| Lignin/FA | 3.0 | 52.3 | 0.19 | 46.1 | 98.6 |
| Lignin/FA | 6.0 | 41.1 | 1.12 | 58.3 | 101 |
| Lignin/Pd/C | 1.0 | 41.3 | 54.4 | 0.84 | 96.5 |
| Lignin/Pd/C | 3.0 | 36.1 | 57.3 | 1.25 | 94.7 |
| Lignin/Pd/C | 6.0 | 35.6 | 59.1 | 2.25 | 97.0 |
| Lignin/FA-Pd/C | 1.0 | 45.8 | 16.3 | 38.7 | 101 |
| Lignin/FA-Pd/C | 3.0 | 38.5 | 18.3 | 49.8 | 107 |
| Lignin/FA-Pd/C | 6.0 | 31.6 | 22.9 | 53.4 | 108 |
| Pd/C | 6.0 | - | 98.5 | - | 98.5 |
| Blank | Ni2% AlSBA | Ni5% AlSBA | Ni10% AlSBA | Pd2% AlSBA | Pt2% AlSBA | Ru2% AlSBA | |
|---|---|---|---|---|---|---|---|
| Mesitol (2) | 0.56 | 2.26 | 1.19 | 1.13 | 0.49 | 1.67 | 1.40 |
| 2,3,6-Trimehtylphenol (3) | 0.19 | 0.76 | 0.22 | 0.28 | 0.18 | 0.17 | 0.45 |
| 6-Ethyl-o-cresol (5) | 0.13 | 0.14 | 0.04 | - | - | 0.32 | 0.19 |
| 4-Ethyl-m-cresol (6) | - | 0.11 | 0.04 | - | - | 0.08 | - |
| Vanillin (7) | 0.20 | 0.20 | 0.19 | 0.20 | - | 0.13 | 0.11 |
| Diethylphthalate (10) | 2.34 | 10.10 | 1.67 | 1.69 | 10.93 | 10.66 | 7.44 |
| 3,4-Dimethoxyphenol (11) | 0.05 | 0.12 | 0.05 | 0.15 | 0.24 | 0.09 | 0.09 |
| Syringaldehyde (12) | 0.47 | 0.68 | 0.57 | 0.56 | 0.15 | 0.43 | 0.43 |
| Butyl-octyl ester phthalic acid (16) | 0.21 | 0.36 | 0.30 | 0.19 | 0.34 | 0.36 | 0.37 |
| Solvent (mL) | Yield of Phenolic Monomers (mg∙g−1) | ||||
|---|---|---|---|---|---|
| H2O | n-BuOH | i-PrOH | 4-Ethyl Guaiacol | Others | Total |
| 25 | / | / | 18.8 ± 0.3 | 37.3 ± 0.5 | 56.1 ± 0.7 |
| 20 | 5.0 | / | 22.0 ± 0.4 | 39.8 ± 0.6 | 61.8 ± 0.8 |
| 15 | 10 | / | 23.5 ± 0.2 | 41.9 ± 0.5 | 65.4 ± 0.7 |
| 10 | 15 | / | 22.3 ± 0.3 | 40.2 ± 0.6 | 62.5 ± 0.8 |
| 20 | / | 5.0 | 21.3 ± 0.5 | 32.1 ± 0.6 | 53.4 ± 1.1 |
| 15 | 5.0 | 5.0 | 24.8 ± 0.4 | 50.2 ± 1.9 | 75.0 ± 2.2 |
| 10 | 5.0 | 10 | 33.8 ± 0.3 | 50.3 ± 2.5 | 84.1 ± 2.8 |
| 10 | 10 | 5.0 | 27.7 ± 0.1 | 52.8 ± 3.0 | 80.5 ± 3.0 |
| 5.0 | 15 | 5.0 | 27.2 ± 0.5 | 55.5 ± 2.4 | 82.7 ± 2.8 |
| 5.0 | 10 | 10 | 29.9 ± 0.6 | 65.5 ± 3.1 | 95.4 ± 3.5 |
| 5.0 | 5.0 | 15 | 39.3 ± 0.7 | 76.8 ± 2.3 | 116.1 ± 2.8 |
| Lignin | Catalyst & Reaction Conditions | Conversion/Yields a (wt.%) | Main Products | Reference |
|---|---|---|---|---|
| Kraft | α-MoC1−x/AC, EtOH, 280 °C | 100/-/- | Esters, alcohols, aromatics | [110] |
| Kraft | Cu/Mo-ZSM-5, H2O:MeOH=1:1, 220 °C, NaOH | 95.7/-/19.9 | Phenolics | [113] |
| Kraft | 10% Ni/Zeolite, H2O:EtOH=1:1, HCOOH, 200 °C | 93.5 b | Aromatics | [114] |
| Kraft | Pd-Nafion SAC-13, H2O, HCOOH, 300 °C | -/-/10.5 | Phenolics | [116] |
| Kraft | MgO/ZrO2, EtOH, 350 °C | -/47.5/4.22 | Phenolics | [117] |
| Kraft | Ru/C+ MgO/ZrO2, EtOH, 350 °C | -/82.7/6.10 | Phenolics, higher alcohols, aliphatic esters | [117] |
| Kraft | HZSM-5, MeOH, 220 °C | 85.1/-/4.2 | Phenolics | [120] |
| Kraft | Iron turnings, MeOH, 220 °C | 44.4/-/1.7 | Phenolics | [120] |
| Soda | CuMgAlOx, EtOH, 300 °C | -/-/17 | Aromatics, furans, hydrogenated cyclics | [121,122,123,124] |
| Soda | 10 wt.% Cu/ZSM-5(30), EtOH, 440 °C | -/-/98.2 | Aromatics | [125] |
| Alkali | CuNiAl-HT, EtOH, Phenol:lignin=0.8, 290 °C | -/72.3/- | - | [126] |
| Alkali | Raney Ni, i-PrOH/H2O, 180 °C | 93.5/-/- | Phenolics-aromatics | [127] |
| Alkali | Pd/C, H2O, HCOOH, 265 °C | -/45.8/- | Phenolics | [128] |
| Organosolv | 10% Ni/Al-SBA-15, Tetralin, 140 °C c | -/16.94/- | Phenolics, esters | [129] |
| Organosolv | 10% Ni/Al-SBA-15, HCOOH, 150 °C c | -/28.89/- | Phenolics | [130] |
| Organosolv | 20 wt.% Pt/C, EtOH, HCOOH, 350 °C | -/-/21 | Phenolics | [131] |
| Organosolv | Ru/C, i-PrOH, HCOOH, 400 °C | -/71.2/- | ketones aromatics, phenolics | [132] |
| Organosolv | Cu20PMO, MeOH, 310 °C | 48.3/-/- | Phenolics, aromatics | [134] |
| Organosolv | FeNiB alloys, EtOH, 320 °C | -/-/- | Phenolics | [136] |
| Enzymatic hydrolysis | NiMo/sulfated alumina, EtOH, HCOOH, 320 °C | -/38.4/- | methoxy, hydroxyl, alkyl benzenes | [138] |
| Acid hydrolysis | 5% Ru/C, EtOH, HCOOH, 300 °C | -/62.9/- | Phenolics, esters | [139] |
| Enzymatic hydrolysis | HUSY+Raney Ni, MeOH/H2O, 250 °C | -/-/27.9 | Phenolics | [140] |
| DES Extracted | Ru/C, i-PrOH, 270 °C | -/36.28/- | Phenolics, acids | [141] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margellou, A.; Triantafyllidis, K.S. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts 2019, 9, 43. https://doi.org/10.3390/catal9010043
Margellou A, Triantafyllidis KS. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts. 2019; 9(1):43. https://doi.org/10.3390/catal9010043
Chicago/Turabian StyleMargellou, Antigoni, and Konstantinos S. Triantafyllidis. 2019. "Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals" Catalysts 9, no. 1: 43. https://doi.org/10.3390/catal9010043
APA StyleMargellou, A., & Triantafyllidis, K. S. (2019). Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts, 9(1), 43. https://doi.org/10.3390/catal9010043