Production of New Isoflavone Glucosides from Glycosylation of 8-Hydroxydaidzein by Glycosyltransferase from Bacillus subtilis ATCC 6633




Abstract
:1. Introduction
2. Results and Discussion
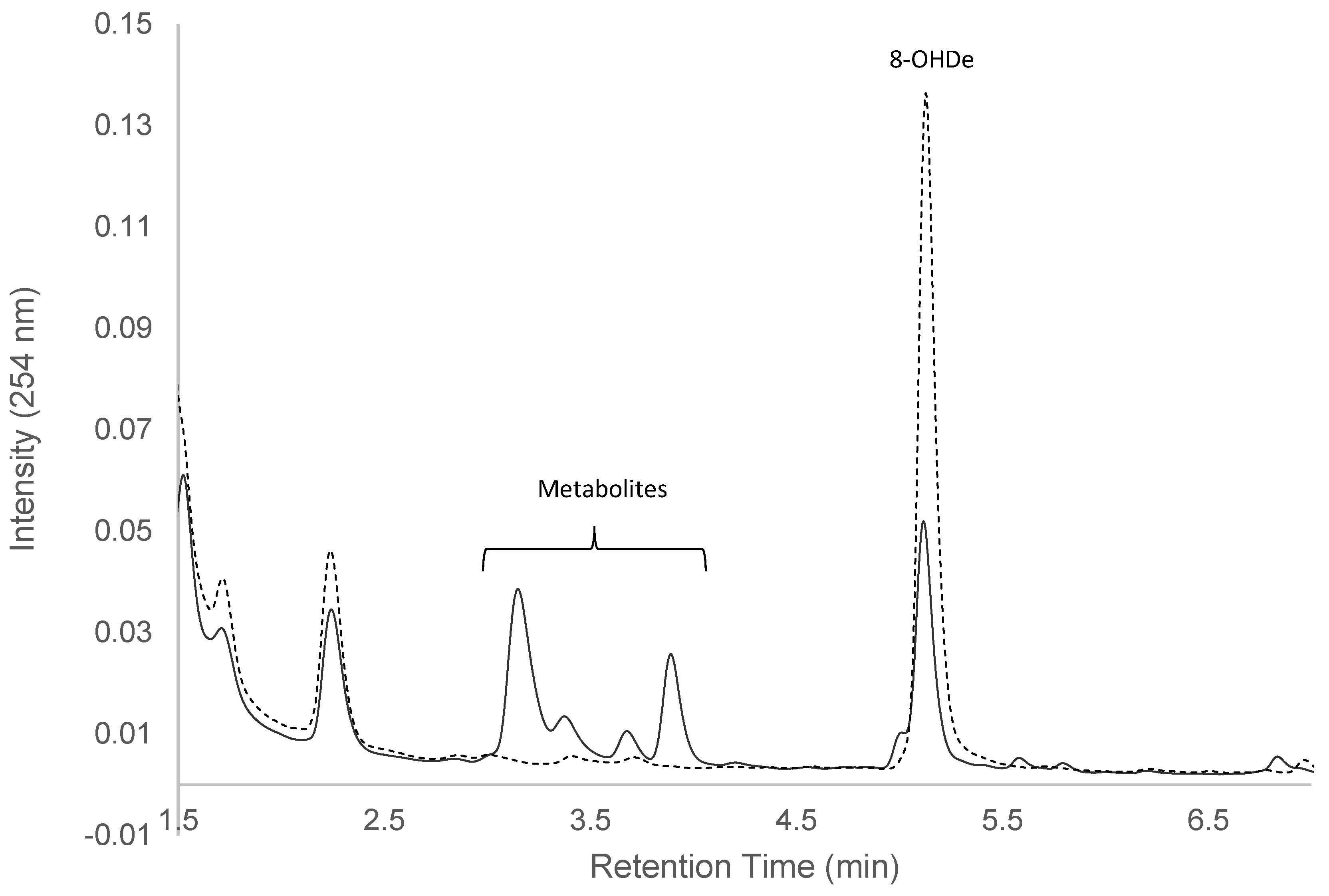
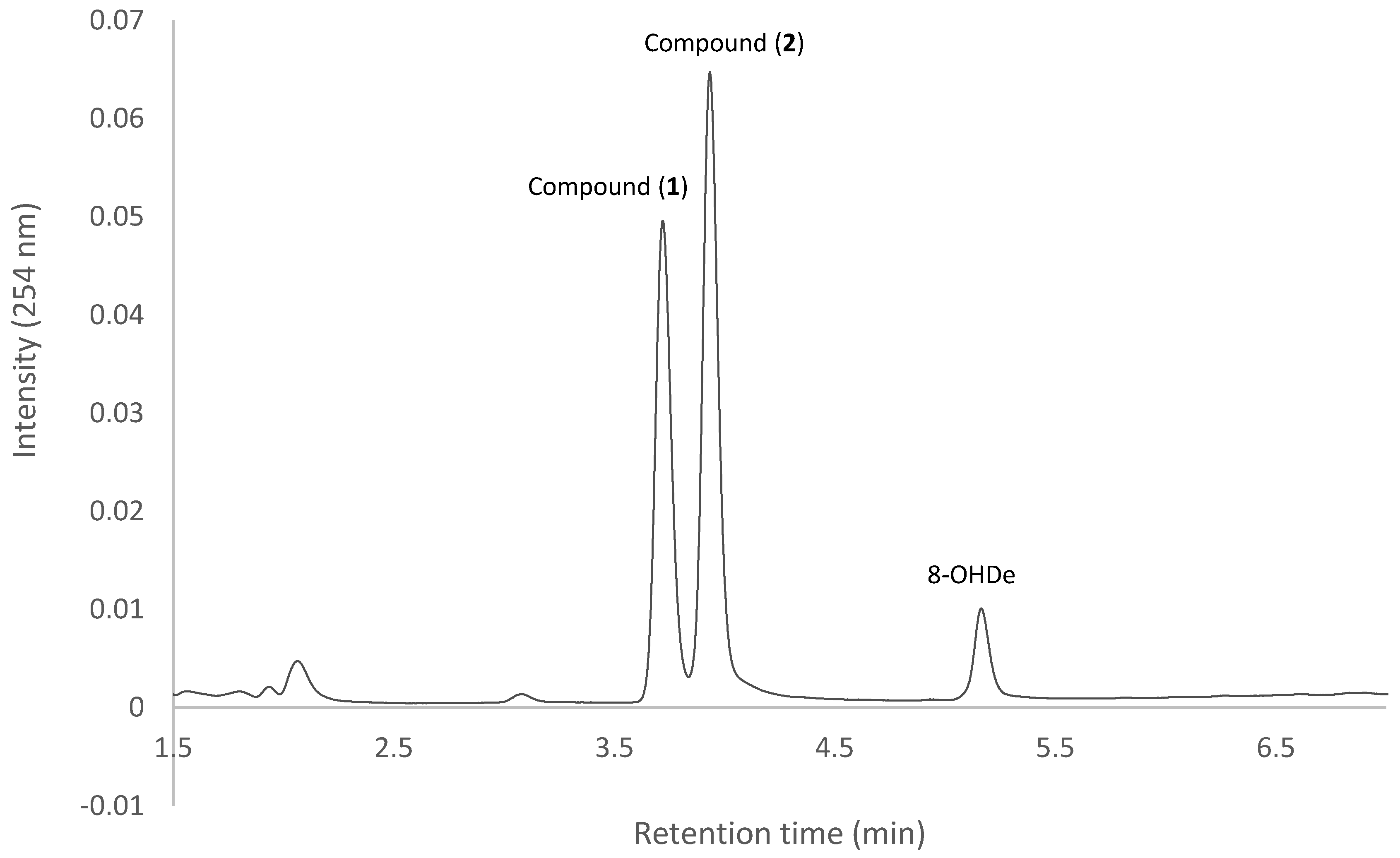
2.1. Confirming Biotransformation of 8-OHDe by Bacillus subtilis ATCC 6633
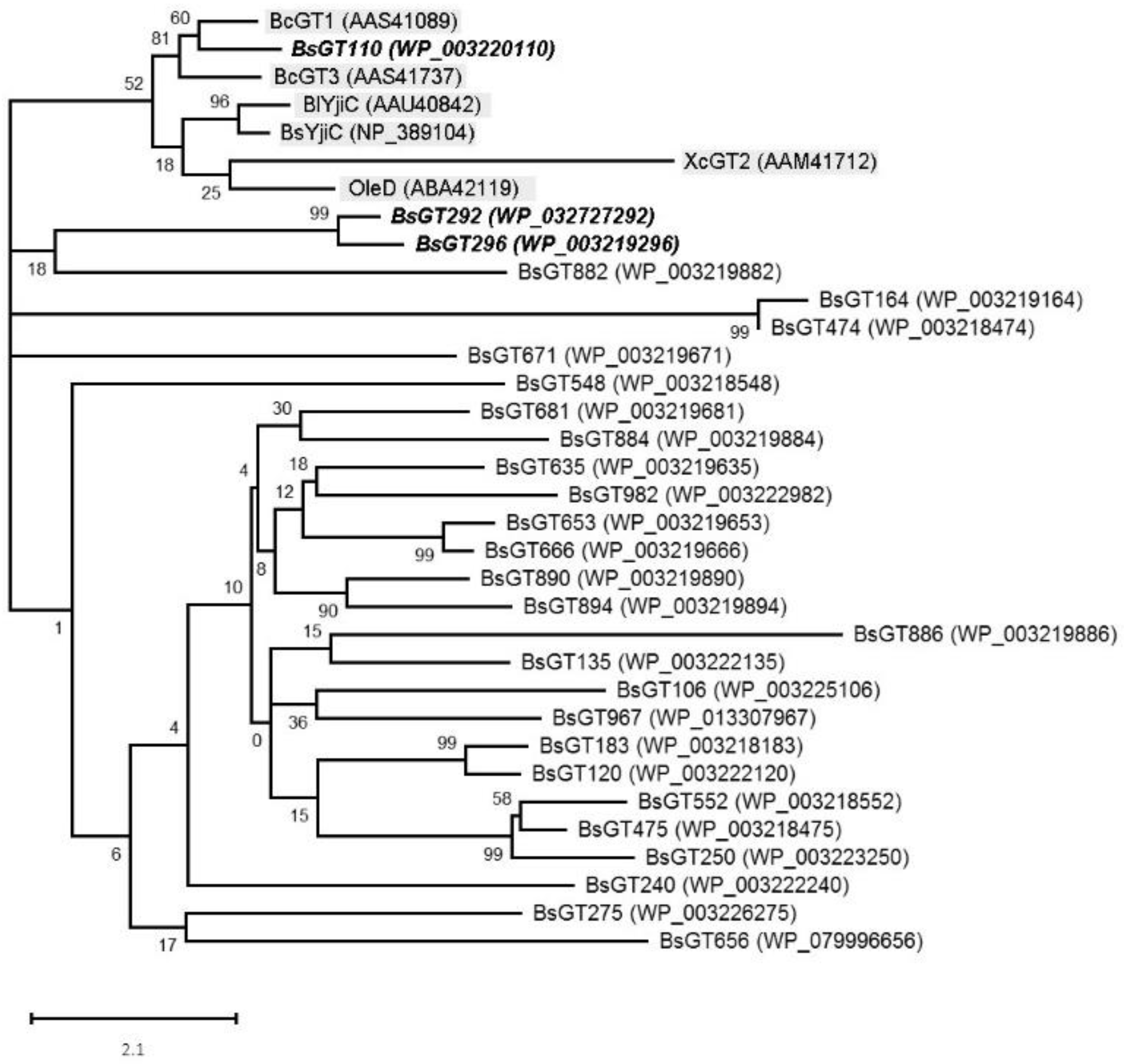
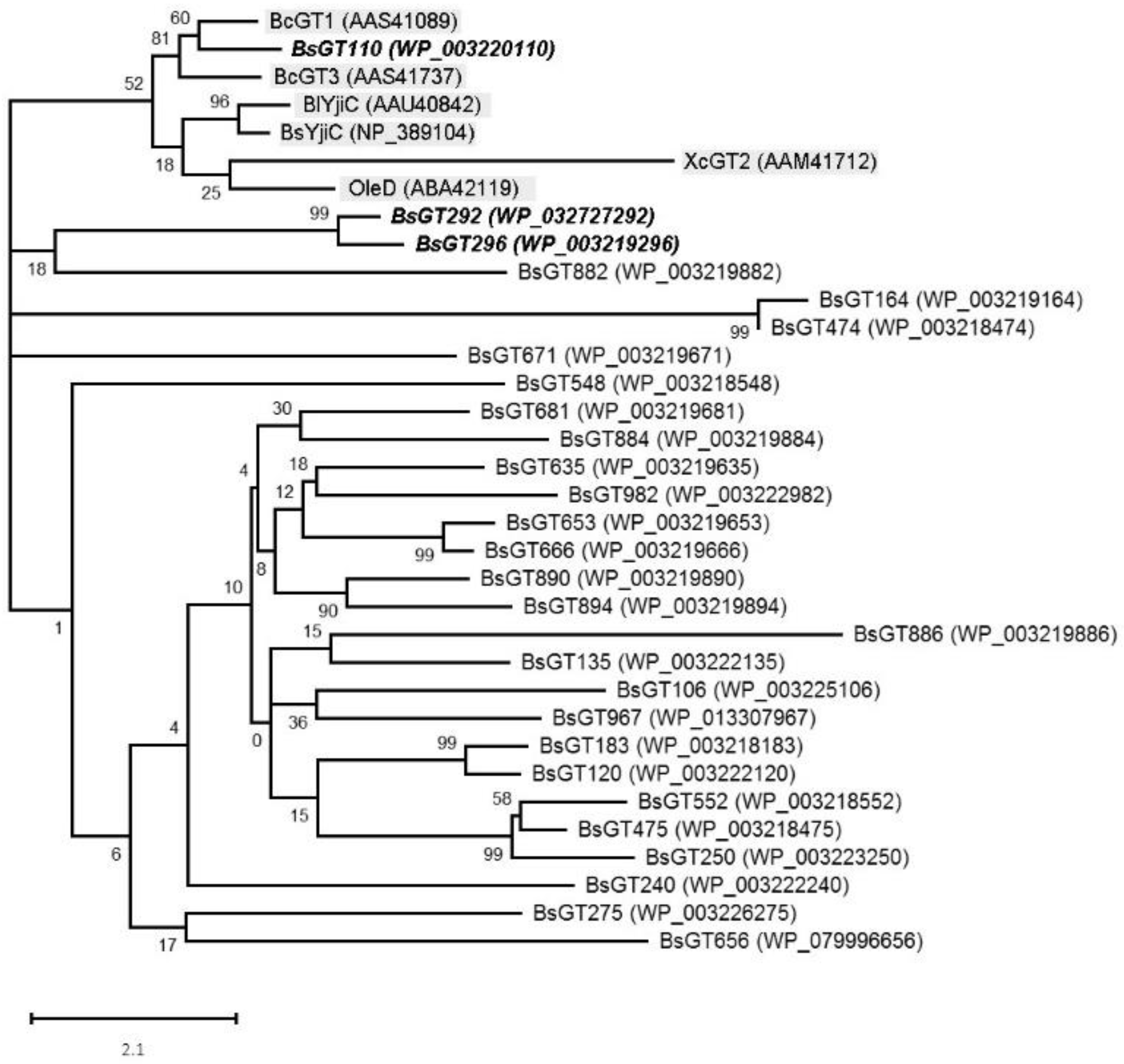
2.2. Phylogenetic Analysis of GTs from B. subtilis ATCC 6633
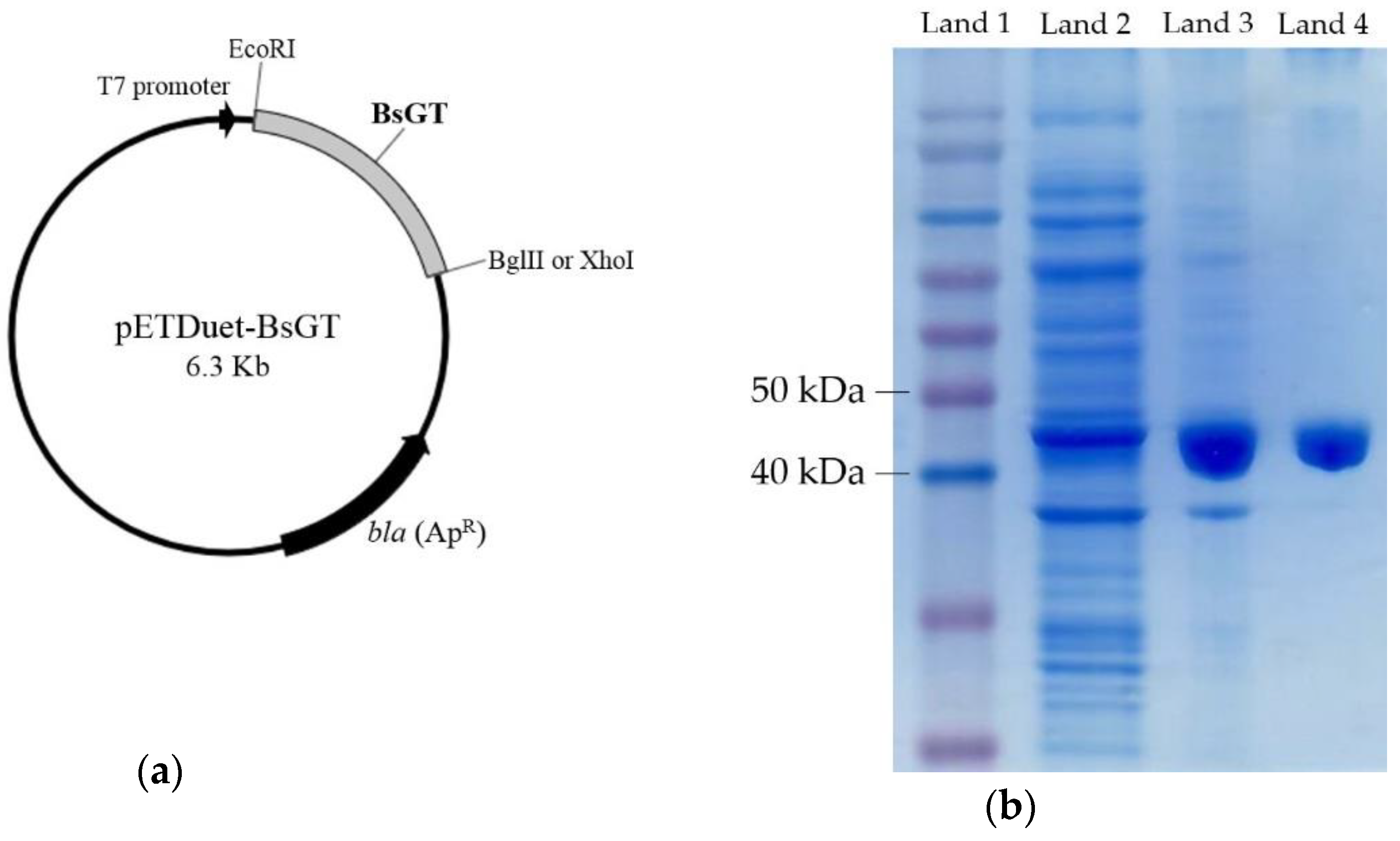
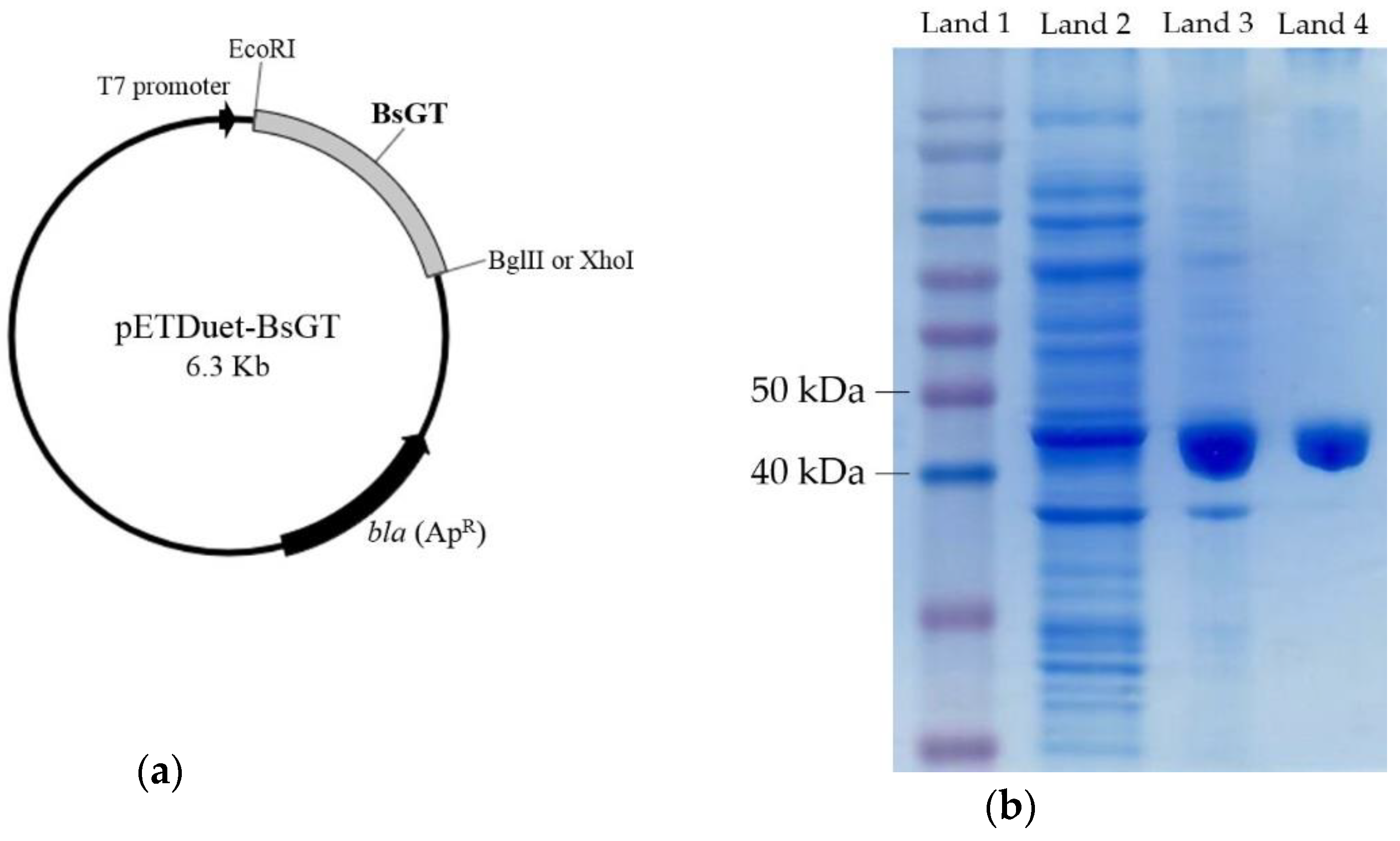
2.3. Cloning, Overexpression, Purification and Activity Assay of BsGTs from B. subtilis ATCC 6633 in E. coli
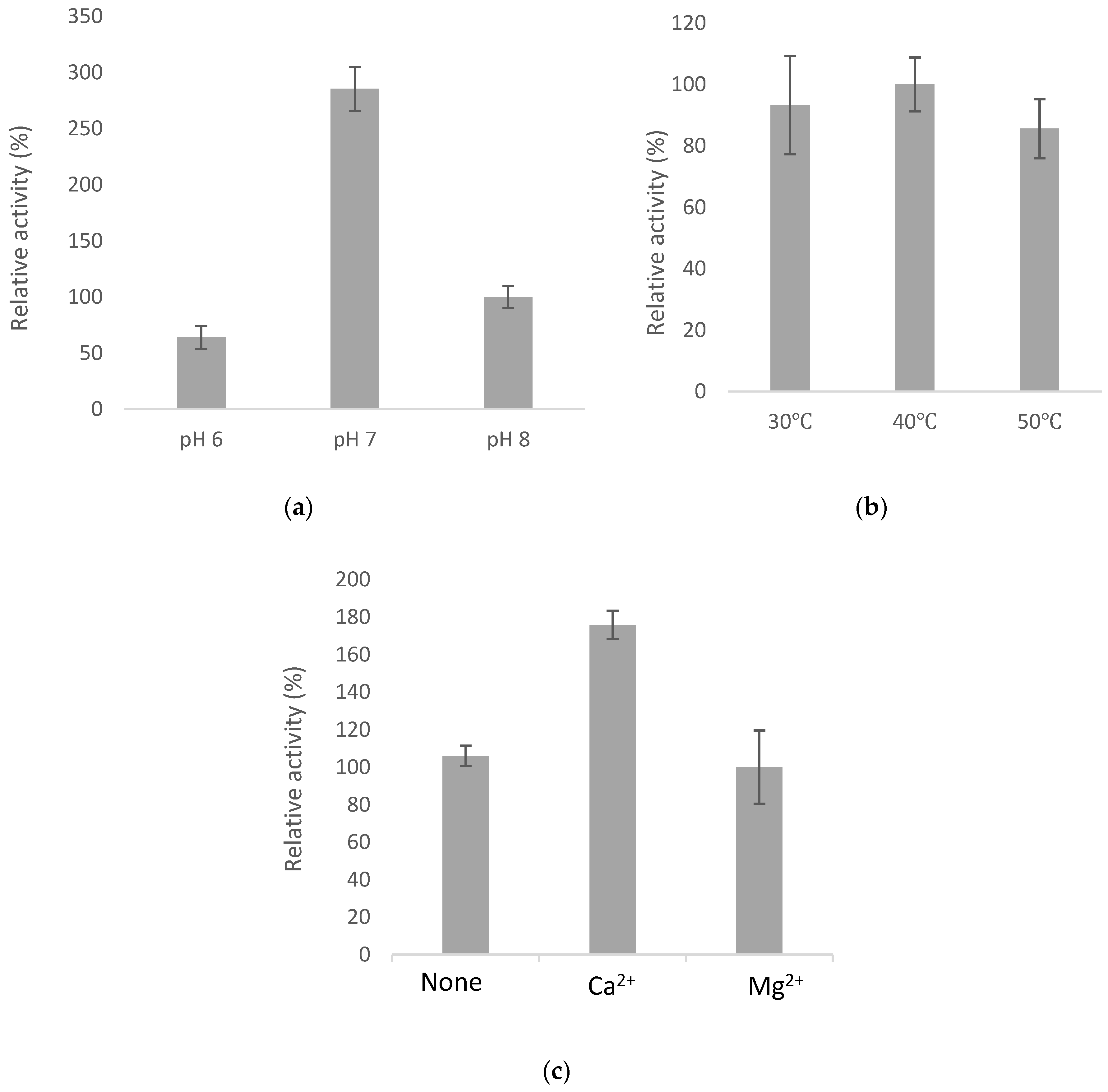
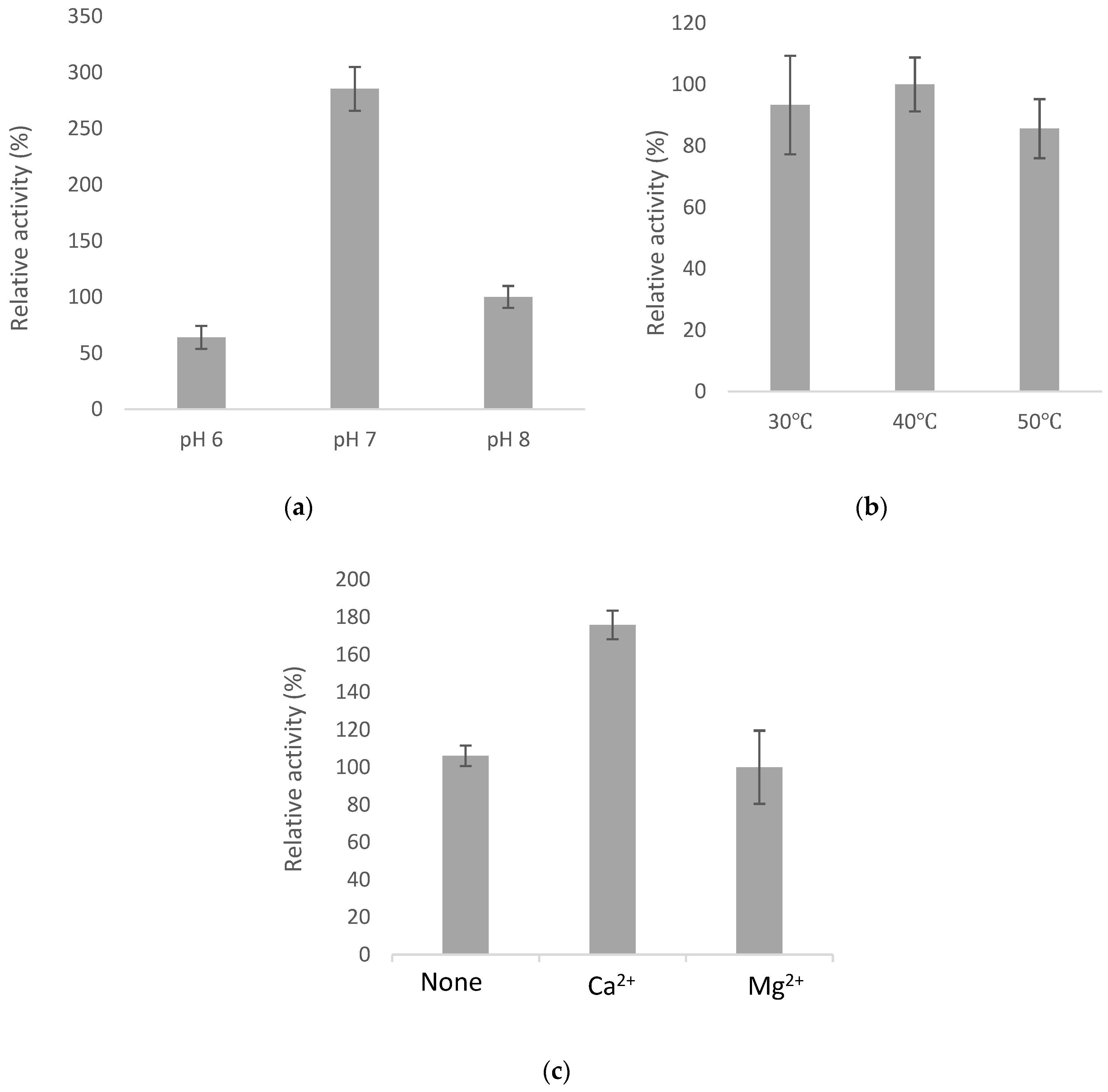
2.4. Optimal Catalyzing Conditions for BsGT110
2.5. Substrate Specificity of BsGT110
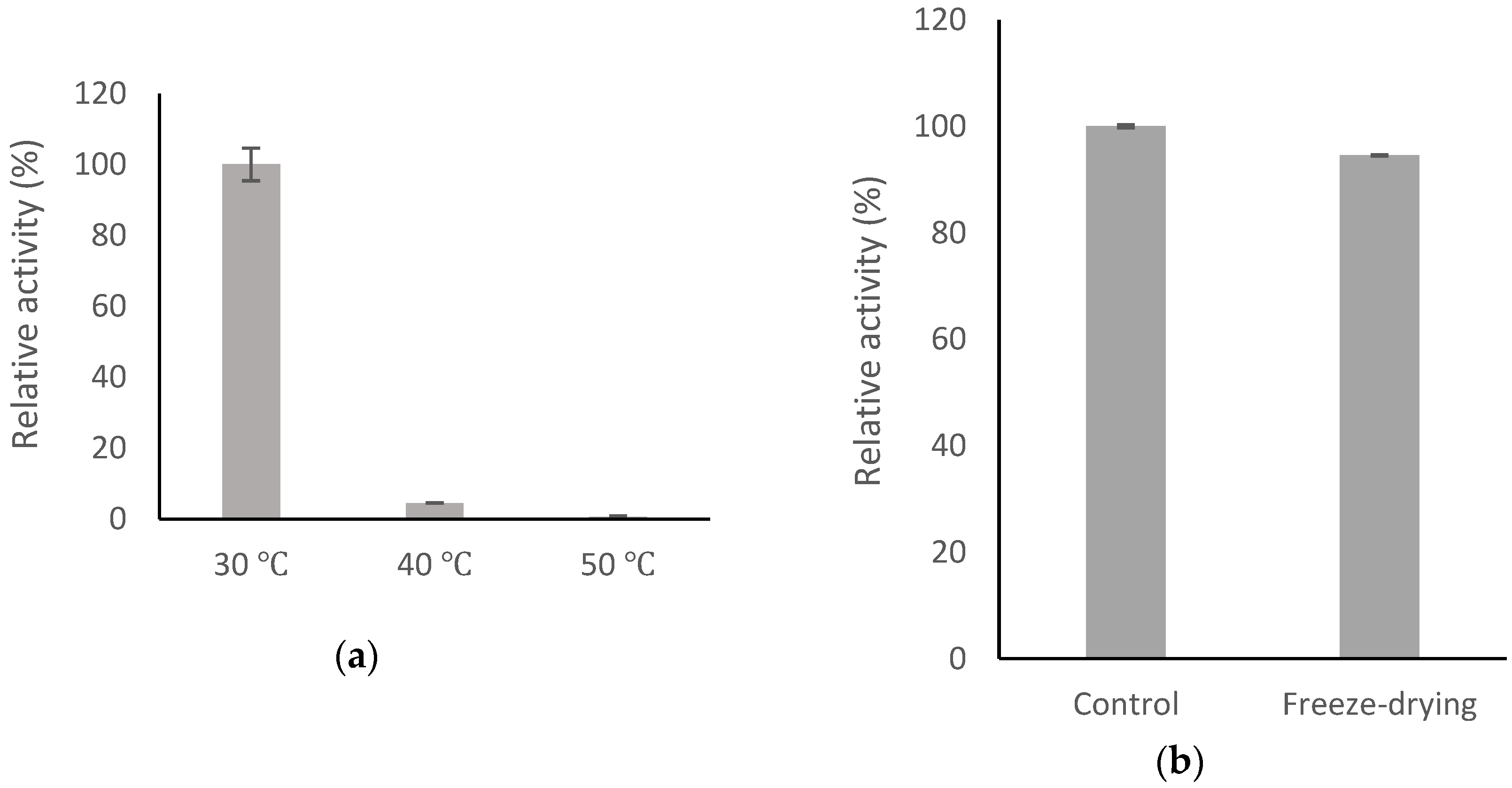
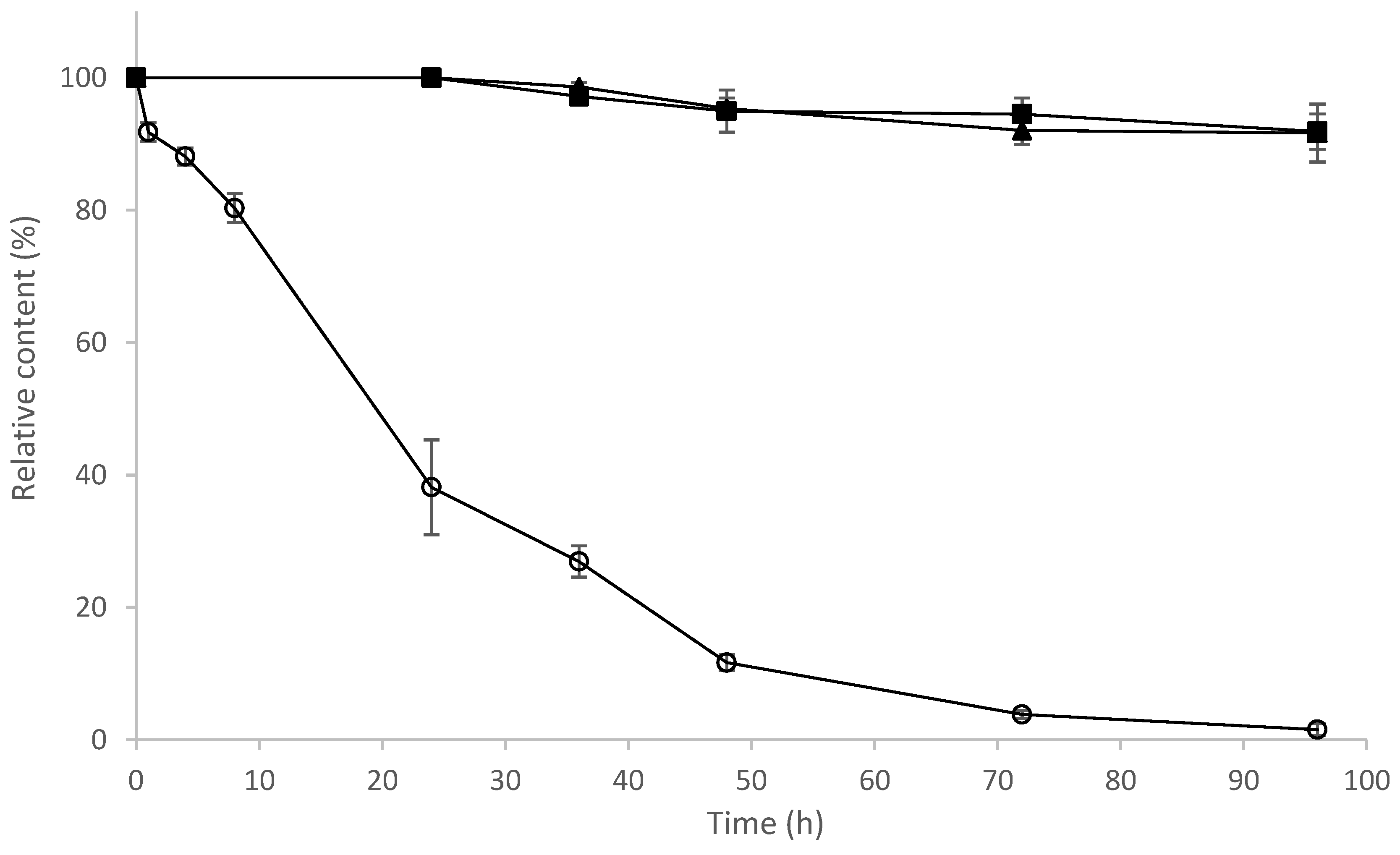
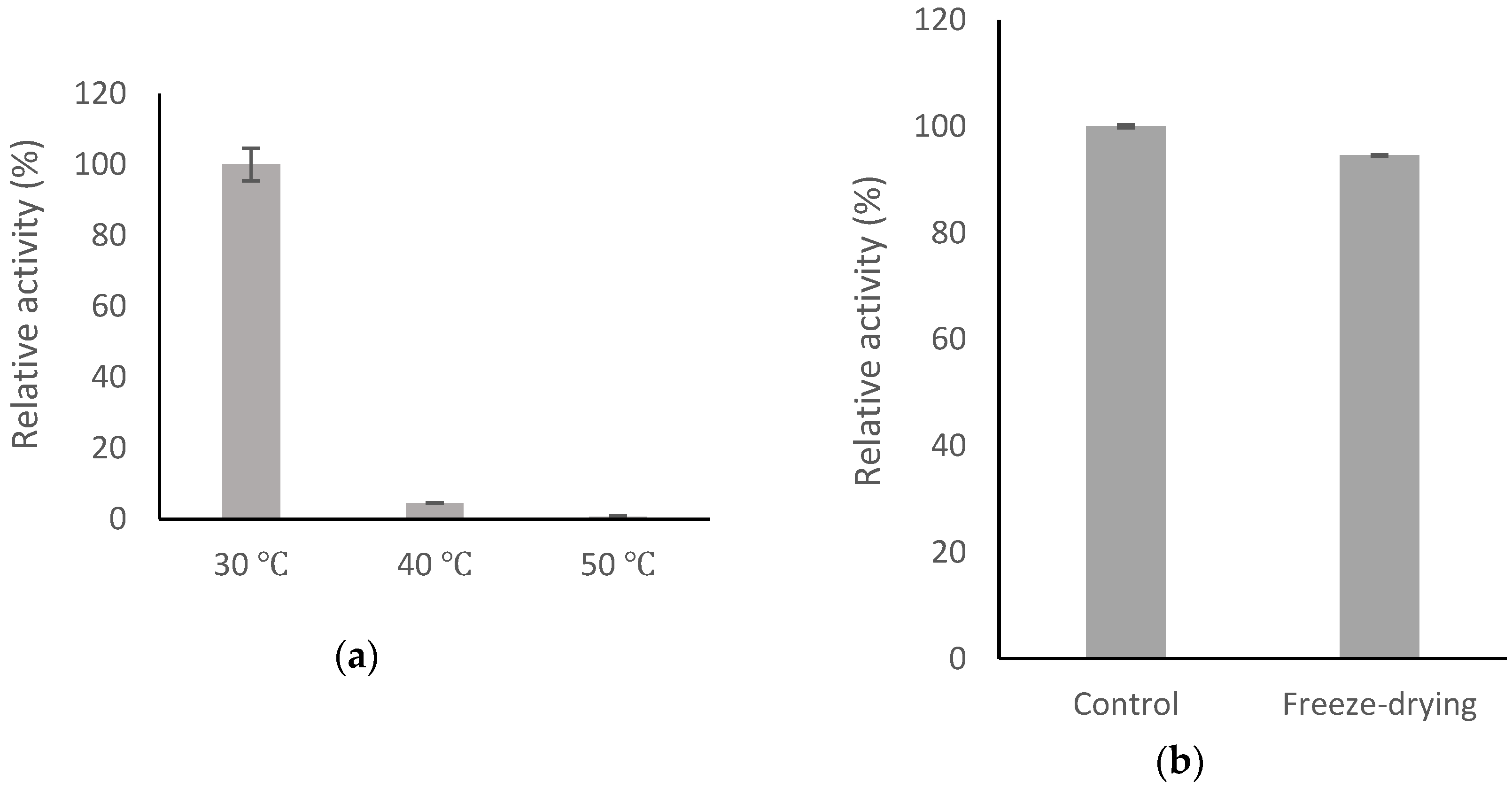
2.6. Stability of BsGT110
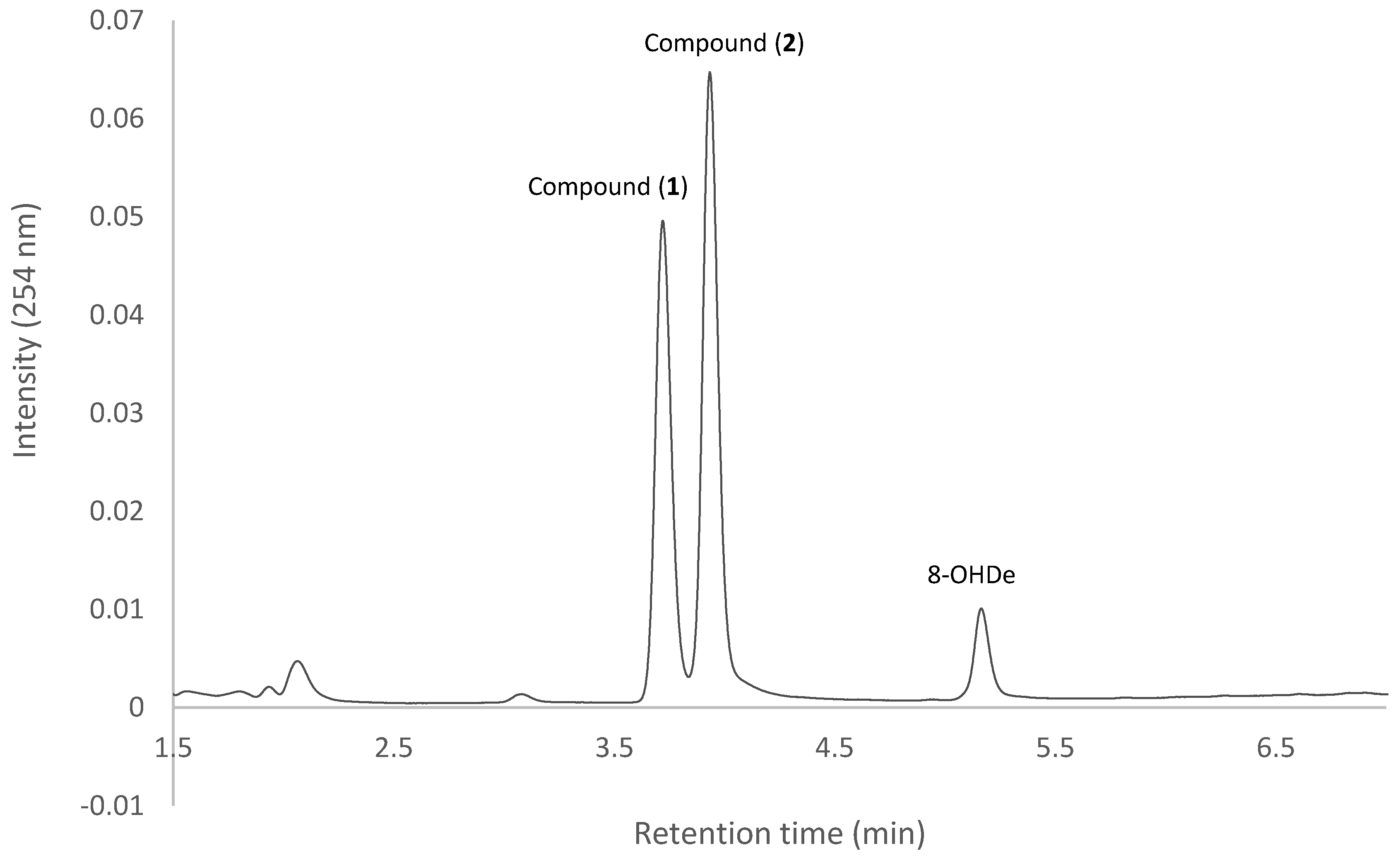
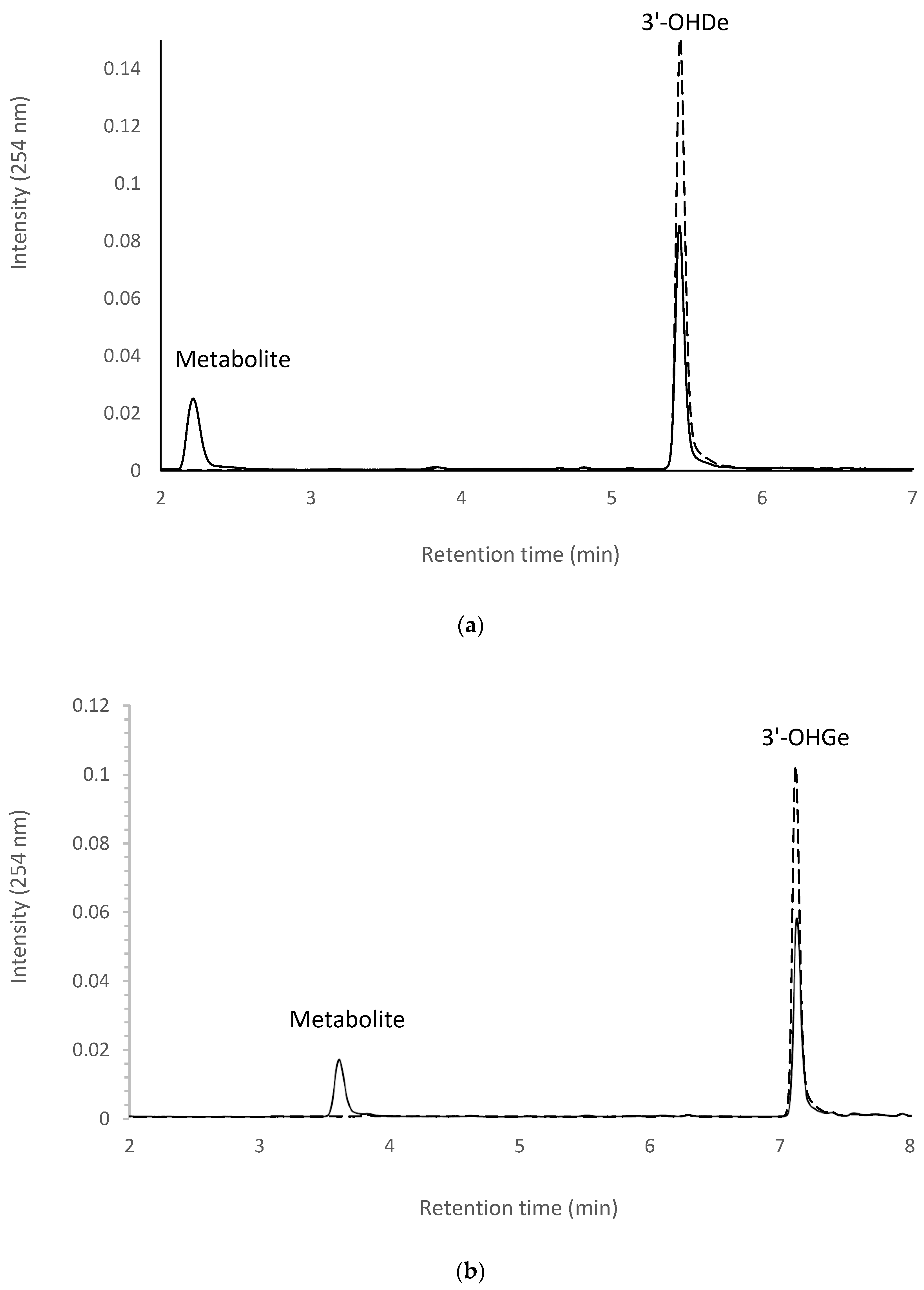
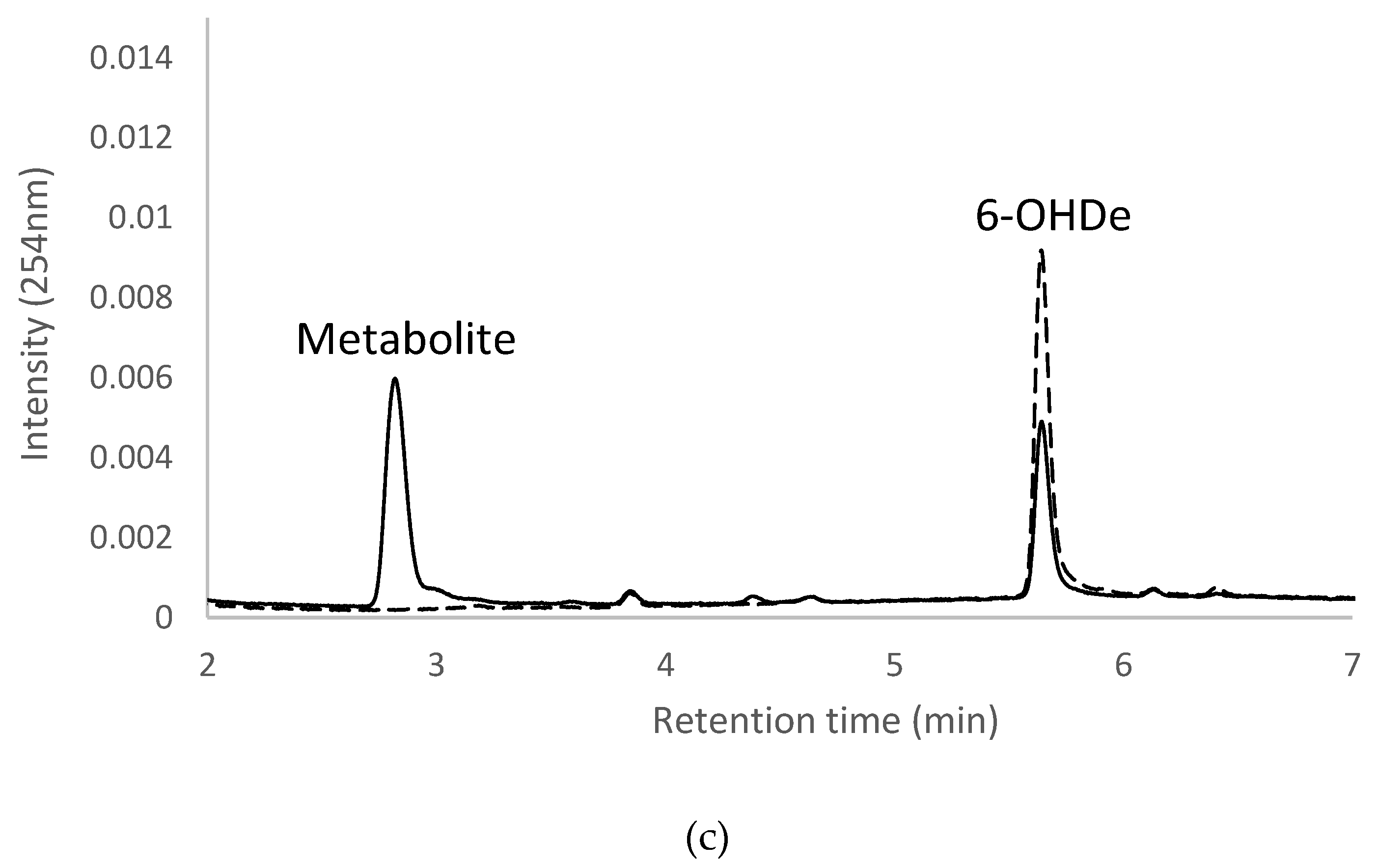
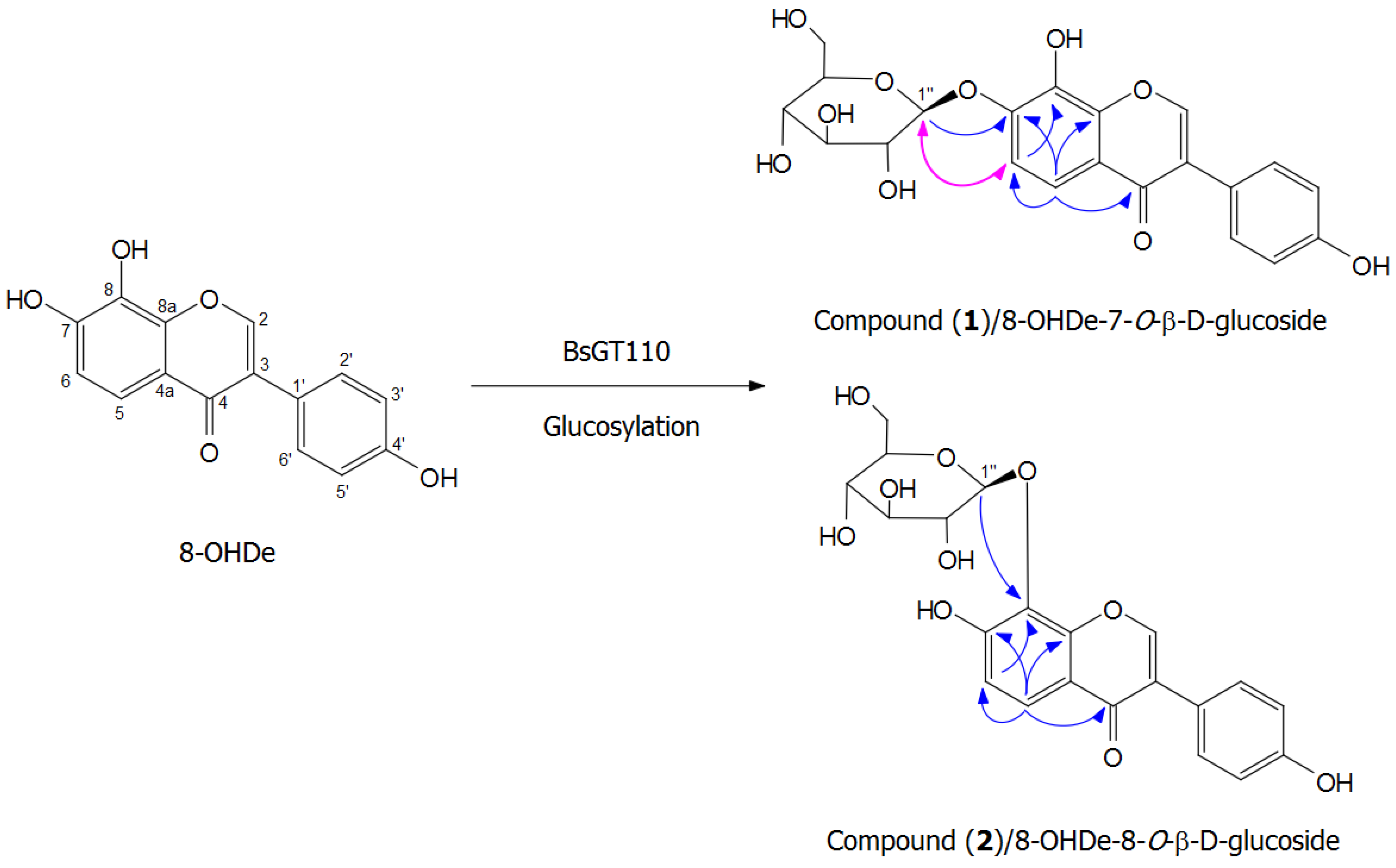
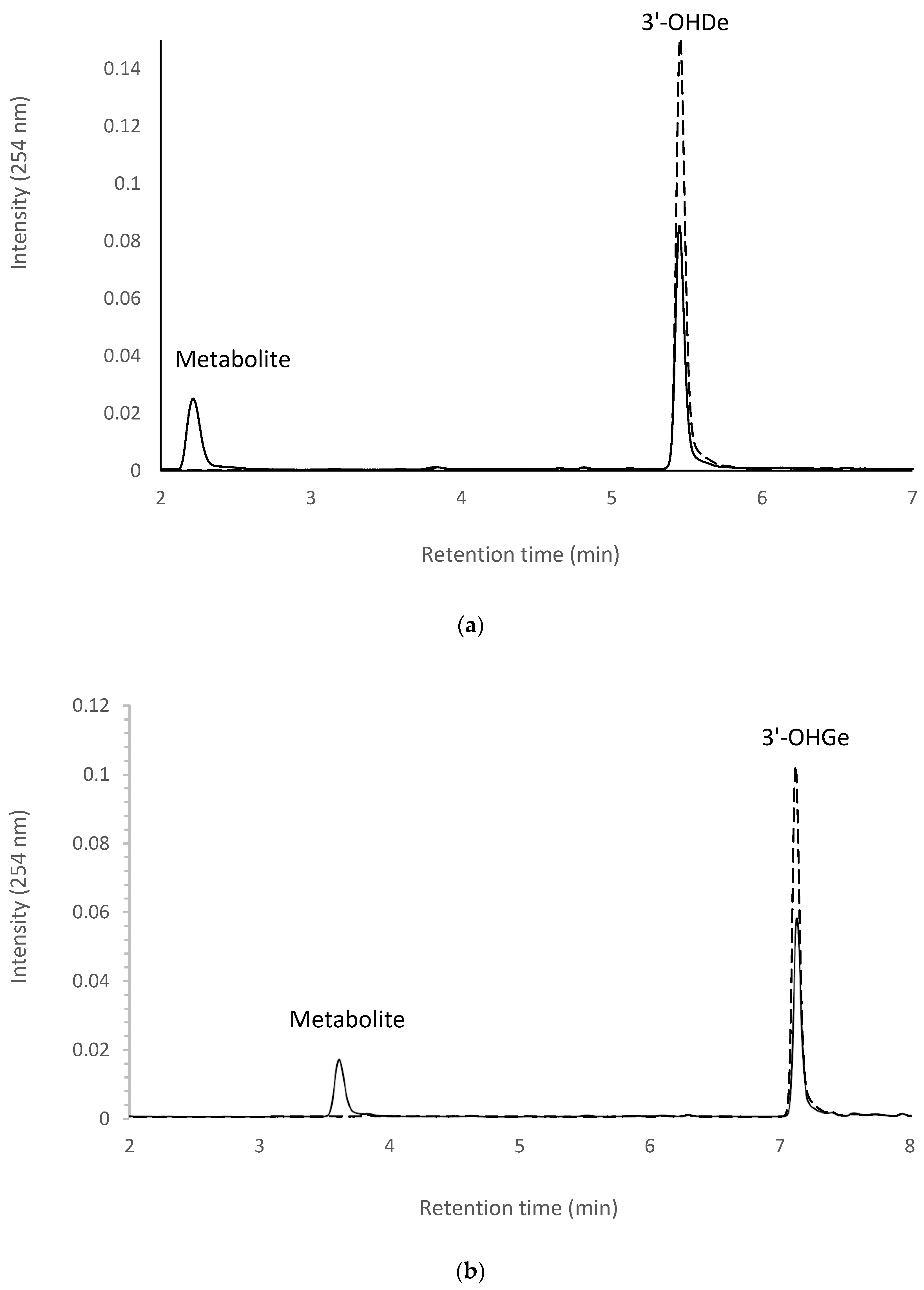
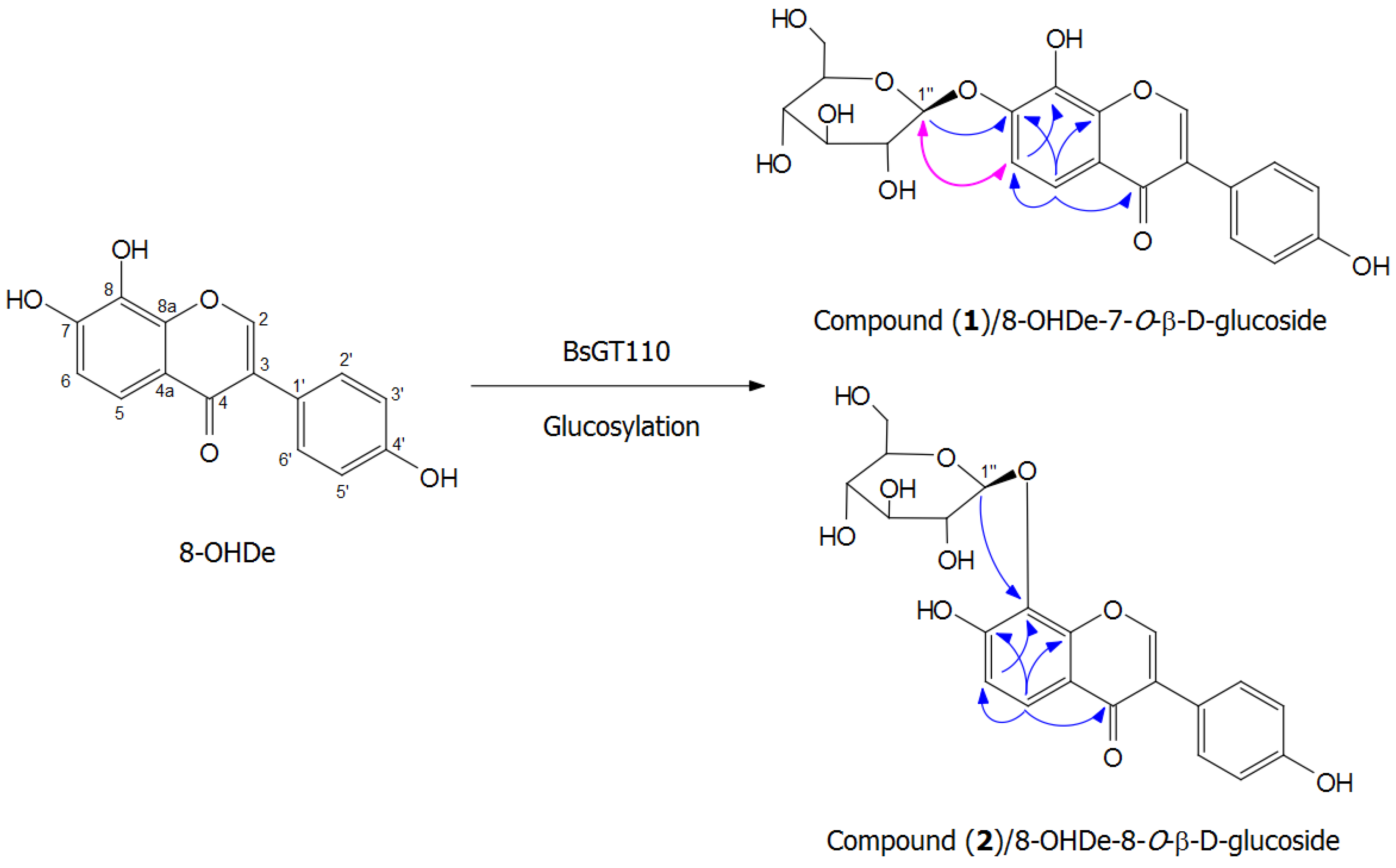
2.7. Isolation and Identification of Biotransformation Metabolites
2.8. Determination of Aqueous Solubility of 8-OHDe and Its Glucosides
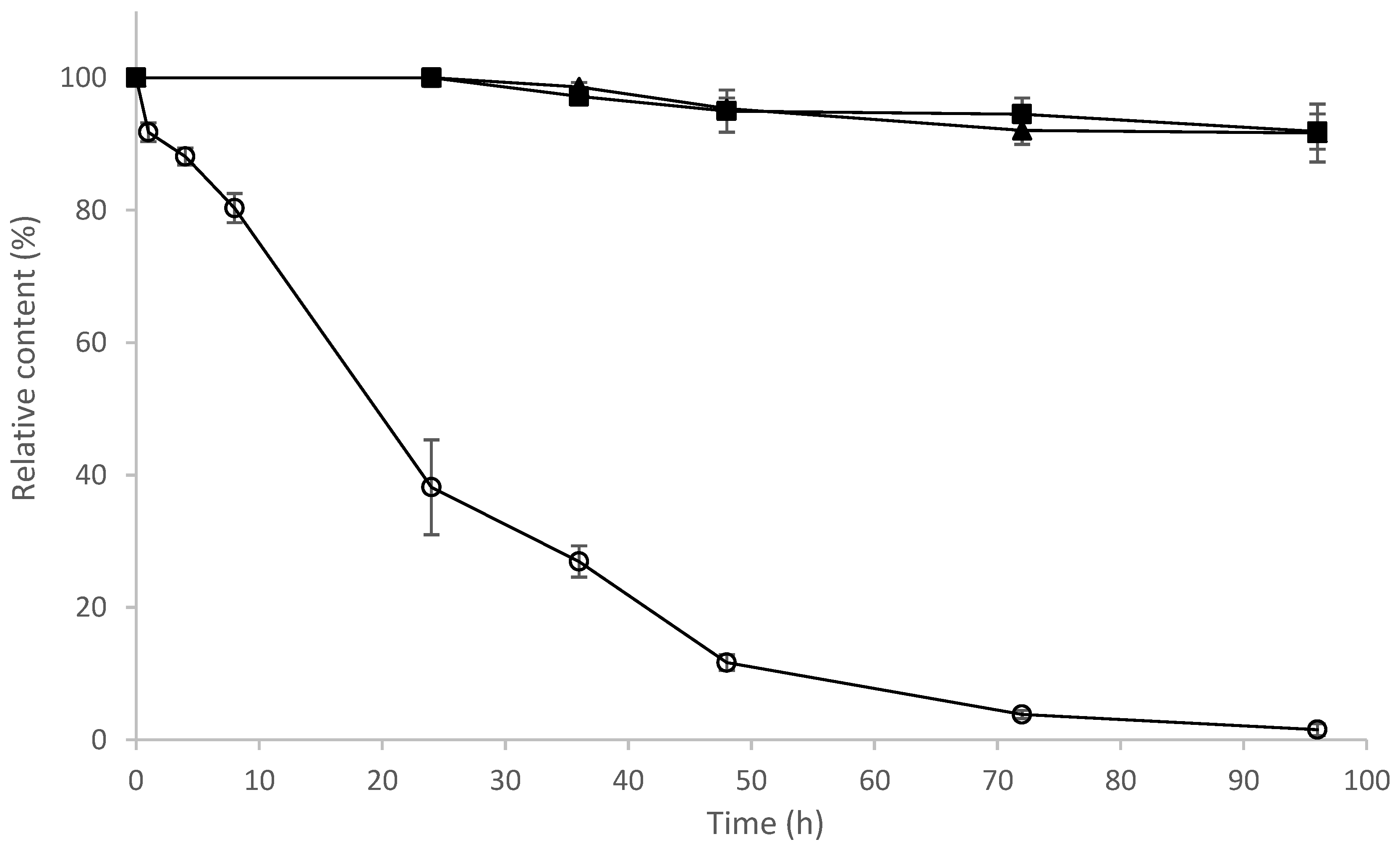
2.9. Determination of Stability of 8-OHDe and Its Glucosides
3. Materials and Methods
3.1. Microorganisms, Animal Cells and Chemicals
3.2. Identification of Bacteria B. subtilis ATCC 6633 with Biotransformation Activity
3.3. UPLC Analysis
3.4. Phylogenetic Analysis of BsGTs
3.5. Expression and Purification of UGT398 and UGT489
3.6. In Vitro Biotransformation Assay
3.7. Scale-Up, Isolation and Identification of the Biotransformation Product
3.8. Determination of Solubility
3.9. Determination of Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Franke, A.A.; Custer, L.J.; Cerna, C.M.; Narala, K.K. Quantitation of phytoestrogens in legumes by HPLC. J. Agric. Food Chem. 1994, 42, 1905–1913. [Google Scholar] [CrossRef]
- Chang, T.S. Isolation, bioactivity, and production of ortho-hydroxydaidzein and ortho-hydroxygenistein. Int. J. Mol. Sci. 2014, 15, 5699–5716. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Ding, H.Y.; Tai, S.S.K.; Wu, C.Y. Metabolism of the soy isoflavone daidzein and genistein by the fungi used for the preparation of various fermented soybean foods. Biosci. Biotechnol. Biochem. 2007, 71, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Chang, C.W.; Lin, C.W.; Hsu, Y.C. Production of 8-hydroxydaidzein polyphenol using biotransformation by Aspergillus oryzae. Food Sci. Technol. Res. 2015, 21, 557–562. [Google Scholar] [CrossRef]
- Seo, M.H.; Kim, B.N.; Kim, K.R.; Lee, K.W.; Lee, C.H.; Oh, D.K. Production of 8-hydroxydaidzein from soybean extract by Aspergillus oryzae KACC 40247. Biosci. Biotechnol. Biochem. 2013, 77, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Chao, S.Y.; Chen, Y.C. Production of ortho-hydroxydaidzein derivatives by a recombinant strain of Pichia pastoris harboring a cytochrome P450 fusion gene. Process. Biochem. 2013, 48, 426–429. [Google Scholar] [CrossRef]
- Roh, C.; Choi, K.Y.; Pandey, B.P.; Kim, B.G. Hydroxylation of daidzein by CYP107H1 from Bacillus subtilis 168. J. Mol. Catal. B Enzym. 2009, 59, 248–253. [Google Scholar] [CrossRef]
- Choi, K.Y.; Jung, E.O.; Jung, D.H.; Pandey, B.P.; Yun, H.; Park, H.Y.; Kazlauskas, R.J.; Kim, B.G. Cloning, expression and characterization of CYP102D1, a self-sufficient P450 monooxygenase from Streptomyces avermitilis. FEBS J. 2012, 279, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Funayama, S.; Anraku, Y.; Mita, A.; Komiyama, K.; Omura, S. Structure study of isoflavonoids possessing antioxidant activity from the fermentation broth of Streptomyces sp. J. Antibiot. 1989, 42, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.L. A potential daidzein derivative enhances cytotoxicity of epirubicin on human colon adenocarcinoma Caco-2 cells. Int. J. Mol. Sci. 2012, 14, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Ding, H.Y.; Tai, S.S.K.; Wu, C.Y. Tyrosinase inhibitors isolated from soygerm koji fermented with Aspergillus oryzae BCRC 32288. Food Chem. 2007, 105, 1430–1438. [Google Scholar] [CrossRef]
- Chang, T.S. Two potent suicide substrates of mushroom tyrosinase: 7,8,4′-trihydroxyisoflavone and 5,7,8,4′-tetrahydroxyisoflavone. J. Agric. Food Chem. 2007, 55, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Goh, M.J.; Park, J.S.; Bae, J.H.; Kim, D.H.; Kim, H.K.; Na, Y.J. Effects of ortho-dihydroxyisoflavone derivatives from Korean fermented soybean paste on melanogenesis in B16 melanoma cells and human skin equivalents. Phytother. Res. 2012, 26, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.S.; Lin, C.G.; Wu, M.H.; Chang, T.S. Evaluation of depigmenting activity by 8-hydroxydaidzein in mouse B16 melanoma cells and human volunteers. Int. J. Mol. Sci. 2009, 10, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Funako, T.; Hayashi, H. 8-Hydroxydaidzein, an aldose reductase inhibitor from okara fermented with Aspergillus sp. HK-388. Biosci. Biotechnol. Biochem. 2004, 68, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.S.; Ding, H.Y.; Yen, J.H.; Chen, S.F.; Lee, K.H.; Wu, M.J. Anti-inflammatory activity of 8-hydroxydaidzein in LPS-stimulated BV2 microglial cells via activation of Nrf2-antioxidant and attenuation of Akt/NF-κB-inflammatory signaling pathways, as well as inhibition of COX-2 activity. J. Agric. Food Chem. 2018, 66, 5790–5801. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kang, Y.G.; Kim, J.H.; Kim, Y.J.; Lee, T.R.; Lee, J.; Kim, D.; Cho, J.Y. The antioxidant and anti-Inflammatory activities of 8-hydroxydaidzein (8-HD) in activated macrophage-Like RAW264.7 Cells. Int. J. Mol. Sci. 2018, 19, 1828. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S. 8-Hydroxydaidzein is unstable in alkaline solutions. J. Cosmet. Sci. 2009, 60, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Hamada, H.; Hamada, H. Synthesis of xylooligosaccharides of daidzein and their anti-oxidant and anti-allergic activities. Int. J. Mol. Sci. 2011, 12, 5616–5625. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Sangwan, R.S.; Sangwan, N.S. Plant secondary metabolism linked glycosyltransferases: An update on expanding knowledge and scopes. Biotechnol. Adv. 2016, 34, 716–739. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Yang, S.M.; Kim, S.Y.; Cha, M.N.; Ahn, J.H. Biosynthesis and production of glycosylated flavonoids in Escherichia coli: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 2979–2988. [Google Scholar] [CrossRef] [PubMed]
- Hofer, B. Recent developments in the enzymatic O-glycosylation of flavonoids. Appl. Microbiol. Biotechnol. 2016, 100, 4269–4281. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Chiang, C.M.; Siao, Y.Y.; Wu, J.Y. Sequential Biotransformation of Antcin K by Bacillus subtilis ATCC 6633. Catalysts 2018, 8, 349. [Google Scholar] [CrossRef]
- Pandey, R.P.; Gurung, R.B.; Parajuli, P.; Koirala, N.; Tuoi, L.T.; Sohng, J.K. Assessing acceptor substrates promiscuity of YjiC-mediated glycosylation towards flavonoids. Car. Res. 2014, 393, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, J.; Yang, J.; Zhu, Y.; Men, Y.; Zeng, Y.; Cai, Y.; Dong, C.; Dai, Z.; Zhang, X.; Sun, Y. Use of a promiscuous glycosyltransferase from Bacillus subtilis 168 for the enzymatic synthesis of novel protopanaxtriol-type ginsenosides. J. Agric. Food Chem. 2017, 66, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, J.; Yao, P.; Zhu, Y.; Men, Y.; Zeng, Y.; Yang, J.; Sun, Y. Exploiting the aglycon promiscuity of glycosyltransferase Bs-YjiC from Bacillus subtilis and its application in synthesis of glycosides. J. Biotechnol. 2017, 248, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Kim, B.G.; Anh, J.H. Glycosylation of flavonoids with a glycosyltransferase from Bacillus cereus. FEMS Mcrobiol. Lett. 2006, 258, 263–268. [Google Scholar]
- Ahn, B.C.; Kim, B.G.; Jeon, Y.M.; Lee, E.J.; Lim, Y.; Ahn, J.H. Formation of flavone di-O-glucosides using a glycosyltransferase from Bacillus cereus. J. Microbiol. Biotechnol. 2009, 19, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Hamza, A.; Zhan, C.G.; Thorson, J.S. Assessing the regioselectivity of OleD-catalyzed glcosylation with a diverse set of acceptors. J. Nat. Prod. 2013, 76, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, B.G.; Kim, J.A.; Park, Y.; Lee, Y.J.; Lim, Y.; Ahn, J.H. Glycosylation of flavonoids with E. coli expression glycosyltransferase from Xanthomonas campestris. J. Microbiol. Biotechnol. 2007, 17, 539–542. [Google Scholar] [PubMed]
- Adachi, J.; Hasegawa, M. Model of amino acid substitution in proteins encoded by mitochondrial DNA. J. Mol. Evol. 1996, 42, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ban, X.; Gu, Z.; Li, Z. Calcium ion contribution to thermostability of cyltrodetrin glycosyltransferase is closely related to calcium-binding site CaIII. J. Agric. Food Chem. 2013, 61, 8836–8841. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Wang, T.S. 3’-Isoflavone Glycosides Having Whitening and Anti-Aging Effects, Preparing Method and Use Thereof. Taiwan Patent I602580, 21 October 2017. [Google Scholar]
- Chiang, C.M.; Wang, T.S.; Chang, T.S. Improving free radical scavenging activity of soy isoflavone glycosides daidzin and genistin by 3’-hydroxylation using recombinant Escherichia coli. Molecules 2016, 21, 1723. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Aqueous Solubility (mg/L) | Fold 1 |
|---|---|---|
| 8-OHDe | 51.3 | 1 |
| 8-OHDe-7-O-β-glucoside | 462.0 | 9.0 |
| 8-OHDe-8-O-β-glucoside | 251.1 | 4.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, C.-M.; Wang, T.-Y.; Yang, S.-Y.; Wu, J.-Y.; Chang, T.-S. Production of New Isoflavone Glucosides from Glycosylation of 8-Hydroxydaidzein by Glycosyltransferase from Bacillus subtilis ATCC 6633. Catalysts 2018, 8, 387. https://doi.org/10.3390/catal8090387
Chiang C-M, Wang T-Y, Yang S-Y, Wu J-Y, Chang T-S. Production of New Isoflavone Glucosides from Glycosylation of 8-Hydroxydaidzein by Glycosyltransferase from Bacillus subtilis ATCC 6633. Catalysts. 2018; 8(9):387. https://doi.org/10.3390/catal8090387
Chicago/Turabian StyleChiang, Chien-Min, Tzi-Yuan Wang, Szu-Yi Yang, Jiumn-Yih Wu, and Te-Sheng Chang. 2018. "Production of New Isoflavone Glucosides from Glycosylation of 8-Hydroxydaidzein by Glycosyltransferase from Bacillus subtilis ATCC 6633" Catalysts 8, no. 9: 387. https://doi.org/10.3390/catal8090387
APA StyleChiang, C.-M., Wang, T.-Y., Yang, S.-Y., Wu, J.-Y., & Chang, T.-S. (2018). Production of New Isoflavone Glucosides from Glycosylation of 8-Hydroxydaidzein by Glycosyltransferase from Bacillus subtilis ATCC 6633. Catalysts, 8(9), 387. https://doi.org/10.3390/catal8090387