Effects of Mesopore Internal Surfaces on the Structure of Immobilized Pd-Bisphosphine Complexes Analyzed by Variable-Temperature XAFS and Their Catalytic Performances

 ,
,
Abstract
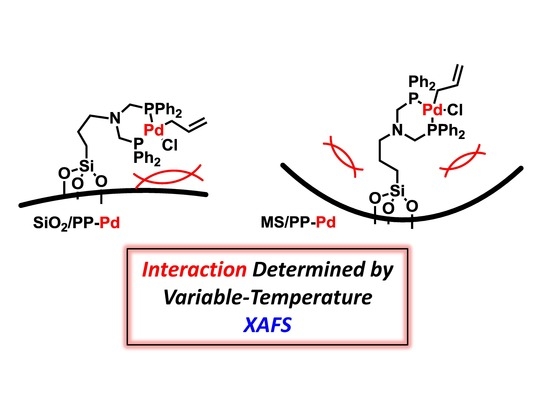
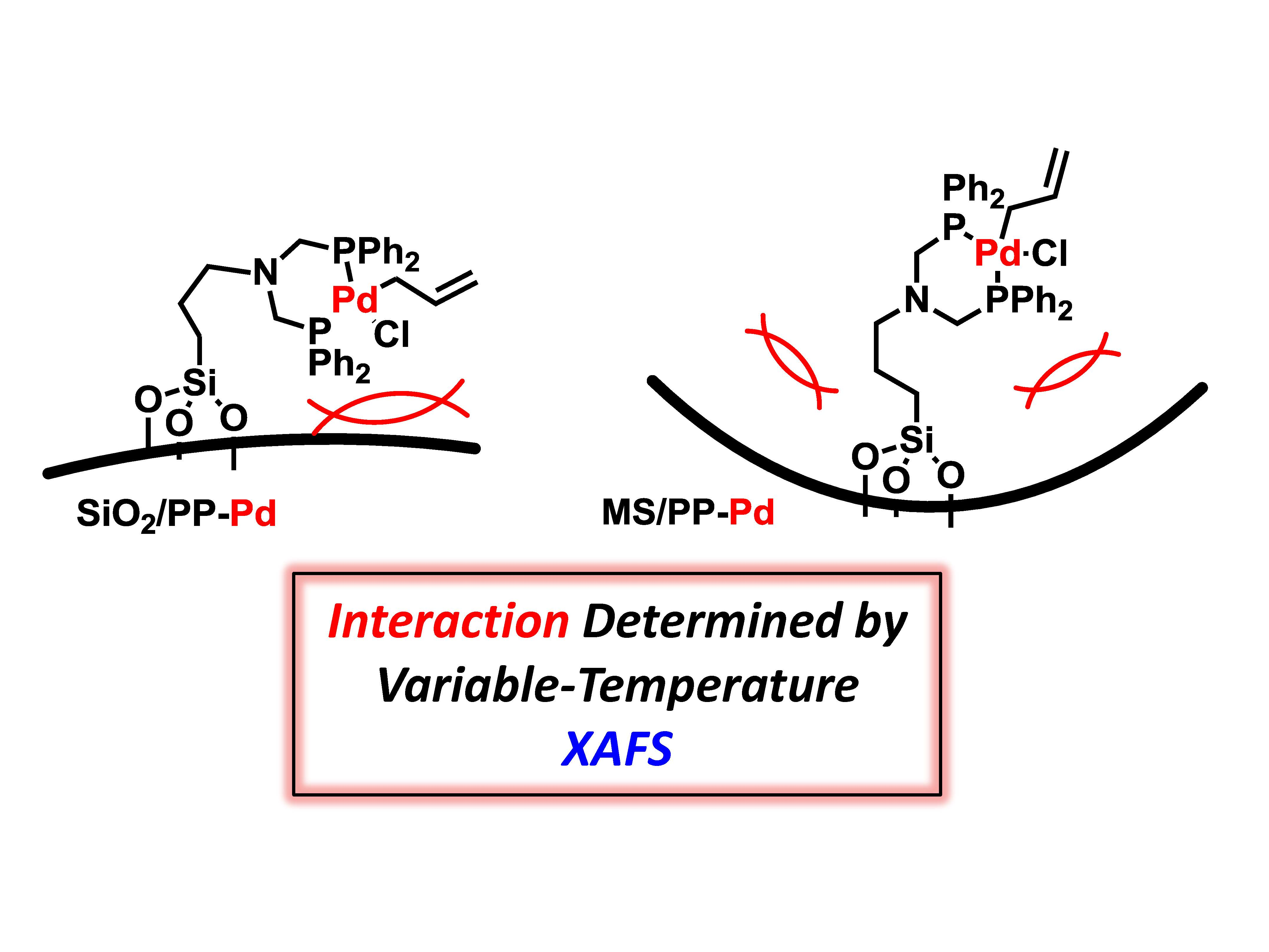
1. Introduction
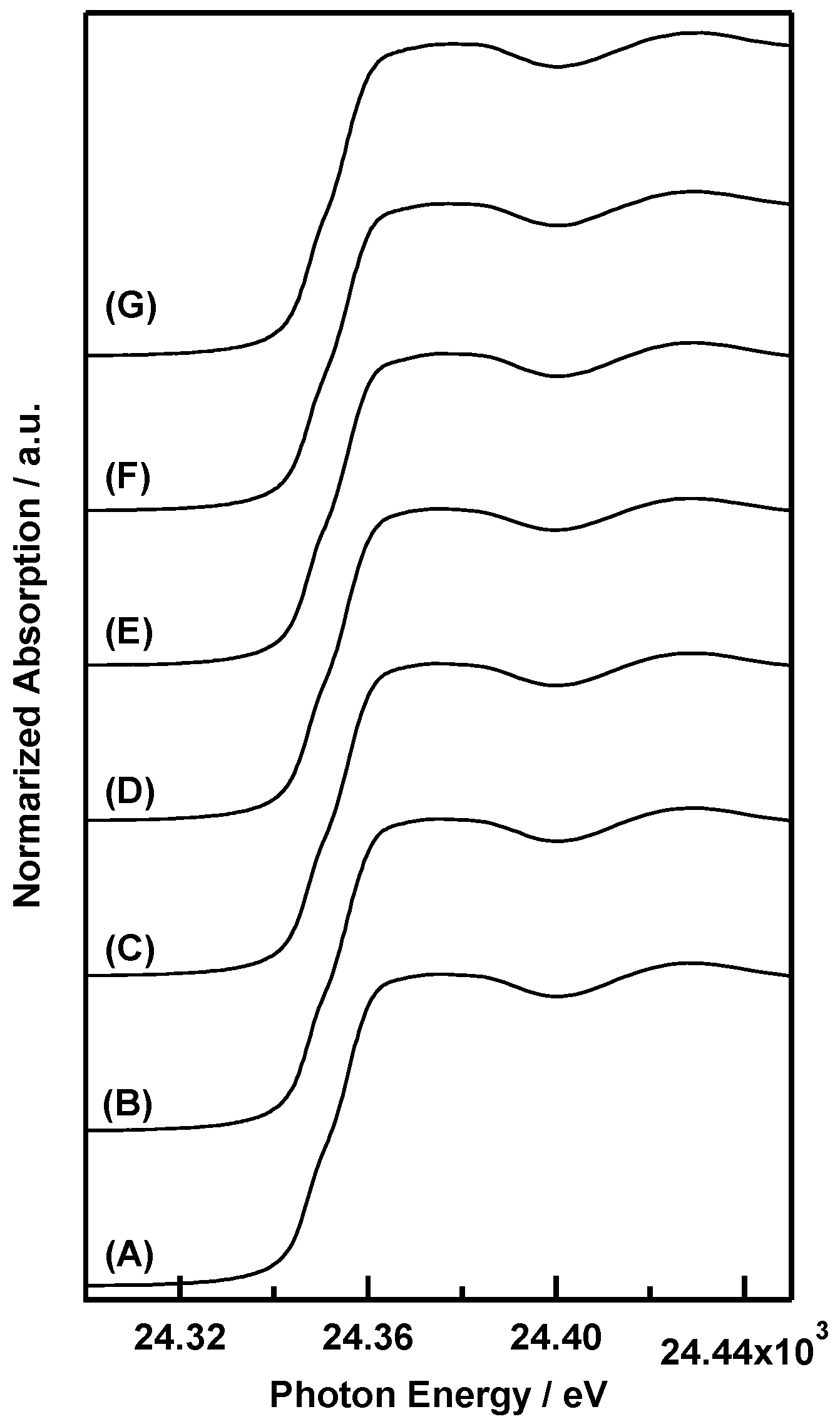
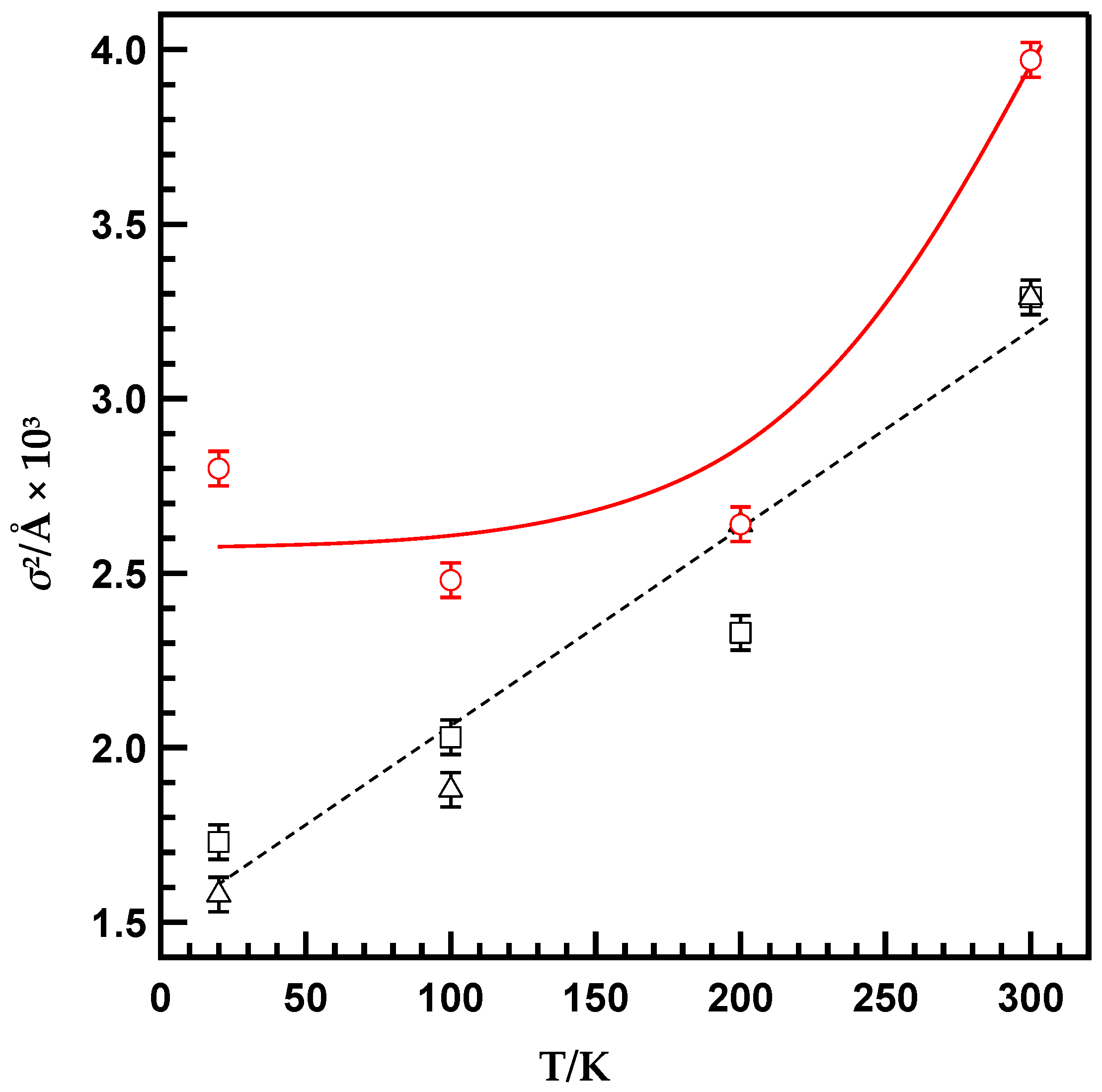
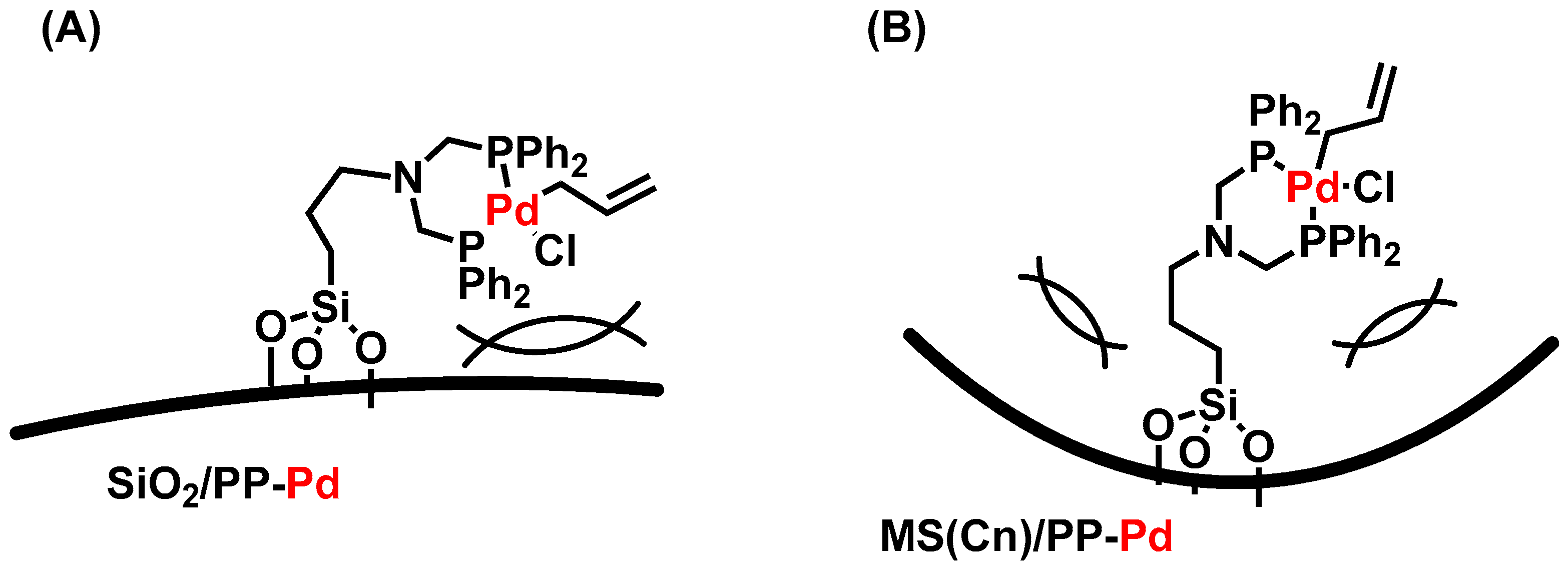
2. Results and Discussion
3. Experimental
3.1. Preparation of Mesoporous Silica (MS)
3.2. Preparation of MS/PP-Pd
3.3. Variable-Temperature XAFS Measurements
3.4. Suzuki-Miyaura Cross Coupling Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cheng, T.; Zhao, Q.; Zhang, D.; Liu, G. Transition-metal-functionalized ordered mesoporous silicas: An overview of sustainable chiral catalysts for enantioselective transformations. Green Chem. 2015, 17, 2100–2122. [Google Scholar] [CrossRef]
- Opanasenko, M.; Štěpnička, P.; Čejika, J. Heterogeneous Pd catalysts supported on silica matrices. RSC Adv. 2014, 4, 65137–65162. [Google Scholar] [CrossRef]
- Rostamnia, S.; Doustkhah, E. Nanoporous silica-supported organocatalyst: A heterogeneous and green hybrid catalyst for organic transformations. RSC Adv. 2014, 4, 28238–28248. [Google Scholar] [CrossRef]
- Conley, M.P.; Copéret, C.; Thieuleux, C. Mesostructured hybrid organic-silica materials: Ideal supports for well-defined heterogeneous organometallic catalysts. ACS Catal. 2014, 4, 1458–1469. [Google Scholar] [CrossRef]
- Yu, C.; He, J. Synergic catalytic effects in confined spaces. Chem. Commun. 2012, 48, 4933–4940. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, T.; Kubota, Y.; Tatsumi, T. Amino-functionalized mesoporous silica as base catalyst and adsorbent. Appl. Catal. A Gen. 2012, 421–422, 14–37. [Google Scholar] [CrossRef]
- Linares, N.; Serrano, E.; Rico, M.; Balu, A.M.; Losada, E.; Luque, R.; García-Martínez, J. Incorporation of chemical functionalities in the framework of mesoporous silica. Chem. Commun. 2011, 47, 9024–9035. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Albiter, M.A.; Zhang, Q.; Ge, J.; Yin, Y.; Zaera, F. New nanostructured heterogeneous catalysts with increased selectivity and stability. Phys. Chem. Chem. Phys. 2011, 13, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Brühwiler, D. Postsynthetic functionalization of mesoporous silica. Nanoscale 2010, 2, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Vivero-Escoto, J.L.; Trewyn, B.G.; Lin, V.S.-Y. Mesoporous silica nanoparticles: Structural design and applications. J. Mater. Chem. 2010, 20, 7924–7937. [Google Scholar] [CrossRef]
- Motokura, K.; Saitoh, K.; Noda, H.; Chun, W.-J.; Miyaji, A.; Yamaguchi, S.; Baba, T. A Pd-bisphosphine complex and organic functionalities immobilized on the same SiO2 surface: Detailed characterization and its use as an efficient catalyst for allylation. Catal. Sci. Technol. 2016, 6, 5380–5388. [Google Scholar] [CrossRef]
- Motokura, K.; Ikeda, M.; Nambo, M.; Chun, W.-J.; Nakajima, K.; Tanaka, S. Concerted catalysis in tight spaces: Palladium-catalyzed allylation reactions accelerated by accumulated active sites in mesoporous silica. ChemCatChem 2017, 9, 2924–2929. [Google Scholar] [CrossRef]
- Tanev, P.T.; Pinnavaia, T.J. A neutral templating route to mesoporous molecular sieves. Science 1995, 267, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, R.S.; Graminha, A.E.; Ellena, J.; Batista, A.A. Weak C–H⋯Cl–Pd interactions toward conformational polymorphism in trans-dichloridobis(triphenylphosphane)palladium(II). Acta Cryst. 2011, C67, m304–m306. [Google Scholar]
- Noda, H.; Motokura, K.; Wakabayashi, Y.; Sasaki, K.; Tajiri, H.; Miyaji, A.; Yamaguchi, S.; Baba, T. Direct estimation of the surface location of immobilized functional groups for concerted catalysis using a probe molecule. Chem. Eur. J. 2016, 22, 5113–5117. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support | Surface Area [m2/g] | Pore Size [Å] |
|---|---|---|
| MS(C8) | 1865 | 16 |
| MS(C12) | 1175 | 23 |
| MS(C18) | 876 | 31 |
| SiO2 a | 300 ± 30 | - |
| Temperature [K] | N [b] | r [c] [Å] | σ2 [d] [Å2 × 10−3] | ∆E [e] [eV] | Rf [f] [%] |
|---|---|---|---|---|---|
| SiO2/PP-Pd | |||||
| 300 | 3 | 2.27 ± 0.01 | 3.97 ± 0.05 | –10.69 ± 2.05 | 1.72 |
| 200 | 2.27 ± 0.01 | 2.64 ± 0.05 | –9.94 ± 2.02 | 3.37 | |
| 100 | 2.27 ± 0.01 | 2.48 ± 0.05 | –11.49 ± 2.08 | 1.90 | |
| 20 | 2.28 ± 0.01 | 2.80 ± 0.05 | –9.71 ± 2.09 | 2.16 | |
| MS(C18)/PP-Pd | |||||
| 300 | 3 | 2.27 ± 0.01 | 3.29 ± 0.05 | –11.46 ± 2.05 | 1.99 |
| 200 | 2.27 ± 0.01 | 2.64 ± 0.05 | –10.79 ± 2.07 | 1.58 | |
| 100 | 2.26 ± 0.01 | 1.88 ± 0.05 | –11.04 ± 2.09 | 1.91 | |
| 20 | 2.27 ± 0.01 | 1.58 ± 0.05 | –10.11 ± 2.09 | 3.56 | |
| MS(C8)/PP-Pd | |||||
| 300 | 3 | 2.27 ± 0.01 | 3.29 ± 0.05 | –11.14 ± 2.05 | 1.96 |
| 200 | 2.27 ± 0.01 | 2.33 ± 0.05 | –10.85 ± 2.06 | 2.11 | |
| 100 | 2.27 ± 0.01 | 2.03 ± 0.05 | –9.67 ± 2.08 | 1.53 | |
| 20 | 2.27 ± 0.01 | 1.73 ± 0.05 | –11.14 ± 2.09 | 2.00 |

| Catalyst | Conv. of Bromobenzene [%] | Yield [%] | TON [Pd−1] |
|---|---|---|---|
| MS(C8)/PP-Pd | 91 | 82 | 126 |
| MS(C12)/PP-Pd | 86 | 86 | 132 |
| MS(C18)/PP-Pd | 93 | 90 | 138 |
| SiO2/PP-Pd | 87 | 83 | 128 |
| PP-Pd | 82 | 47 | 72 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motokura, K.; Fukuda, T.; Uemura, Y.; Matsumura, D.; Ikeda, M.; Nambo, M.; Chun, W.-J. Effects of Mesopore Internal Surfaces on the Structure of Immobilized Pd-Bisphosphine Complexes Analyzed by Variable-Temperature XAFS and Their Catalytic Performances. Catalysts 2018, 8, 106. https://doi.org/10.3390/catal8030106
Motokura K, Fukuda T, Uemura Y, Matsumura D, Ikeda M, Nambo M, Chun W-J. Effects of Mesopore Internal Surfaces on the Structure of Immobilized Pd-Bisphosphine Complexes Analyzed by Variable-Temperature XAFS and Their Catalytic Performances. Catalysts. 2018; 8(3):106. https://doi.org/10.3390/catal8030106
Chicago/Turabian StyleMotokura, Ken, Takuma Fukuda, Yohei Uemura, Daiju Matsumura, Marika Ikeda, Masayuki Nambo, and Wang-Jae Chun. 2018. "Effects of Mesopore Internal Surfaces on the Structure of Immobilized Pd-Bisphosphine Complexes Analyzed by Variable-Temperature XAFS and Their Catalytic Performances" Catalysts 8, no. 3: 106. https://doi.org/10.3390/catal8030106
APA StyleMotokura, K., Fukuda, T., Uemura, Y., Matsumura, D., Ikeda, M., Nambo, M., & Chun, W.-J. (2018). Effects of Mesopore Internal Surfaces on the Structure of Immobilized Pd-Bisphosphine Complexes Analyzed by Variable-Temperature XAFS and Their Catalytic Performances. Catalysts, 8(3), 106. https://doi.org/10.3390/catal8030106