Computational Design of SCS Nickel Pincer Complexes for the Asymmetric Transfer Hydrogenation of 1-Acetonaphthone


Abstract
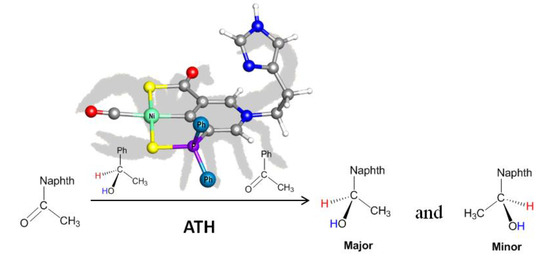
1. Introduction
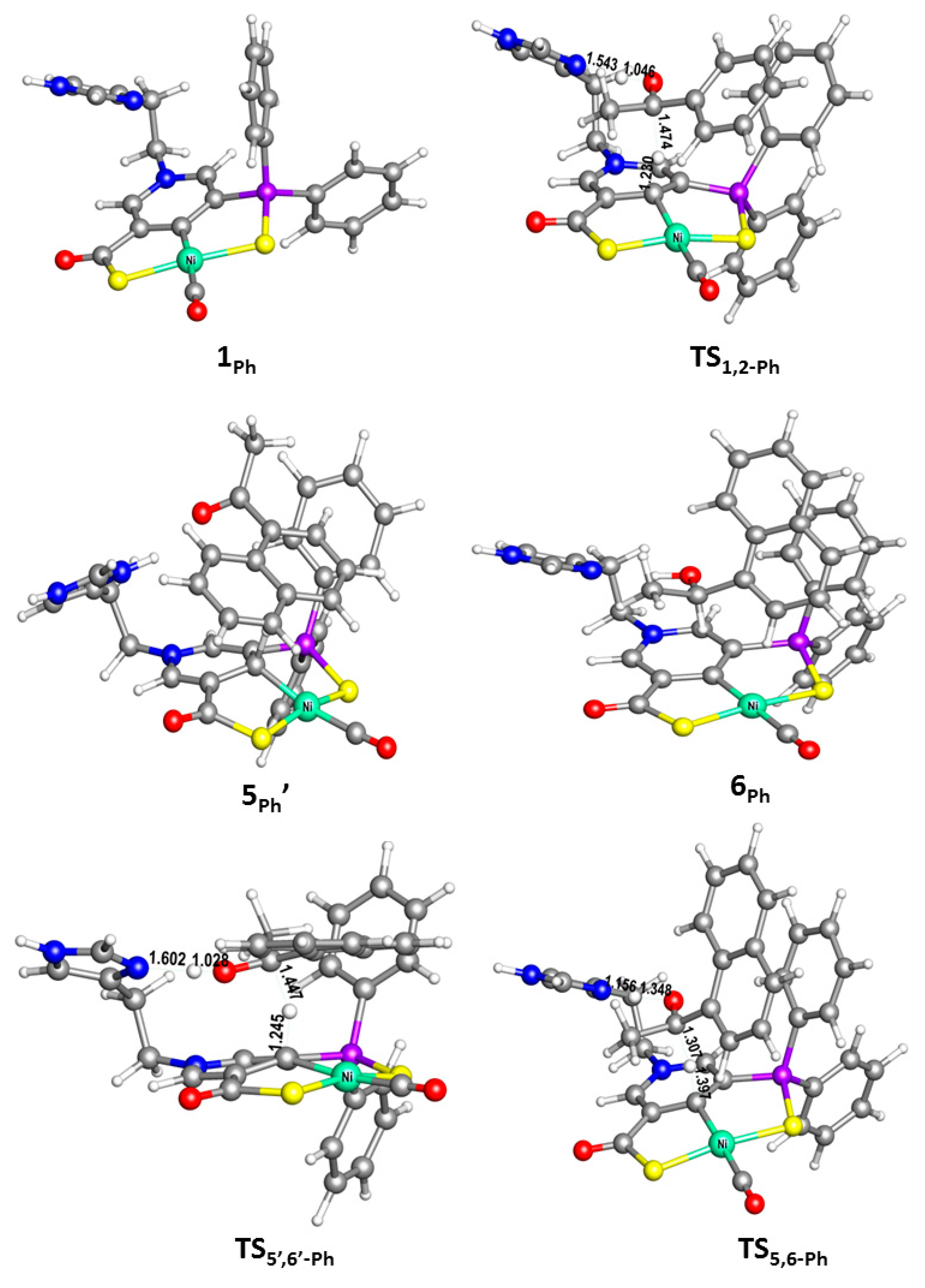
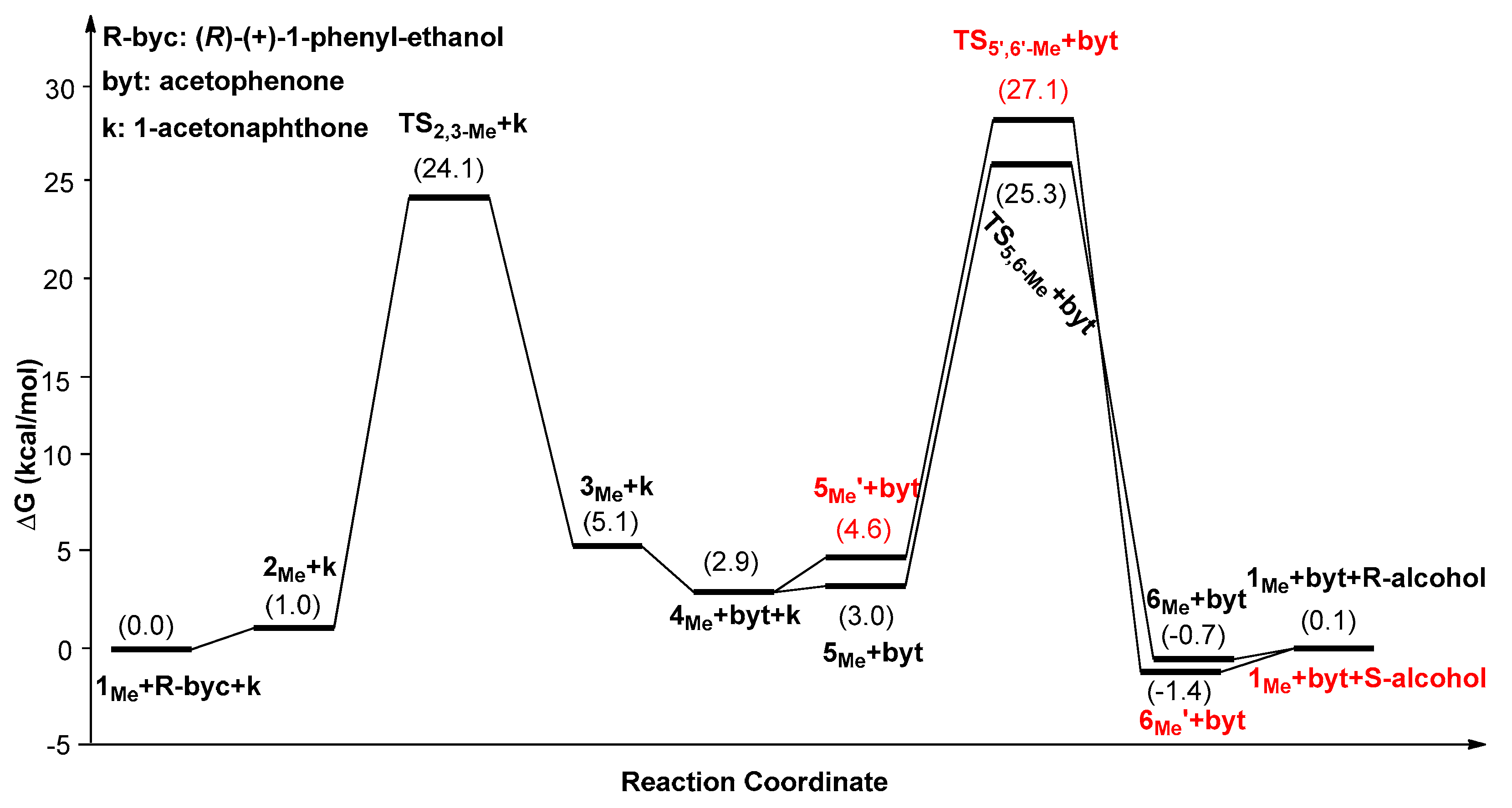
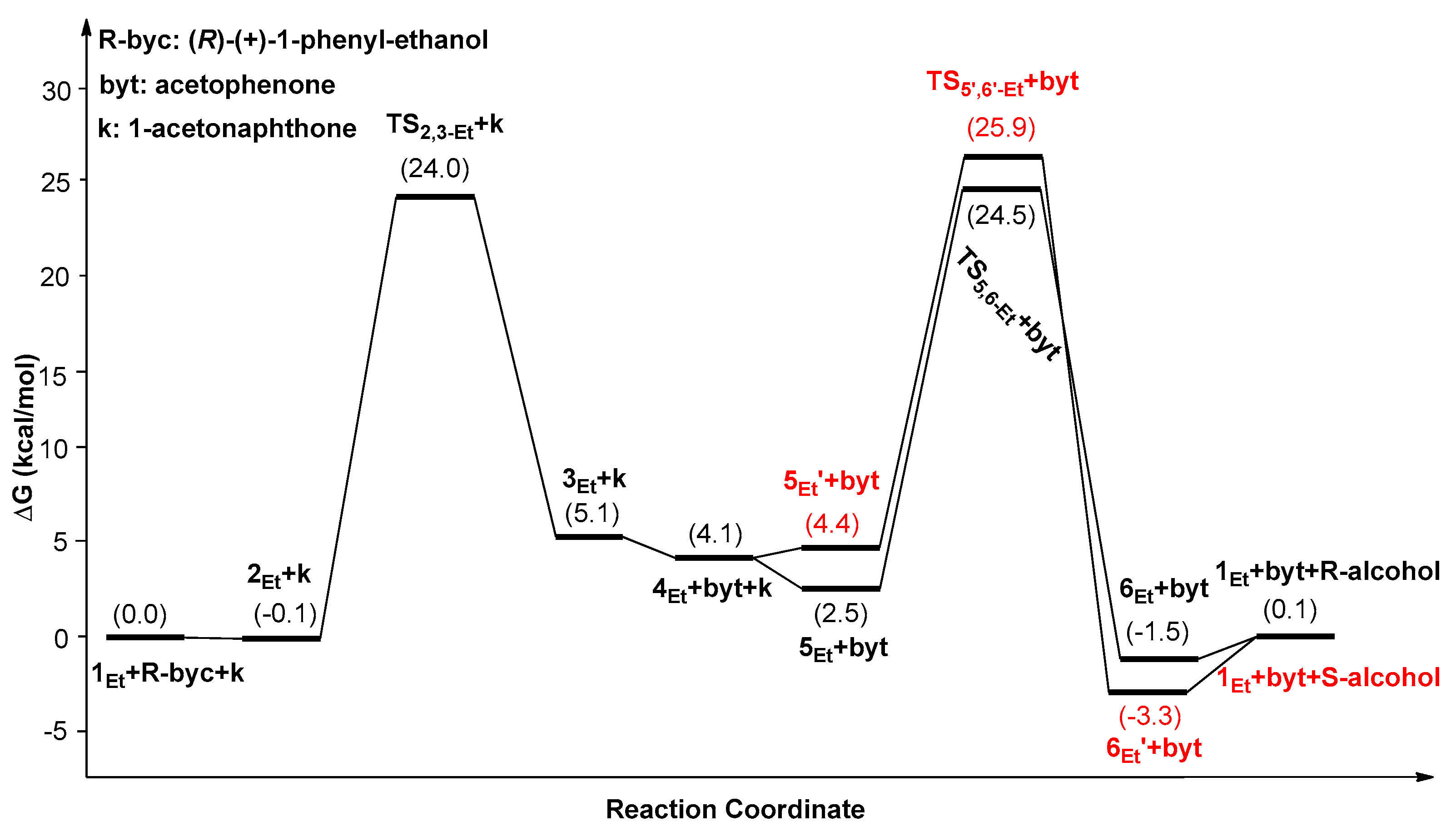
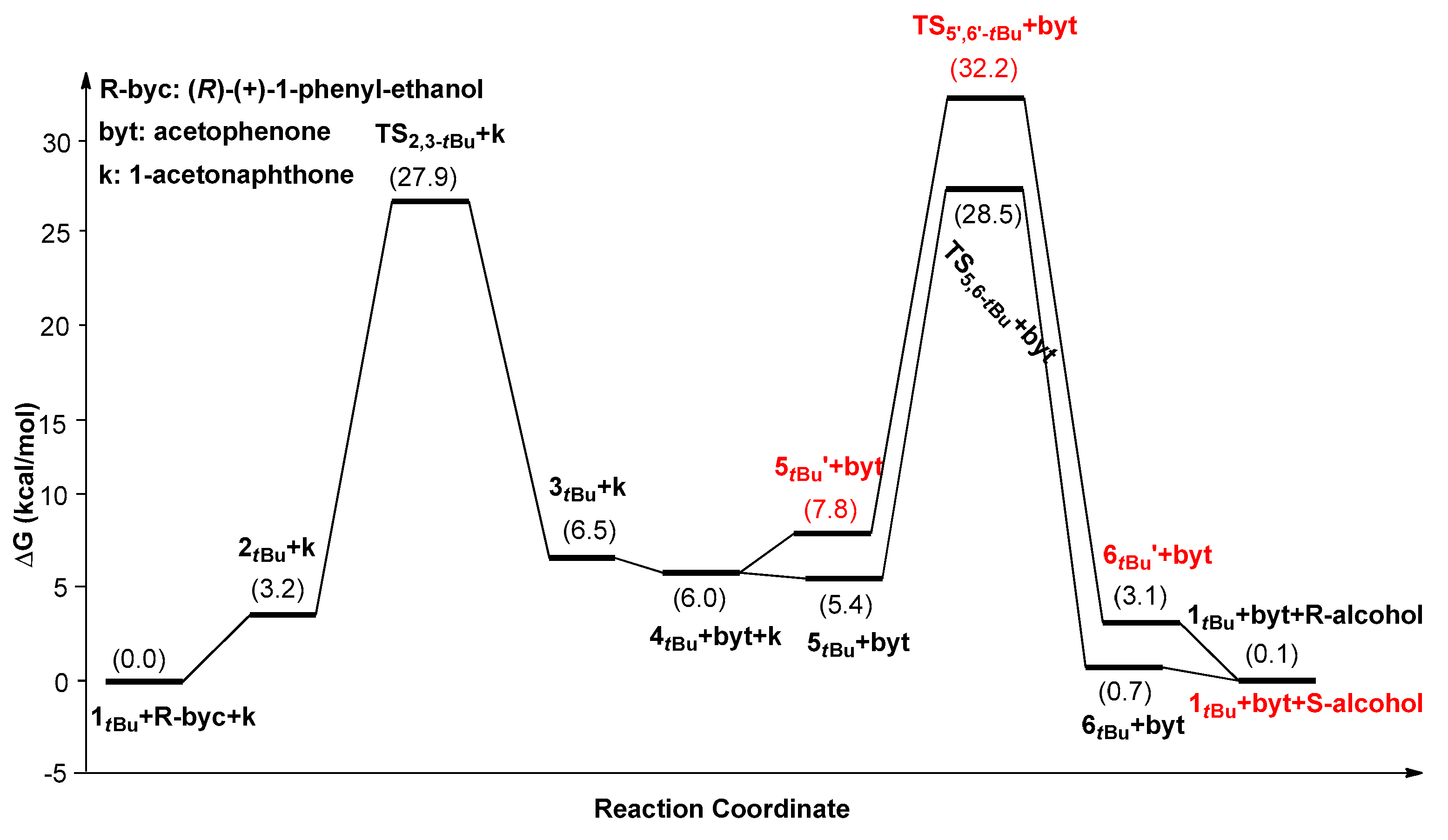
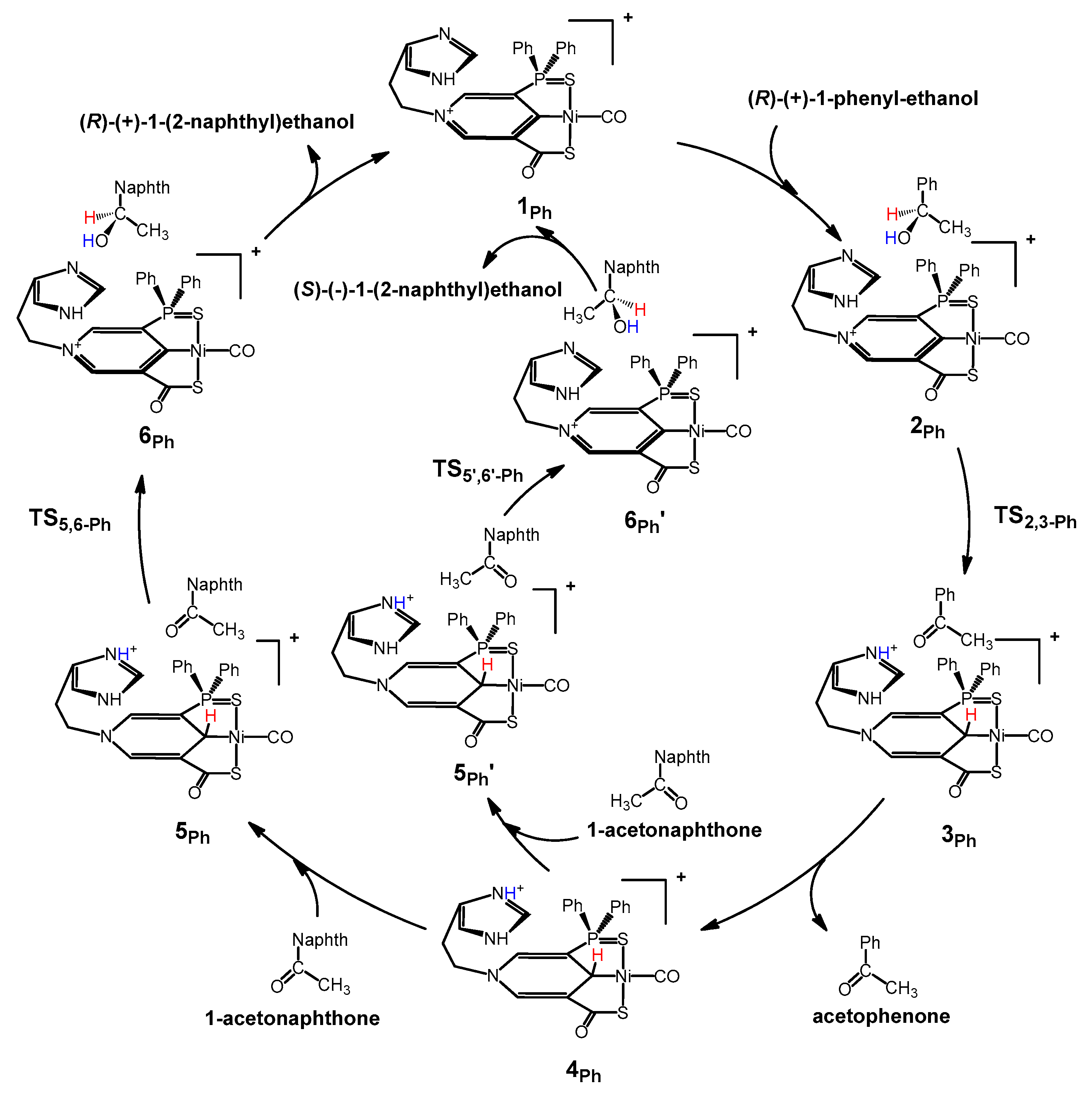
2. Results and Discussion
3. Computational Methods
3.1. DFT Calculation Details
3.2. Quantitative Estimation of Enantiomeric Excess
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hansen, K.B.; Hsiao, Y.; Xu, F.; Rivera, N.; Clausen, A.; Kubryk, M.; Krska, S.; Rosner, T.; Simmons, B.; Balsells, J.; et al. Highly Efficient Asymmetric Synthesis of Sitagliptin. J. Am. Chem. Soc. 2009, 131, 8798–8804. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.U.; Pugin, B.; Spindler, F.; Thommen, M. From a Chiral Switch to a Ligand Portfolio for Asymmetric Catalysis. Acc. Chem. Res. 2007, 40, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Saudan, L.A. Hydrogenation Processes in the Synthesis of Perfumery Ingredients. Acc. Chem. Res. 2007, 40, 1309–1319. [Google Scholar] [CrossRef]
- Blaser, H.U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Gopalaiah, K. Chiral Iron Catalysts for Asymmetric Synthesis. Chem. Rev. 2013, 113, 3248–3296. [Google Scholar] [CrossRef]
- Pellissier, H.; Clavier, H. Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev. 2014, 114, 2775–2823. [Google Scholar] [CrossRef]
- Morris, R.H. Exploiting Metal-Ligand Bifunctional Reactions in the Design of Iron Asymmetric Hydrogenation Catalysts. Acc. Chem. Res. 2015, 48, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Yu, S.L.; Shen, W.Y.; Gao, J.X. Iron-, Cobalt-, and Nickel-Catalyzed Asymmetric Transfer Hydrogenation and Asymmetric Hydrogenation of Ketones. Acc. Chem. Res. 2015, 48, 2587–2598. [Google Scholar] [CrossRef]
- Zuo, W.W.; Lough, A.J.; Li, Y.F.; Morris, R.H. Amine(imine)diphosphine Iron Catalysts for Asymmetric Transfer Hydrogenation of Ketones and Imines. Science 2013, 342, 1080–1083. [Google Scholar] [CrossRef]
- Zuo, W.W.; Morris, R.H. Synthesis and Use of an Asymmetric Transfer Hydrogenation Catalyst Based on Iron(II) for the Synthesis of Enantioenriched Alcohols and Amines. Nat. Protoc. 2015, 10, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.W.; Prokopchuk, D.E.; Lough, A.J.; Morris, R.H. Details of the Mechanism of the Asymmetric Transfer Hydrogenation of Acetophenone Using the Amine(imine)diphosphine Iron Precatalyst: The Base Effect and the Enantiodetermining Step. ACS Catal. 2016, 6, 301–314. [Google Scholar] [CrossRef]
- Smith, S.A.M.; Lagaditis, P.O.; Lupke, A.; Lough, A.J.; Morris, R.H. Unsymmetrical Iron P-NH-P′ Catalysts for the Asymmetric Pressure Hydrogenation of Aryl Ketones. Chem. Eur. J. 2017, 23, 7212–7216. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Koseki, Y.; Fujii, T.; Maeda, T.; Hibino, T.; Makino, K. Catalytic Asymmetric Hydrogenation of α-Amino-β-Keto Ester Hydrochlorides Using Homogeneous Chiral Nickel-Bisphosphine Complexes through DKR. Chem. Commum. 2008, 0, 6206–6208. [Google Scholar] [CrossRef] [PubMed]
- Hibino, T.; Makino, K.; Sugiyama, T.; Hamada, Y. Homogeneous Chiral Nickel-Catalyzed Asymmetric Hydrogenation of Substituted Aromatic α-Aminoketone Hydrochlorides through Dynamic Kinetic Resolution. ChemCatChem 2009, 1, 237–240. [Google Scholar] [CrossRef]
- Dong, Z.R.; Li, Y.Y.; Yu, S.L.; Sun, G.S.; Gao, J.X. Asymmetric Transfer Hydrogenation of Ketones Catalyzed by Nickel Complex with New PNO-Type Ligands. Chin. Chem. Lett. 2012, 23, 533–536. [Google Scholar] [CrossRef]
- Desguin, B.; Zhang, T.; Soumillion, P.; Hols, P.; Hu, J.; Hausinger, R.P. A Tethered Niacin-Derived Pincer Complex with a Nickel-Carbon Bond in Lactate Racemase. Science 2015, 349, 66–69. [Google Scholar] [CrossRef]
- Meguro, H.; Koizumi, T.; Yamamoto, T.; Kanbara, T. Synthesis, Structure, and Quaternization and Complexation Reactions of κ3SCS Pincer Palladium Complexes Having 3,5-Pyridinediyl Unit. J. Organomet. Chem. 2008, 693, 1109–1116. [Google Scholar] [CrossRef]
- Qiu, B.; Yang, X.Z. A Bio-Inspired Design and Computational Prediction of Scorpion-Like SCS Nickel Pincer Complexes for Lactate Racemization. Chem. Commun. 2017, 53, 11410–11413. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation Consistent Valence Basis Sets for Use with the Stuttgart-Dresden-Bonn Relativistic Effective Core Potentials: The Atoms Ga-Kr and In-Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Manson, J.; Webster, C.E.; Hall, M.B. JIMP2, Version 0.091, A Free Program for Visualizing and Manipulating Molecules; Texas A&M University: College Station, TX, USA, 2006. [Google Scholar]
- Scheneebeli, S.T.; Hall, M.L.; Breslow, R.; Friesner, R. Quantitative DFT Modeling of the Enantiomeric Excess for Dioxirane-Catalyzed Epoxidations. J. Am. Chem. Soc. 2009, 131, 3965–3973. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | ΔG1 (kcal/mol) | ΔG2 (kcal/mol) | ΔG3 (kcal/mol) | ΔΔG(ee) = ΔG2 − ΔG3 |
|---|---|---|---|---|
| 1Ph | 25.1(5Ph’→TS2,3-Ph) | 23.8(5Ph’→TS5,6-Ph) | 27.3(5Ph’→TS5’,6’-Ph) | −3.5 (99.5%) |
| 1Me | 25.6(6Me’→TS2,3-Me) | 26.8(6Me’→TS5,6-Me) | 28.6(6Me’→TS5’,6’-Me) | −1.8 (90.9%) |
| 1Et | 27.4(6Et’→TS2,3-Et) | 27.9(6Et’→TS5,6-Et) | 29.3(6Et’→TS5’,6’-Et) | −1.4 (82.8%) |
| 1tBu | 27.9(1tBu→TS2,3-tBu) | 28.5(1tBu→TS5,6-tBu) | 32.2(1tBu→TS5’,6’-tBu) | −3.7 (99.6%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, B.; Wang, W.; Yang, X. Computational Design of SCS Nickel Pincer Complexes for the Asymmetric Transfer Hydrogenation of 1-Acetonaphthone. Catalysts 2019, 9, 101. https://doi.org/10.3390/catal9010101
Qiu B, Wang W, Yang X. Computational Design of SCS Nickel Pincer Complexes for the Asymmetric Transfer Hydrogenation of 1-Acetonaphthone. Catalysts. 2019; 9(1):101. https://doi.org/10.3390/catal9010101
Chicago/Turabian StyleQiu, Bing, Wan Wang, and Xinzheng Yang. 2019. "Computational Design of SCS Nickel Pincer Complexes for the Asymmetric Transfer Hydrogenation of 1-Acetonaphthone" Catalysts 9, no. 1: 101. https://doi.org/10.3390/catal9010101
APA StyleQiu, B., Wang, W., & Yang, X. (2019). Computational Design of SCS Nickel Pincer Complexes for the Asymmetric Transfer Hydrogenation of 1-Acetonaphthone. Catalysts, 9(1), 101. https://doi.org/10.3390/catal9010101