TiO2 Photocatalyzed C–H Bond Transformation for C–C Coupling Reactions


 ,
, 
Abstract
1. Introduction
2. C–H Activation Available to Convert Simple Molecules into Desired Complex C–C Coupling Products
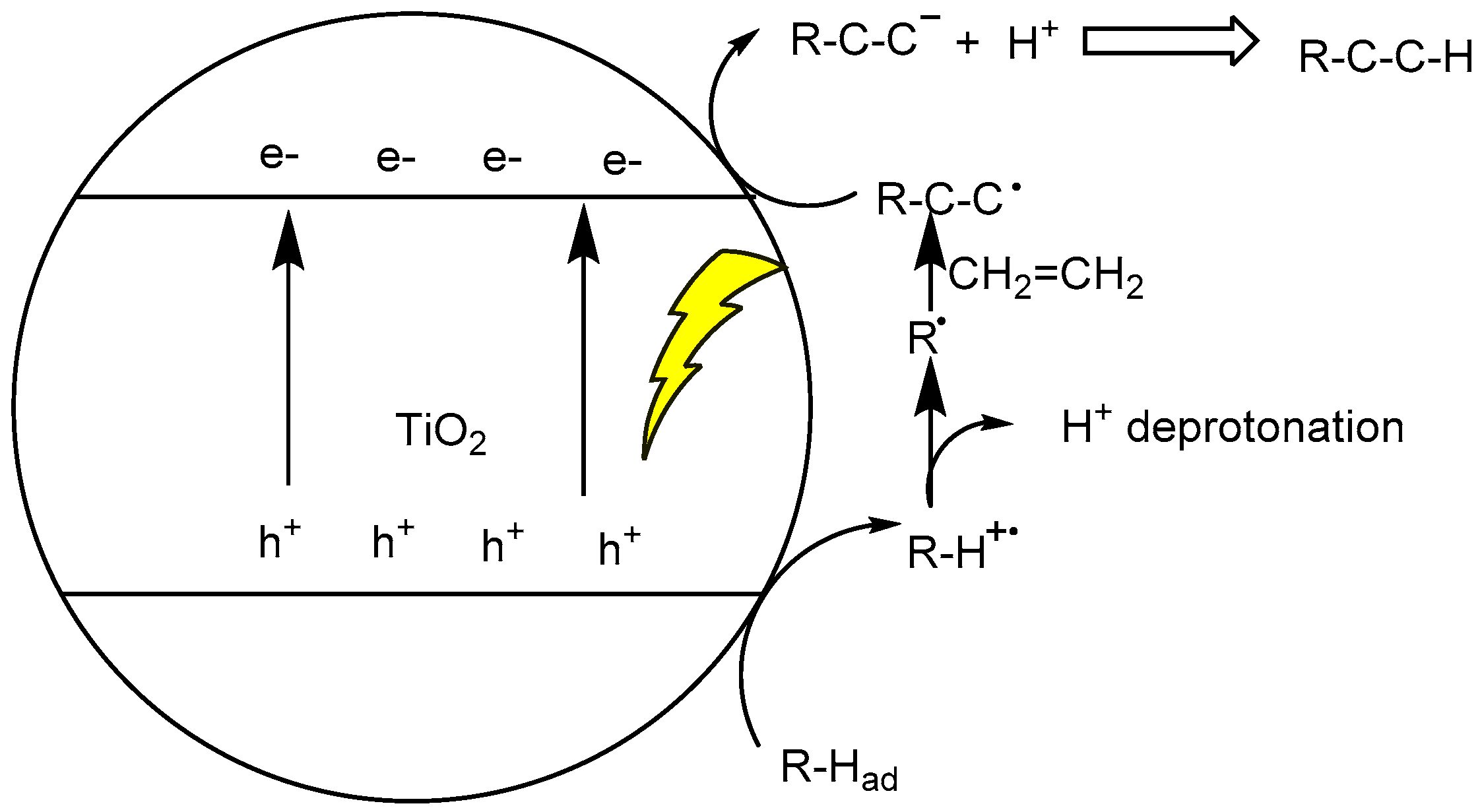
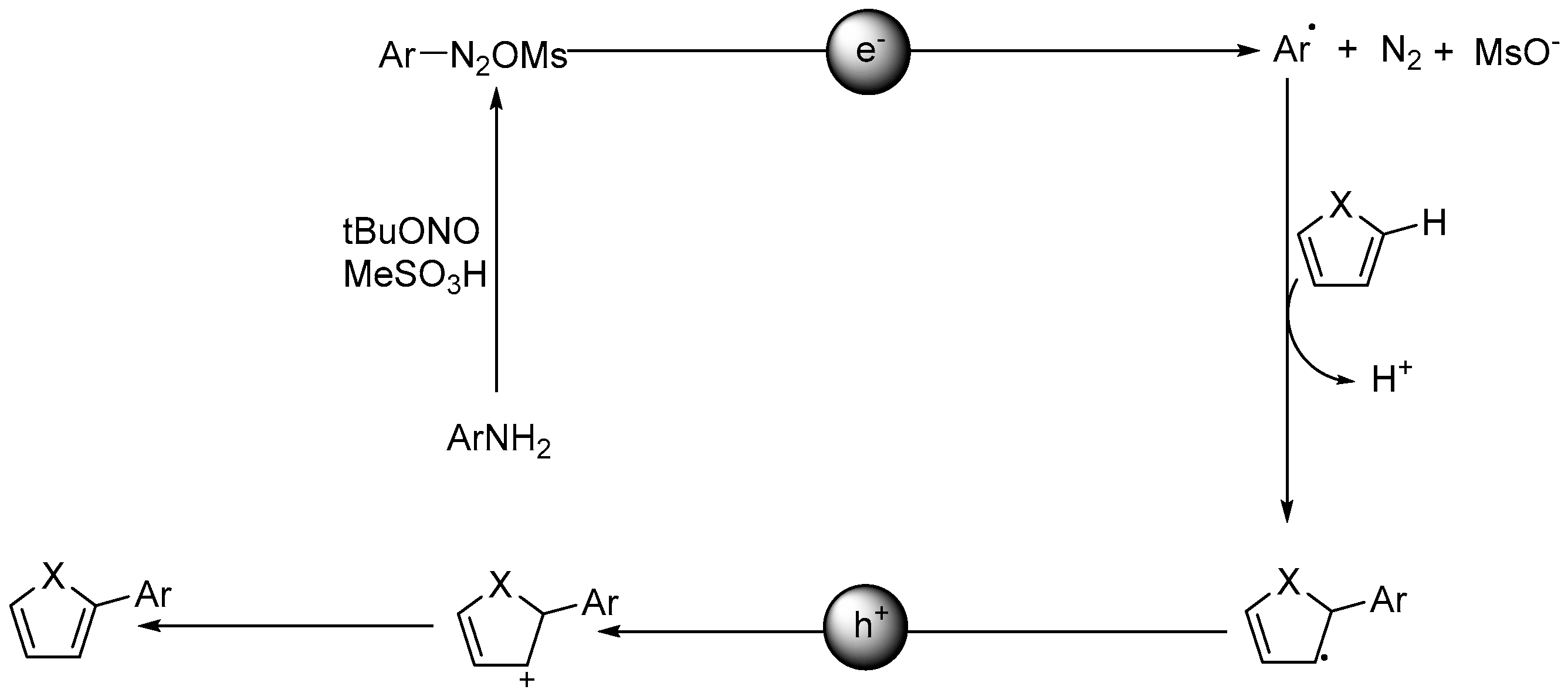
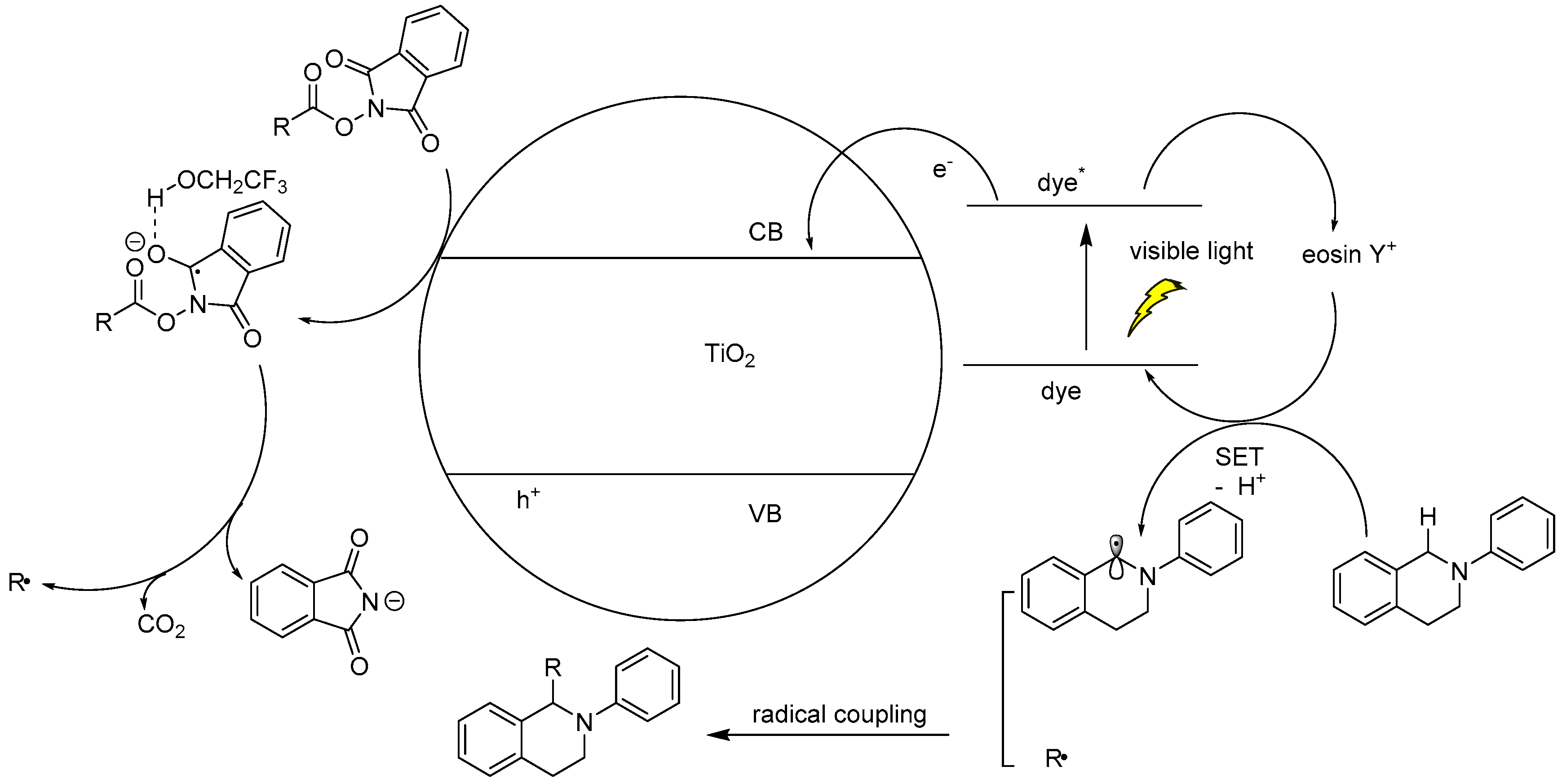
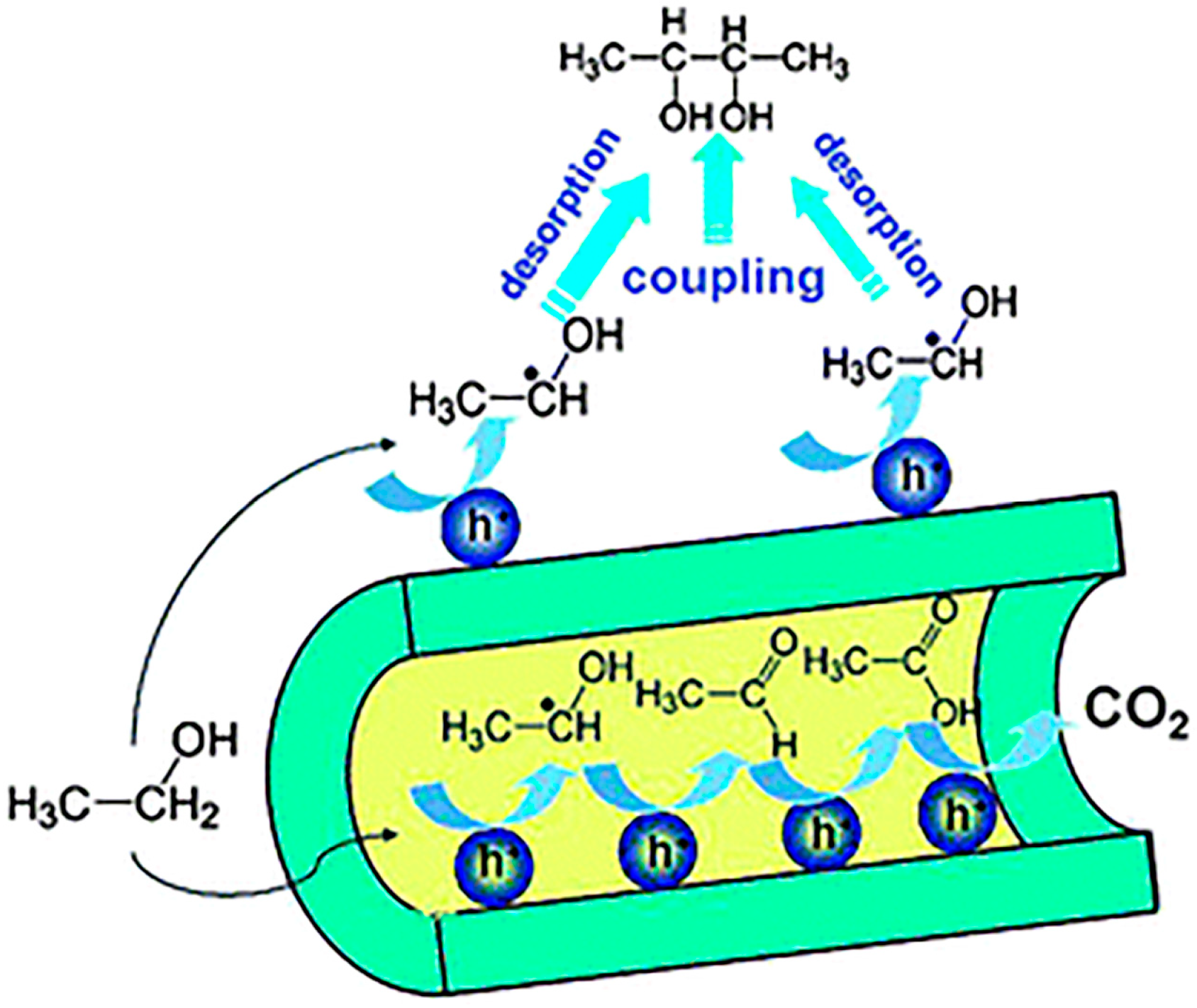
2.1. Activating C–H Bond to React with C=C or C–X Bond for C–C Coupling Reactions
2.2. C–H Bond Functionalization for C–C Bond Coupling Reactions
3. Conclusions
Funding
Conflicts of Interest
References
- Tian, C.; Massignan, L.; Meyer, T.H.; Ackermann, L. Electrochemical C–H/N–H Activation by Water-Tolerant Cobalt Catalysis at Room Temperature. Angew. Chem. Int. Ed. 2018, 57, 2383–2387. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-Metal-Catalyzed Direct Arylation of (Hetero)Arenes by C-H Bond Cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.; Wang, L.; Ding, Q.; Peng, Y. Nitrile as a Versatile Directing Group for C(sp2)–H Functionalizations. Adv. Synth. Catal. 2017, 359, 3274–3291. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, O.; Yu, J.-Q. Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.J.; Lin, D.W.; Miura, M.; Zhu, R.Y.; Gong, W.; Wasa, M.; Yu, J.Q. Palladium(II)-catalyzed enantioselective C(sp3) –H activation using a chiral hydroxamic acid ligand. J. Am. Chem. Soc. 2014, 136, 8138–8142. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.F.; Maugel, N.; Zhang, Y.H.; Yu, J.Q. Pd(II)-catalyzed enantioselective activation of C(sp2)–H and C(sp3) –H bonds using monoprotected amino acids as chiral ligands. Angew. Chem. Int. Ed. 2008, 47, 4882–4886. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Ren, Z.; Thompson, S.J.; Xu, Y.; Dong, G. Transition-Metal-Catalyzed C-H Alkylation Using Alkenes. Chem. Rev. 2017, 117, 9333–9403. [Google Scholar] [CrossRef] [PubMed]
- Daugulis, O.; Do, H.-Q.; Shabashov, D. Palladium- and Copper-Catalyzed Arylation of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2009, 42, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Bergman, R.G. Organometallic chemistry—C–H activation. Nature 2007, 446, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Cherney, A.H.; Kadunce, N.T.; Reisman, S.E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C–C Bonds. Chem. Rev. 2015, 115, 9587–9652. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Melchor, M.; Braga, A.A.C.; Lledos, A.; Ujaque, G.; Maseras, F. Computational Perspective on Pd-Catalyzed C–C Cross-Coupling Reaction Mechanisms. Acc. Chem. Res. 2013, 46, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jin, H.; Hashmi, A.S.K. The recent achievements of redox-neutral radical C–C cross-coupling enabled by visible-light. Chem. Soc. Rev. 2017, 46, 5193–5203. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.C.; Vila, I.C.; Neild, E. Ecotoxicity assessment of metals and wastewater using multitrophic assays. Environ. Toxicol. 2000, 15, 370–375. [Google Scholar] [CrossRef]
- Santoro, S.; Kozhushkov, S.I.; Ackermann, L.; Vaccaro, L. Heterogeneous catalytic approaches in C–H activation reactions. Green Chem. 2016, 18, 3471–3493. [Google Scholar] [CrossRef]
- Takanabe, K.; Domen, K. Preparation of Inorganic Photocatalytic Materials for Overall Water Splitting. ChemCatChem 2012, 4, 1485–1497. [Google Scholar] [CrossRef]
- Han, F.; Kambala, V.S.R.; Srinivasan, M.; Rajarathnam, D.; Naidu, R. Tailored titanium dioxide photocatalysts for the degradation of organic dyes in wastewater treatment: A review. Appl. Catal. A Gen. 2009, 359, 25–40. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Huang, F. Black titanium dioxide (TiO2) nanomaterials. Chem. Soc. Rev. 2015, 44, 1861–1885. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Zhang, X.T.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Rueping, M.; Zoller, J.; Fabry, D.C.; Poscharny, K.; Koenigs, R.M.; Weirich, T.E.; Mayer, J. Light-Mediated Heterogeneous Cross Dehydrogenative Coupling Reactions: Metal Oxides as Efficient, Recyclable, Photoredox Catalysts in C-C Bond-Forming Reactions. Chem. Eur. J. 2012, 18, 3478–3481. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shang, J.; Ai, Z.; Zhang, L. Efficient Visible Light Nitrogen Fixation with BiOBr Nanosheets of Oxygen Vacancies on the Exposed {001} Facets. J. Am. Chem. Soc. 2015, 137, 6393–6399. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhang, L.; Wang, J.; Li, Q.; He, W.; Yin, J.J. Surface Structure-Dependent Molecular Oxygen Activation of BiOCl Single-Crystalline Nanosheets. J. Am. Chem. Soc. 2013, 135, 15750–15753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhao, K.; Xiao, X.; Zhang, L. Synthesis and Facet-Dependent Photoreactivity of BiOCl Single-Crystalline Nanosheets. J. Am. Chem. Soc. 2012, 134, 4473–4476. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ho, W.; Ai, Z.; Song, X.; Zhang, L.; Lee, S. Aerosol-assisted flow synthesis of B-doped, Ni-doped and B-Ni-codoped TiO2 solid and hollow microspheres for photocatalytic removal of NO. Appl. Catal. B Environ. 2009, 89, 398–405. [Google Scholar] [CrossRef]
- Huang, Y.; Ho, W.; Lee, S.; Zhang, L.; Li, G.; Yu, J.C. Effect of carbon doping on the mesoporous structure of nanocrystalline titanium dioxide and its solar-light-driven photocatalytic degradation of NOx. Langmuir 2008, 24, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, Y.; Zhang, H.; Duan, R.; Chen, C.; Song, W.; Zhao, J. Localized TiIII mediated dissociative electron transfer for carbon halogen bond activation on TiO2. Appl. Catal. B Environ. 2017, 219, 322–328. [Google Scholar] [CrossRef]
- Lang, X.J.; Leow, W.R.; Zhao, J.C.; Chen, X.D. Synergistic photocatalytic aerobic oxidation of sulfides and amines on TiO2 under visible-light irradiation. Chem. Sci. 2015, 6, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lang, X.J.; Ma, W.H.; Ji, H.W.; Chen, C.C.; Zhao, J.C. Selective aerobic oxidation of amines to imines by TiO2 photocatalysis in water. Chem. Commun. 2013, 49, 5034–5036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.A.; Chen, C.C.; Ma, W.H.; Zhao, J.C. Visible-Light-Induced Aerobic Oxidation of Alcohols in a Coupled Photocatalytic System of Dye-Sensitized TiO2 and TEMPO. Angew. Chem. Int. Ed. 2008, 47, 9730–9733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, C.; Ma, W.; Zhu, H.; Zhao, J. Pivotal Role of Fluorine in Tuning Band Structure and Visible-Light Photocatalytic Activity of Nitrogen-Doped TiO2. Chem. Eur. J. 2009, 15, 4765–4769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Q.; Chen, C.C.; Zang, L.; Ma, W.H.; Zhao, J.C. Oxygen Atom Transfer in the Photocatalytic Oxidation of Alcohols by TiO2: Oxygen Isotope Studies. Angew. Chem. Int. Ed. 2009, 48, 6081–6084. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Ma, W.H.; Zhao, J.C. Semiconductor-mediated photodegradation of pollutants under visible-light irradiation. Chem. Soc. Rev. 2010, 39, 4206–4219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, M.A.; Chen, C.C.; Ma, W.H.; Zhao, J.C. Photocatalytic Aerobic Oxidation of Alcohols on TiO2: The Acceleration Effect of a Brønsted Acid. Angew. Chem. Int. Ed. 2010, 49, 7976–7979. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.J.; Ji, H.W.; Chen, C.C.; Ma, W.H.; Zhao, J.C. Selective Formation of Imines by Aerobic Photocatalytic Oxidation of Amines on TiO2. Angew. Chem. Int. Ed. 2011, 50, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, A.; Lu, C.; Chen, C. Photocatalytic Dehydrogenation of Primary Alcohols: Selectivity Goes against Adsorptivity. ACS Omega 2017, 2, 4161–4172. [Google Scholar] [CrossRef]
- Ma, D.; Yan, Y.; Ji, H.W.; Chen, C.C.; Zhao, J.C. Photocatalytic activation of pyridine for addition reactions: An unconventional reaction feature between a photo-induced hole and electron on TiO2. Chem. Commun. 2015, 51, 17451–17454. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, A.; Li, S.; Lu, C.; Chen, C. TiO2 photocatalysis for C-C bond formation. Catal. Sci. Technol. 2018, 8, 2030–2045. [Google Scholar] [CrossRef]
- Augugliaro, V.; Camera-Roda, G.; Loddo, V.; Palmisano, G.; Palmisano, L.; Soria, J.; Yurdakal, S. Heterogeneous Photocatalysis and Photoelectrocatalysis: From Unselective Abatement of Noxious Species to Selective Production of High-Value Chemicals. J. Phys. Chem. Lett. 2015, 6, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Augugliaro, V.; Bellardita, M.; Loddo, V.; Palmisano, G.; Palmisano, L.; Yurdakal, S. Overview on oxidation mechanisms of organic compounds by TiO2 in heterogeneous photocatalysis. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 224–245. [Google Scholar] [CrossRef]
- Palmisano, G.; Garcia-Lopez, E.; Marci, G.; Loddo, V.; Yurdakal, S.; Augugliaro, V.; Palmisano, L. Advances in selective conversions by heterogeneous photocatalysis. Chem. Commun. 2010, 46, 7074–7089. [Google Scholar] [CrossRef] [PubMed]
- Gambarotti, C.; Punta, C.; Recupero, F.; Caronna, T.; Palmisano, L. TiO2 in Organic Photosynthesis: Sunlight Induced Functionalization of Heterocyclic Bases. Curr. Org. Chem. 2010, 14, 1153–1169. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor Photocatalysis for Chemoselective Radical Coupling Reactions. Acc. Chem. Res. 2017, 50, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Kisch, H. Semiconductor Photocatalysis—Mechanistic and Synthetic Aspects. Angew. Chem. Int. Ed. 2013, 52, 812–847. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.J.; Zhao, J.C.; Chen, X.D. Cooperative photoredox catalysis. Chem. Soc. Rev. 2016, 45, 3026–3038. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.J.; Ma, W.H.; Chen, C.C.; Ji, H.W.; Zhao, J.C. Selective Aerobic Oxidation Mediated by TiO2 Photocatalysis. Acc. Chem. Res. 2014, 47, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.J.; Chen, X.D.; Zhao, J.C. Heterogeneous visible light photocatalysis for selective organic transformations. Chem. Soc. Rev. 2014, 43, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lang, X. Visible light photocatalysis of dye-sensitized TiO2: The selective aerobic oxidation of amines to imines. Appl. Catal. B Environ. 2018, 224, 404–409. [Google Scholar] [CrossRef]
- Lang, X.; Zhao, J. Integrating TEMPO and Its Analogues with Visible-Light Photocatalysis. Chem. Asian J. 2018, 13, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Lang, X. Merging visible light photocatalysis of dye-sensitized TiO2 with TEMPO: The selective aerobic oxidation of alcohols. Catal. Sci. Technol. 2017, 7, 4955–4963. [Google Scholar] [CrossRef]
- Ravelli, D.; Fagnoni, M.; Dondi, D.; Albini, A. Significance of TiO2 Photocatalysis for Green Chemistry. J. Adv. Oxid. Technol. 2011, 14, 40–46. [Google Scholar] [CrossRef]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Howe, R.F.; Rhydderch, S.; Walton, J.C. Unconventional Titania Photocatalysis: Direct Deployment of Carboxylic Acids in Alkylations and Annulations. J. Am. Chem. Soc. 2012, 134, 13580–13583. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N. Combining Photoredox and Metal Catalysis. Chemcatchem 2015, 7, 393–394. [Google Scholar] [CrossRef]
- Hoffmann, N. Photocatalysis with TiO2 Applied to Organic Synthesis. Aust. J. Chem. 2015, 68, 1621–1639. [Google Scholar] [CrossRef]
- Cherevatskaya, M.; Koenig, B. Heterogeneous photocatalysts in organic synthesis. Russ. Chem. Rev. 2014, 83, 183–195. [Google Scholar] [CrossRef]
- Vila, C.; Rueping, M. Visible-light mediated heterogeneous C-H functionalization: Oxidative multi-component reactions using a recyclable titanium dioxide (TiO2) catalyst. Green Chem. 2013, 15, 2056–2059. [Google Scholar] [CrossRef]
- Scandura, G.; Palmisano, G.; Yurdakal, S.; Tek, B.S.; Ozcan, L.; Loddo, V.; Augugliaro, V. Selective photooxidation of ortho-substituted benzyl alcohols and the catalytic role of ortho-methoxybenzaldehyde. J. Photochem. Photobiol. A Chem. 2016, 328, 122–128. [Google Scholar] [CrossRef]
- Palmisano, G.; Scandura, G.; Augugliaro, V.; Loddo, V.; Pace, A.; Tek, B.S.; Yurdakal, S.; Palmisano, L. Unexpectedly ambivalent O2 role in the autocatalytic photooxidation of 2-methoxybenzyl alcohol in water. J. Mol. Catal. A Chem. 2015, 403, 37–42. [Google Scholar] [CrossRef]
- Ozcan, L.; Yurdakal, S.; Augugliaro, V.; Loddo, V.; Palmas, S.; Palmisano, G.; Palmisano, L. Photoelectrocatalytic selective oxidation of 4-methoxybenzyl alcohol in water by TiO2 supported on titanium anodes. Appl. Catal. B Environ. 2013, 132, 535–542. [Google Scholar] [CrossRef]
- Yurdakal, S.; Augugliaro, V.; Loddo, V.; Palmisano, G.; Palmisano, L. Enhancing selectivity in photocatalytic formation of p-anisaldehyde in aqueous suspension under solar light irradiation via TiO2 N-doping. New J. Chem. 2012, 36, 1762–1768. [Google Scholar] [CrossRef]
- Palmisano, L.; Augugliaro, V.; Bellardita, M.; Di Paola, A.; Lopez, E.G.; Loddo, V.; Marci, G.; Palmisano, G.; Yurdakal, S. Titania Photocatalysts for Selective Oxidations in Water. Chemsuschem 2011, 4, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Augugliaro, V.; El Nazer, H.A.H.; Loddo, V.; Mele, A.; Palmisano, G.; Palmisano, L.; Yurdakal, S. Partial photocatalytic oxidation of glycerol in TiO2 water suspensions. Catal. Today 2010, 151, 21–28. [Google Scholar] [CrossRef]
- Augugliaro, V.; Kisch, H.; Loddo, V.; Lopez-Munoz, M.J.; Marquez-Alvarez, C.; Palmisano, G.; Palmisano, L.; Parrino, F.; Yurdakal, S. Photocatalytic oxidation of aromatic alcohols to aldehydes in aqueous suspension of home prepared titanium dioxide 2. Intrinsic and surface features of catalysts. Appl. Catal. A Gen. 2008, 349, 189–197. [Google Scholar] [CrossRef]
- Yurdakal, S.; Palmisano, G.; Loddo, V.; Augugliaro, V.; Palmisano, L. Nanostructured rutile TiO2 for selective photocatalytic oxidation of aromatic alcohols to aldehydes in water. J. Am. Chem. Soc. 2008, 130, 1568–1569. [Google Scholar] [CrossRef] [PubMed]
- Augugliaro, V.; Caronna, T.; Loddo, V.; Marci, G.; Palmisano, G.; Palmisano, L.; Yurdakal, S. Oxidation of aromatic alcohols in irradiated aqueous suspensions of commercial and home-prepared ruffle TiO2: A selectivity study. Chem. Eur. J. 2008, 14, 4640–4646. [Google Scholar] [CrossRef] [PubMed]
- Yurdakal, S.; Palmisano, G.; Loddo, V.; Alagoz, O.; Augugliaro, V.; Palmisano, L. Selective photocatalytic oxidation of 4-substituted aromatic alcohols in water with rutile TiO2 prepared at room temperature. Green Chem. 2009, 11, 510–516. [Google Scholar] [CrossRef]
- Zhang, H.X.; Zhu, Z.P.; Wu, Y.P.; Zhao, T.J.; Li, L. TiO2-photocatalytic acceptorless dehydrogenation coupling of primary alkyl alcohols into acetals. Green Chem. 2014, 16, 4076–4080. [Google Scholar] [CrossRef]
- Lang, X.J.; Ma, W.H.; Zhao, Y.B.; Chen, C.C.; Ji, H.W.; Zhao, J.C. Visible-Light-Induced Selective Photocatalytic Aerobic Oxidation of Amines into Imines on TiO2. Chem. Eur. J. 2012, 18, 2624–2631. [Google Scholar] [CrossRef] [PubMed]
- Teruhisa, O.; Takayo, K.; Keizo, N.; Michio, M. Stereospecific Epoxidation of 2-Hexene with Molecular Oxygen on Photoirradiated Titanium Dioxide Powder. Chem. Lett. 1998, 27, 877–878. [Google Scholar]
- Ohno, T.; Nakabeya, K.; Matsumura, M. Epoxidation of Olefins on Photoirradiated Titanium Dioxide Powder Using Molecular Oxygen as an Oxidant. J. Catal. 1998, 176, 76–81. [Google Scholar] [CrossRef]
- Zoller, J.; Fabry, D.C.; Rueping, M. Unexpected Dual Role of Titanium Dioxide in the Visible Light Heterogeneous Catalyzed C-H Arylation of Heteroarenes. ACS Catal. 2015, 5, 3900–3904. [Google Scholar] [CrossRef]
- Fabry, D.C.; Ho, Y.A.; Zapf, R.; Tremel, W.; Panthofer, M.; Rueping, M.; Rehm, T.H. Blue light mediated C-H arylation of heteroarenes using TiO2 as an immobilized photocatalyst in a continuous-flow microreactor. Green Chem. 2017, 19, 1911–1918. [Google Scholar] [CrossRef]
- Cermenati, L.; Richter, C.; Albini, A. Solar light induced carbon-carbon bond formation via TiO2 photocatalysis. Chem. Commun. 1998, 805–806. [Google Scholar] [CrossRef]
- Cermenati, L.; Mella, M.; Albini, A. Titanium dioxide photocatalysed alkylation of maleic acid derivatives. Tetrahedron 1998, 54, 2575–2582. [Google Scholar] [CrossRef]
- Cermenati, L.; Albini, A. Titanium dioxide photocatalysis for radical alkylation. J. Adv. Oxid. Technol. 2002, 5, 58–66. [Google Scholar] [CrossRef]
- Cermenati, L.; Fagnoni, M.; Albini, A. TiO2-photocatalyzed reactions of some benzylic donors. Canad. J. Chem. 2003, 81, 560–566. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, M.; Tung, C.-H.; Wang, Y. TiO2 Photocatalytic Cyclization Reactions for the Syntheses of Aryltetralones. ACS Catal. 2016, 6, 8389–8394. [Google Scholar] [CrossRef]
- Qiao, X.; Biswas, S.; Wu, W.; Zhu, F.; Tung, C.-H.; Wang, Y. Selective endoperoxide formation by heterogeneous TiO2 photocatalysis with dioxygen. Tetrahedron 2018, 74, 2421–2427. [Google Scholar] [CrossRef]
- Selvam, K.; Sakamoto, H.; Shiraishi, Y.; Hirai, T. Photocatalytic secondary amine synthesis from azobenzenes and alcohols on TiO2 loaded with Pd nanoparticles. New J. Chem. 2015, 39, 2856–2860. [Google Scholar] [CrossRef]
- Hirakawa, H.; Katayama, M.; Shiraishi, Y.; Sakamoto, H.; Wang, K.L.; Ohtani, B.; Ichikawa, S.; Tanaka, S.; Hirai, T. One-Pot Synthesis of Imines from Nitroaromatics and Alcohols by Tandem Photocatalytic and Catalytic Reactions on Degussa (Evonik) P25 Titanium Dioxide. ACS Appl. Mater. Interface 2015, 7, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Adolph, C.M.; Werth, J.; Selvaraj, R.; Wegener, E.C.; Uyeda, C. Dehydrogenative Transformations of Imines Using a Heterogeneous Photocatalyst. J. Org. Chem. 2017, 82, 5959–5965. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Fujiwara, K.; Sugano, Y.; Ichikawa, S.; Hirai, T. N-Monoalkylation of Amines with Alcohols by Tandem Photocatalytic and Catalytic Reactions on TiO2 Loaded with Pd Nanoparticles. ACS Catal. 2013, 3, 312–320. [Google Scholar] [CrossRef]
- Tsarev, V.N.; Morioka, Y.; Caner, J.; Wang, Q.; Ushimaru, R.; Kudo, A.; Naka, H.; Saito, S. N-Methylation of Amines with Methanol at Room Temperature. Org. Lett. 2015, 17, 2530–2533. [Google Scholar] [CrossRef] [PubMed]
- Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. Photocatalysis for the Formation of the C−C Bond. Chem. Rev. 2007, 107, 2725–2756. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, G.; Augugliaro, V.; Pagliaro, M.; Palmisano, L. Photocatalysis: A promising route for 21st century organic chemistry. Chem. Commun. 2007, 2007, 3425–3437. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Lu, C.; Wang, J.; Chen, Y.; Xu, Z.; Varma, R.S. Selectivity Enhancement in Heterogeneous Photocatalytic Transformations. Chem. Rev. 2017, 117, 1445–1514. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.L. Friedel-Crafts Acylations. Ind. Eng. Chem. 1959, 51, 1099–1101. [Google Scholar] [CrossRef]
- You, S.; Cai, Q.; Zeng, M. Chiral Bronsted Acid Catalyzed Friedel-Crafts Alkylation Reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. [Google Scholar] [CrossRef] [PubMed]
- Lacey, H.T. Friedel-Crafts Reactions. Ind. Eng. Chem. 1954, 46, 1827–1835. [Google Scholar] [CrossRef]
- Watson, W.J. Book Review of The Mizoroki−Heck Reaction. Org. Process Res. Dev. 2010, 14, 748. [Google Scholar] [CrossRef]
- Ozawa, F.; Kubo, A.; Hayashi, T. Catalytic Asymmetric Heck Reaction. In Selectivity in Catalysis; ACS Symposium Series 517; American Chemical Society: Washington, DC, USA, 1993; Volume 517, pp. 75–85. [Google Scholar]
- Calloway, N.O. The Friedel-Crafts Syntheses. Chem. Rev. 1935, 17, 327–392. [Google Scholar] [CrossRef]
- Sartori, G.; Maggi, R. Use of Solid Catalysts in Friedel−Crafts Acylation Reactions. Chem. Rev. 2006, 106, 1077–1104. [Google Scholar] [CrossRef] [PubMed]
- Sartori, G.; Maggi, R. Update 1 of: Use of Solid Catalysts in Friedel−Crafts Acylation Reactions. Chem. Rev. 2011, 111, 181–214. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.B.; Jørgensen, K.A. Catalytic Asymmetric Friedel−Crafts Alkylation Reactions—Copper Showed the Way. Chem. Rev. 2008, 108, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Cheprakov, A.V. The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Xu, Y.J.; Yuan, W.C.; Cui, X.; Cun, L.F.; Gong, L.Z. Rhodium-catalyzed asymmetric nitroallylation of arylmetallics with cyclic nitroallyl acetates and applications in organic synthesis. Eur. J. Org. Chem. 2006, 18, 4093–4105. [Google Scholar] [CrossRef]
- Wasa, M.; Engle, K.M.; Lin, D.W.; Yoo, E.J.; Yu, J.Q. Pd(II)-catalyzed enantioselective C–H activation of cyclopropanes. J. Am. Chem. Soc. 2011, 133, 19598–19601. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Yang, C.; Ji, H.; Chen, C.; Ma, W.; Zhao, J. Mechanistic Studies of TiO2 Photocatalysis and Fenton Degradation of Hydrophobic Aromatic Pollutants in Water. Chem. Asian J. 2016, 11, 3568–3574. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental Applications of Semiconductor Photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Gilbert, J.C.; Giamalva, D.H. Alkylidenecarbenes from 1-diazo-1-alkenes: Their electrophilicity and stereochemistry of [2 + 2]-cycloaddition. J. Org. Chem. 1992, 57, 4185–4188. [Google Scholar] [CrossRef]
- Qiu, H.; Srinivas, H.D.; Zavalij, P.Y.; Doyle, M.P. Unprecedented Intramolecular [4 + 2]-Cycloaddition between a 1,3-Diene and a Diazo Ester. J. Am. Chem. Soc. 2016, 138, 1808–1811. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shen, J.; Yang, S.; Liu, W.; Chen, Q.; He, M. C-H arylation reactions through aniline activation catalysed by a PANI-g-C3N4-TiO2 composite under visible light in aqueous medium. Green Chem. 2018, 20, 1290–1296. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Chen, Q.; He, M. Photocatalyzed facile synthesis of 2,5-diaryl 1,3,4-oxadiazoles with polyaniline-g-C3N4-TiO2 composite under visible light. Tetrahedron Lett. 2018, 59, 1489–1492. [Google Scholar] [CrossRef]
- Cherevatskaya, M.; Neumann, M.; Fueldner, S.; Harlander, C.; Kuemmel, S.; Dankesreiter, S.; Pfitzner, A.; Zeitler, K.; Koenig, B. Visible-Light-Promoted Stereoselective Alkylation by Combining Heterogeneous Photocatalysis with Organocatalysis. Angew. Chem. Int. Ed. 2012, 51, 4062–4066. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Yoshida, M. Redox system for perfluoroalkylation of arenes and α-methylstyrene derivatives using titanium oxide as photocatalyst. J. Fluorine Chem. 2009, 130, 926–932. [Google Scholar] [CrossRef]
- Iizuka, M.; Fukushima, S.; Yoshida, M. Perfluoroalkylation of alpha-methylstyrene using titanium oxide as a photocatalyst. Chem. Lett. 2007, 36, 1042–1043. [Google Scholar] [CrossRef]
- Tang, J.; Grampp, G.; Liu, Y.; Wang, B.-X.; Tao, F.-F.; Wang, L.-J.; Liang, X.-Z.; Xiao, H.-Q.; Shen, Y.-M. Visible Light Mediated Cyclization of Tertiary Anilines with Maleimides Using Nickel(II) Oxide Surface-Modified Titanium Dioxide Catalyst. J. Org. Chem. 2015, 80, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.M.; Wu, T.X.; Zhao, J.C.; Hidaka, H.; Serpone, N. Photoassisted degradation of dye pollutants. 8. Irreversible degradation of alizarin red under visible light radiation in air-equilibrated aqueous TiO2 dispersions. Environ. Sci. Technol. 1999, 33, 2081–2087. [Google Scholar] [CrossRef]
- Wu, T.X.; Lin, T.; Zhao, J.C.; Hidaka, H.; Serpone, N. TiO2-assisted photodegradation of dyes. 9. Photooxidation of a squarylium cyanine dye in aqueous dispersions under visible light irradiation. Environ. Sci. Technol. 1999, 33, 1379–1387. [Google Scholar] [CrossRef]
- Baghbanzadeh, M.; Glasnov, T.N.; Kappe, C.O. Continuous-flow production of photocatalytically active titanium dioxide nanocrystals and its application to the photocatalytic addition of n, n-dimethylaniline to n-methylmaleimide. J. Flow Chem. 2013, 3, 109–113. [Google Scholar] [CrossRef]
- Guan, B.-T.; Hou, Z. Rare-earth-catalyzed C-H bond addition of pyridines to olefins. J. Am. Chem. Soc. 2011, 133, 18086–18089. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Wylie, W.N.O.; Hou, Z. Enantioselective C-H bond addition of pyridines to alkenes catalyzed by chiral half-sandwich rare-earth complexes. J. Am. Chem. Soc. 2014, 136, 12209–12212. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Cong, H. Visible-light-driven decarboxylative alkylation of C-H bond catalyzed by dye-sensitized semiconductor. Org. Lett. 2018, 20, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- Caronna, T.; Gambarotti, C.; Palmisano, L.; Punta, C.; Recupero, F. Sunlight induced functionalisation of some heterocyclic bases in the presence of polycrystalline TiO2. Chem. Commun. 2003, 2003, 2350–2351. [Google Scholar] [CrossRef]
- Caronna, T.; Gambarotti, C.; Palmisano, L.; Punta, C.; Recupero, F. Sunlight-induced reactions of some heterocyclic bases with ethers in the presence of TiO2—A green route for the synthesis of heterocyclic aldehydes. J. Photochem. Photobiol A Chem. 2005, 171, 237–242. [Google Scholar] [CrossRef]
- Yang, P.; Zhao, J.; Cao, B.; Li, L.; Wang, Z.; Tian, X.; Jia, S.; Zhu, Z. Selective photocatalytic cc coupling of bioethanol into 2,3-butanediol over Pt-decorated hydroxyl-group-tunable TiO2 photocatalysts. Chemcatchem 2015, 7, 2384–2390. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, J.; Li, L.; Gong, L.; Zheng, J.; Zhang, L.; Wang, Z.; Zhang, J.; Zhu, Z. Selective oxidation of sacrificial ethanol over TiO2-based photocatalysts during water splitting. Energ Environ. Sci. 2011, 4, 3384–3388. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
 Yield 94% |  Yield 79% |  Yield 90% |  Yield 90% |
 Yield 83% |  Yield 77% |  Yield 80% |  Yield 64% |
 Yield 80% |  Yield 96% |  Yield 88% |  Yield 72% |
 Yield 67% |  Yield 55% |  Yield 53% |  Yield 54% |
 Yield 79% |  Yield 53% | ||
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | 1 | R3 | 2 | 3 | Yield (%) |
| 1 | Me | H | 1a | Ph | 2a | 3a | 75 |
| 2 | Me | H | 1a | 4-MeOPh | 2b | 3b | 64 |
| 3 | Me | H | 1a | 4-MePh | 2c | 3c | 89 |
| 4 | Me | H | 1a | 4-FPh | 2d | 3d | 81 |
| 5 | Me | H | 1a | 4-ClPh | 2e | 3e | 62 |
| 6 | Me | H | 1a | Me | 2f | 3f | 67 |
| 7 | Me | H | 1a | t-Bu | 2g | 3g | 69 |
| 8 | Me | H | 1a | Bn | 2h | 3h | 68 |
| 9 | Me | 4-Me | 1b | Ph | 2a | 3i | 85 |
| 10 | Me | 4-Me | 1b | 4-MeOPh | 2b | 3j | 68 |
| 11 | Me | 4-Me | 1b | 4-MePh | 2c | 3k | 93 |
| 12 | Me | 4-Me | 1b | 4-FPh | 2d | 3l | 86 |
| 13 | Me | 4-Me | 1b | 4-ClPh | 2e | 3m | 73 |
| 14 | Me | 4-Me | 1b | Me | 2f | 3n | 77 |
| 15 | Me | 4-Me | 1b | Bn | 2h | 3o | 77 |
| 16 | Me | 4-MeO | 1c | Ph | 2a | 3p | 69 |
| 17 | Me | 2-Me | 1d | Ph | 2a | 3q | 64 |
| 18 | Me | 4-F | 1e | Ph | 2a | 3r | 86 |
| 19 | Me | 4-Cl | 1f | Ph | 2a | 3s | 75 |
| 20 | Me | 4-Br | 1g | Ph | 2a | 3t | 61 |
| 21 | Me | 4-Br | 1g | 4-MePh | 2c | 3u | 68 |
| 22 | Me | 4-Br | 1g | 4-ClPh | 2e | 3v | 52 |
| 23 | Me | 4-Br | 1g | Me | 2f | 3w | 65 |
| 24 | Me | 4-Br | 1g | Bn | 2h | 3x | 45 |
| 25 | Et | H | 1h | Ph | 2a | 3y | 71 |
| 26 | Ph | H | 1i | 4-MePh | 2c | 3z | 48 |
 | |||
 Yield 85% |  Yield 82% |  Yield 82% |  Yield 71% |
 Yield 77% |  Yield 62% |  Yield 87% |  Yield 69% |
 Yield 66% |  Yield 75% |  Yield 85% |  Yield 90% |
 Yield 76% |  Yield 44% |  Yield 79% |  Yield 74% |
 Yield 62% |  Yield 54% | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, A.; Ma, D.; Li, S.; Lu, C.; Li, T.; Chen, C. TiO2 Photocatalyzed C–H Bond Transformation for C–C Coupling Reactions. Catalysts 2018, 8, 355. https://doi.org/10.3390/catal8090355
Wang Y, Liu A, Ma D, Li S, Lu C, Li T, Chen C. TiO2 Photocatalyzed C–H Bond Transformation for C–C Coupling Reactions. Catalysts. 2018; 8(9):355. https://doi.org/10.3390/catal8090355
Chicago/Turabian StyleWang, Yi, Anan Liu, Dongge Ma, Shuhong Li, Chichong Lu, Tao Li, and Chuncheng Chen. 2018. "TiO2 Photocatalyzed C–H Bond Transformation for C–C Coupling Reactions" Catalysts 8, no. 9: 355. https://doi.org/10.3390/catal8090355
APA StyleWang, Y., Liu, A., Ma, D., Li, S., Lu, C., Li, T., & Chen, C. (2018). TiO2 Photocatalyzed C–H Bond Transformation for C–C Coupling Reactions. Catalysts, 8(9), 355. https://doi.org/10.3390/catal8090355