
The Catalytic Hydrogenation of Maleic Anhydride on CeO2−δ-Supported Transition Metal Catalysts

 ,
,
Abstract
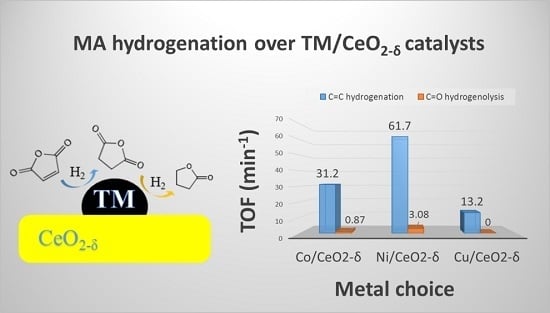
:
1. Introduction
2. Results and Discussion
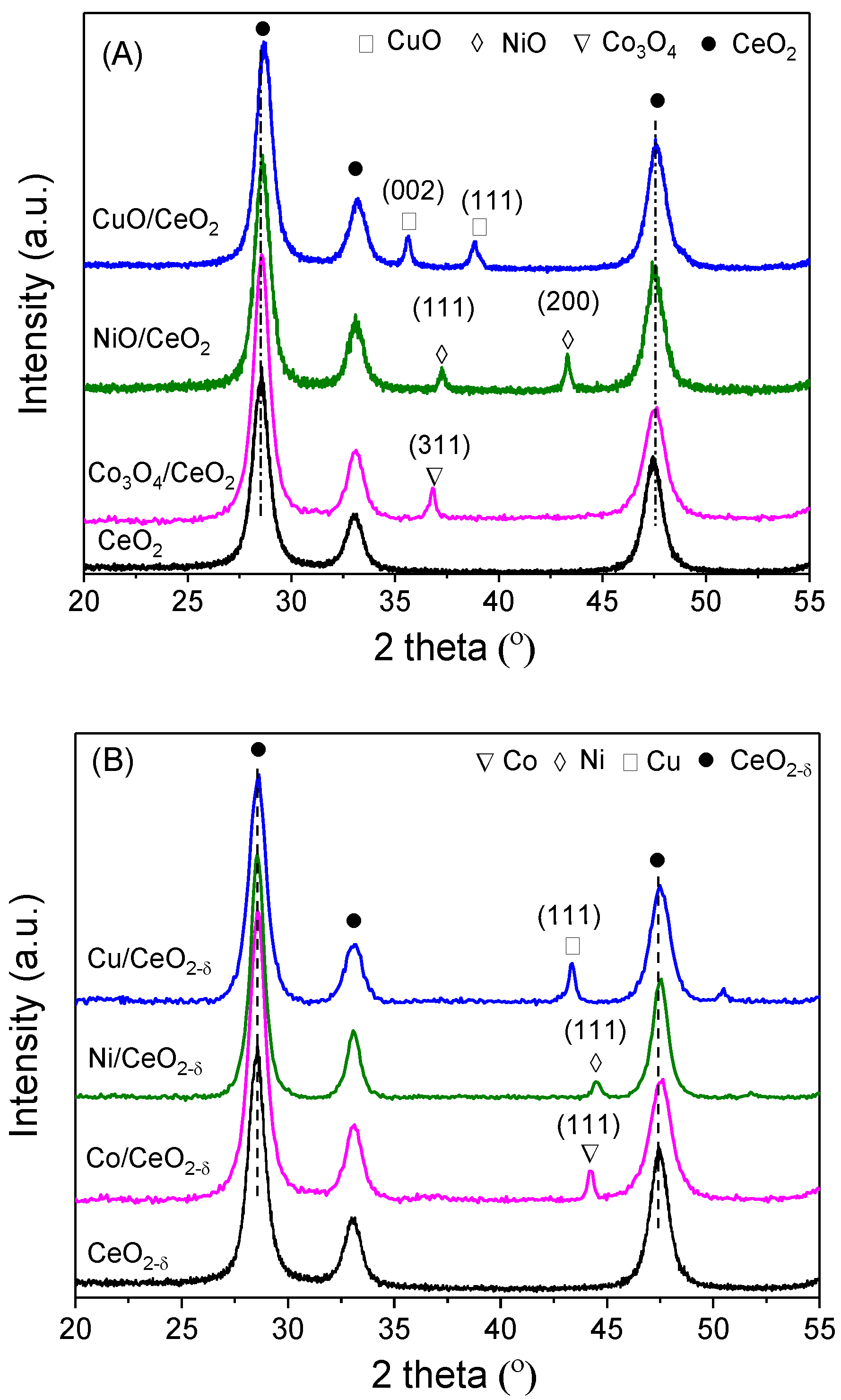
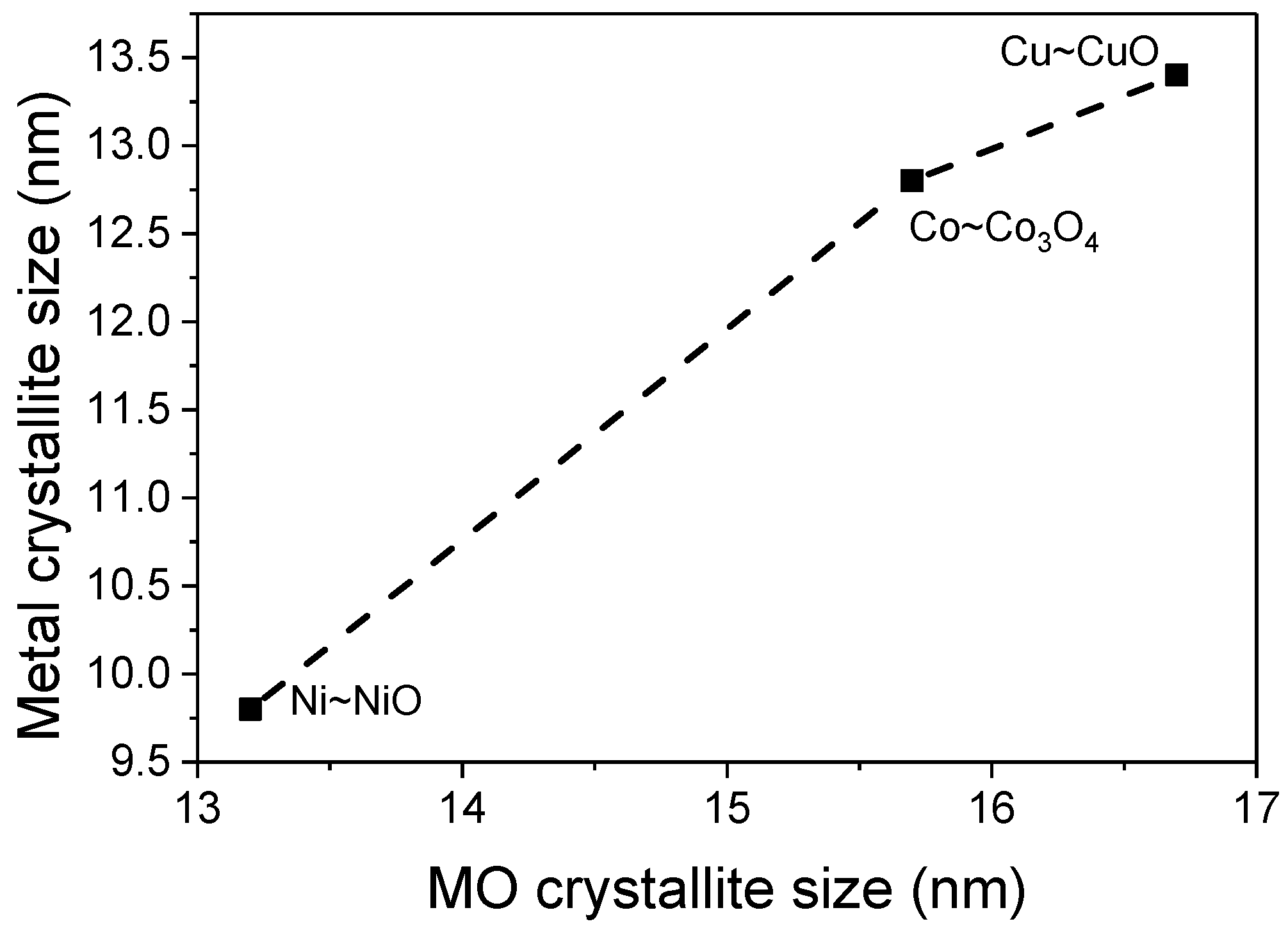
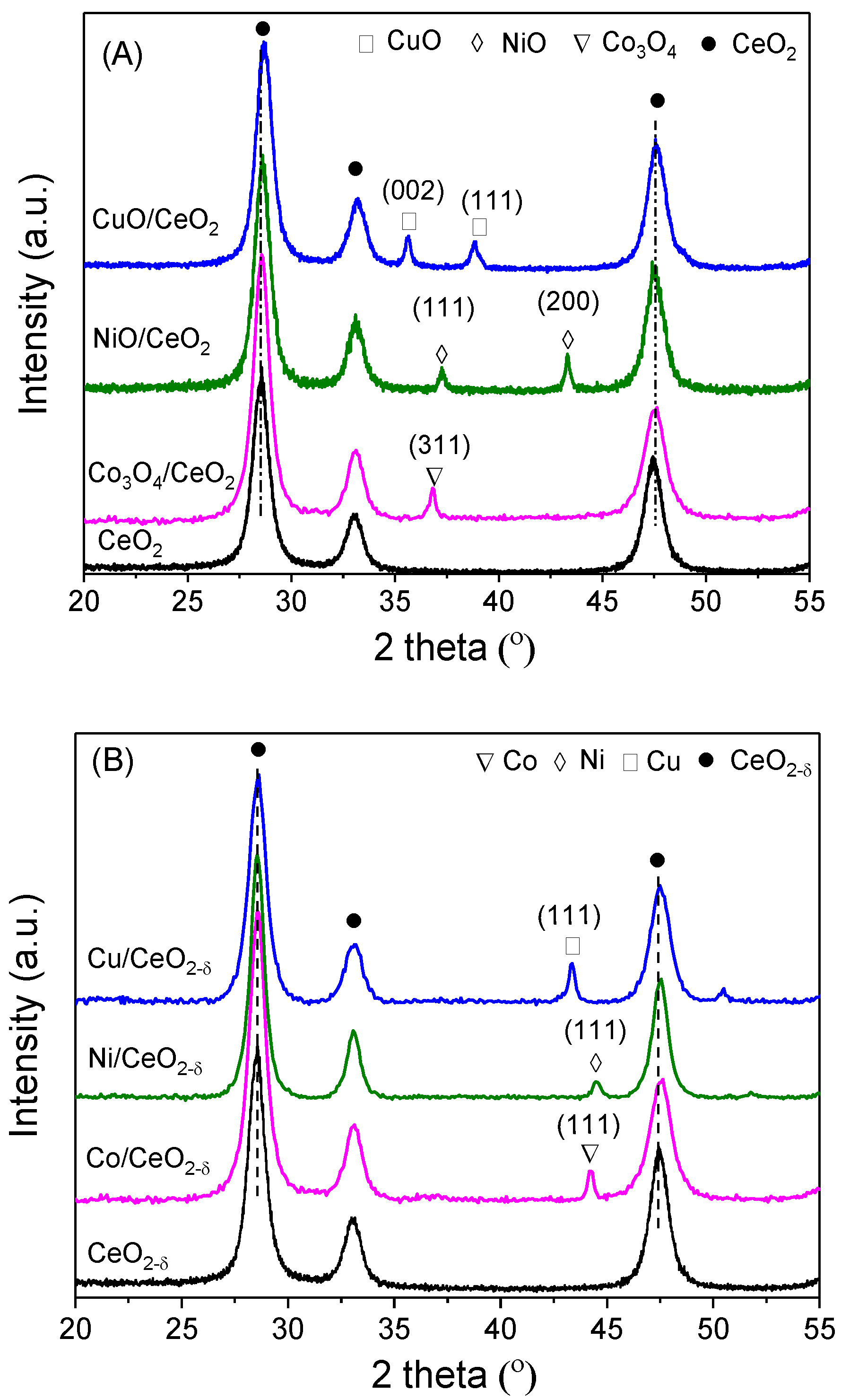
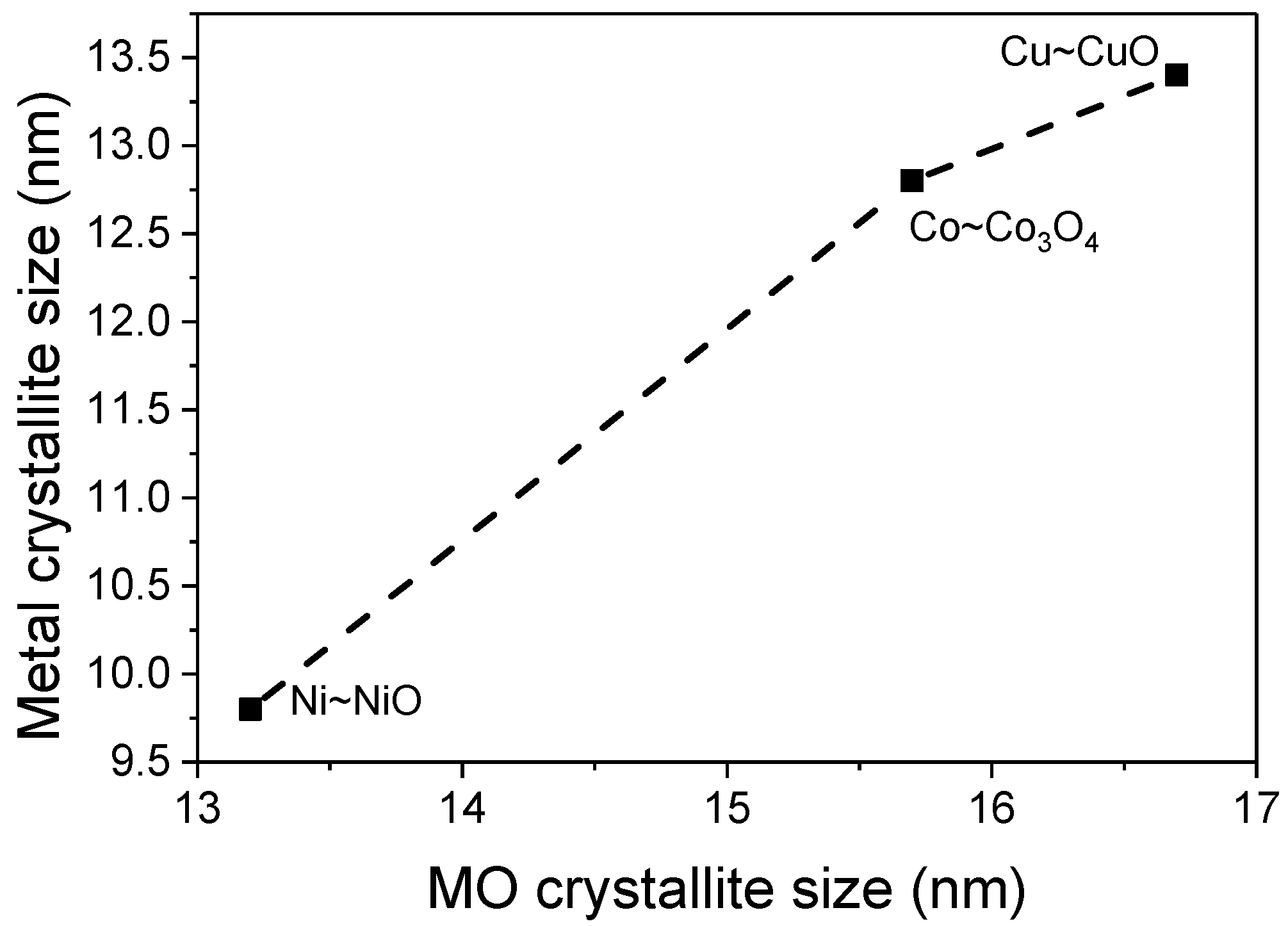
2.1. Structure and Morphology
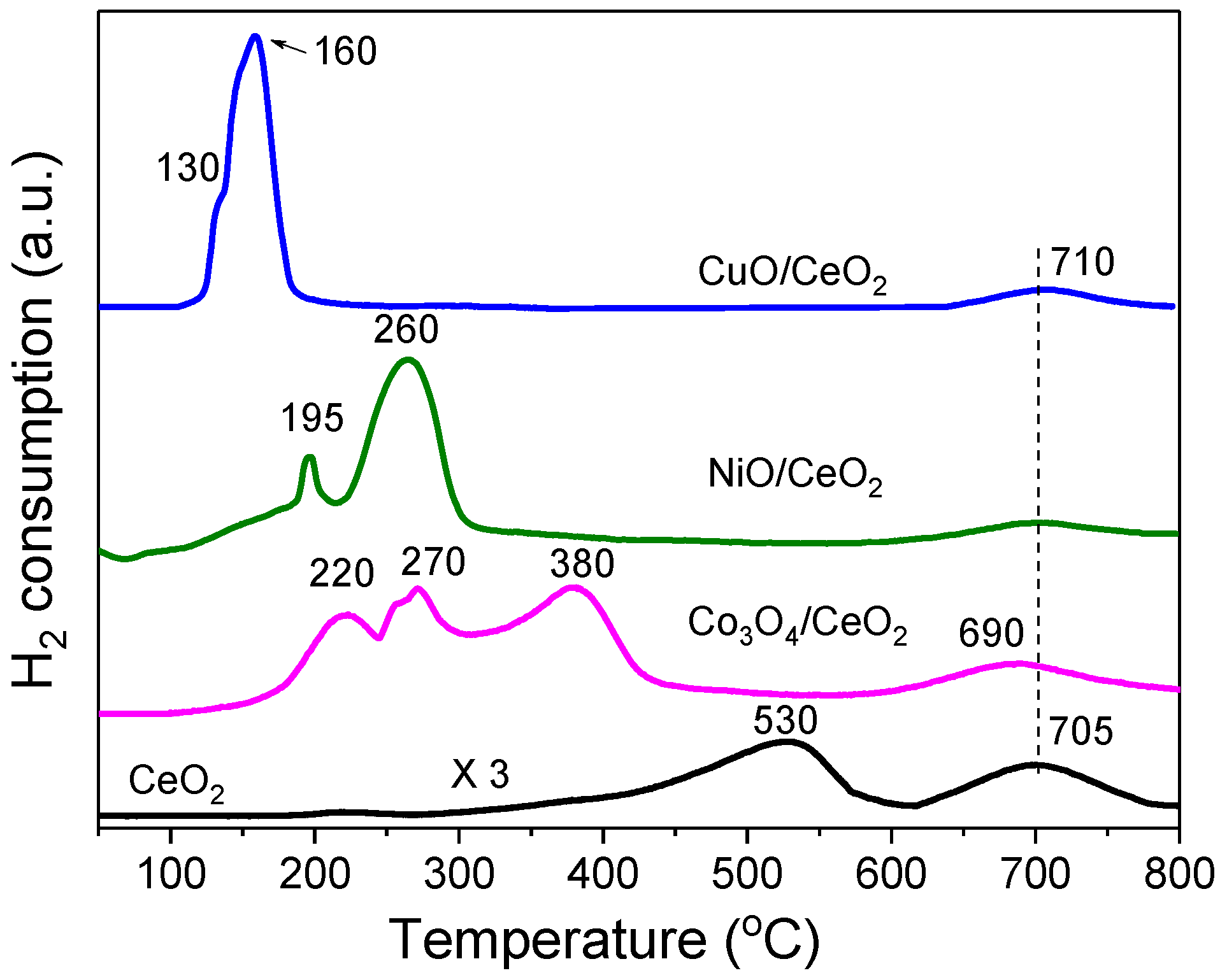
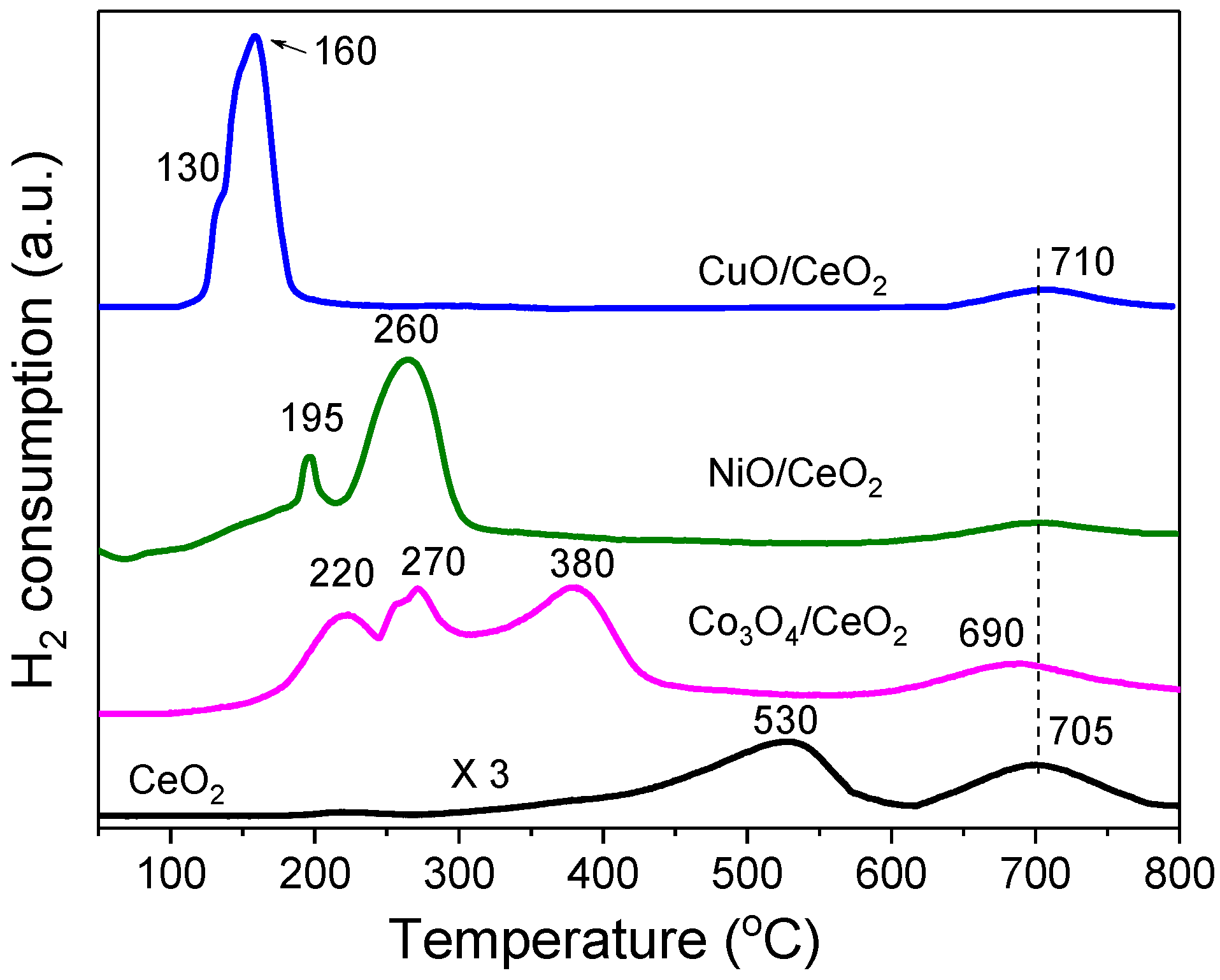
2.2. Reduction Behaviour of the MOx/CeO2 Catalyst Precursors
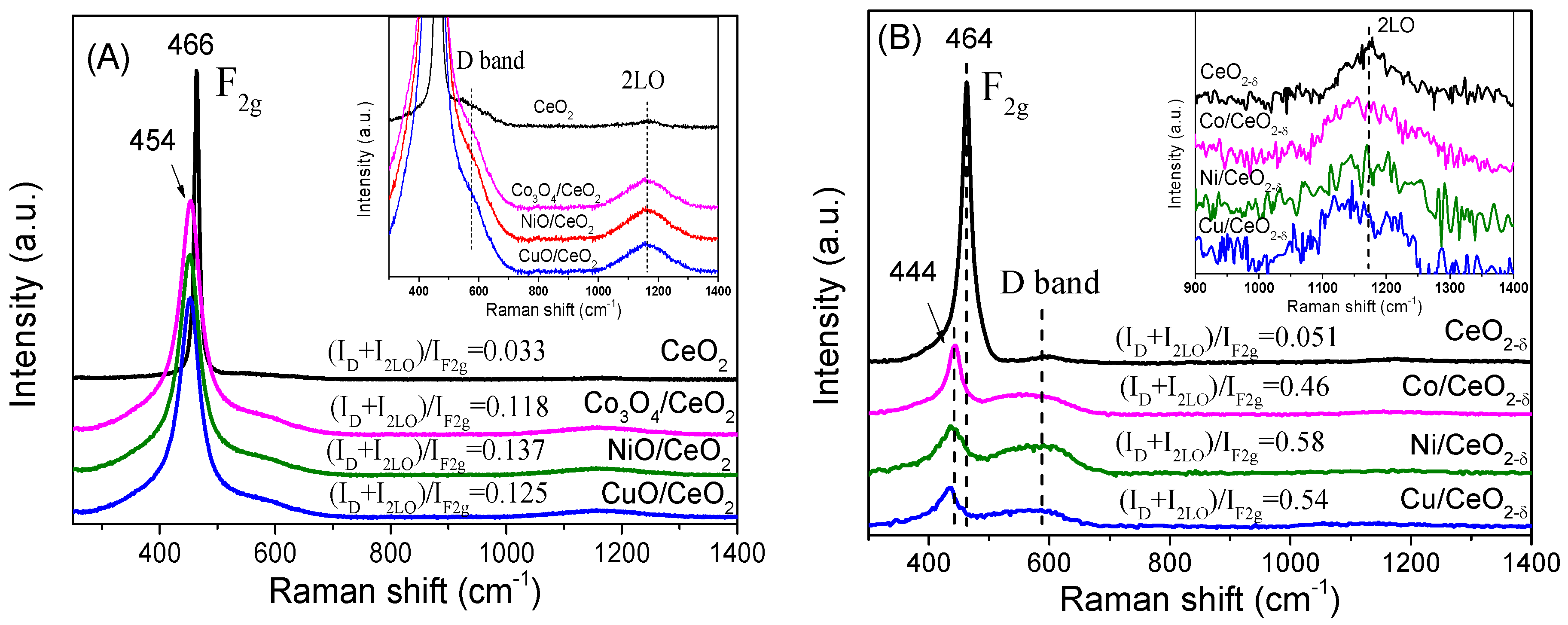
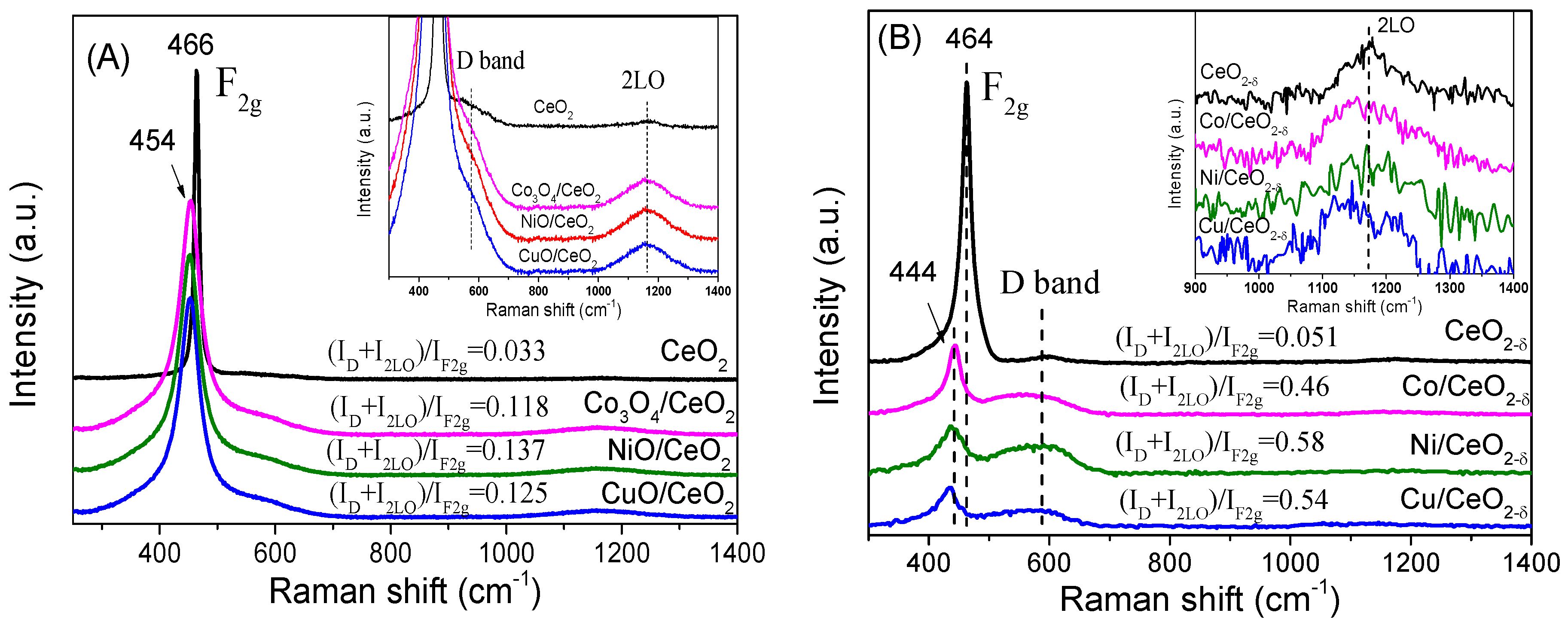
2.3. Surface Structural Analysis
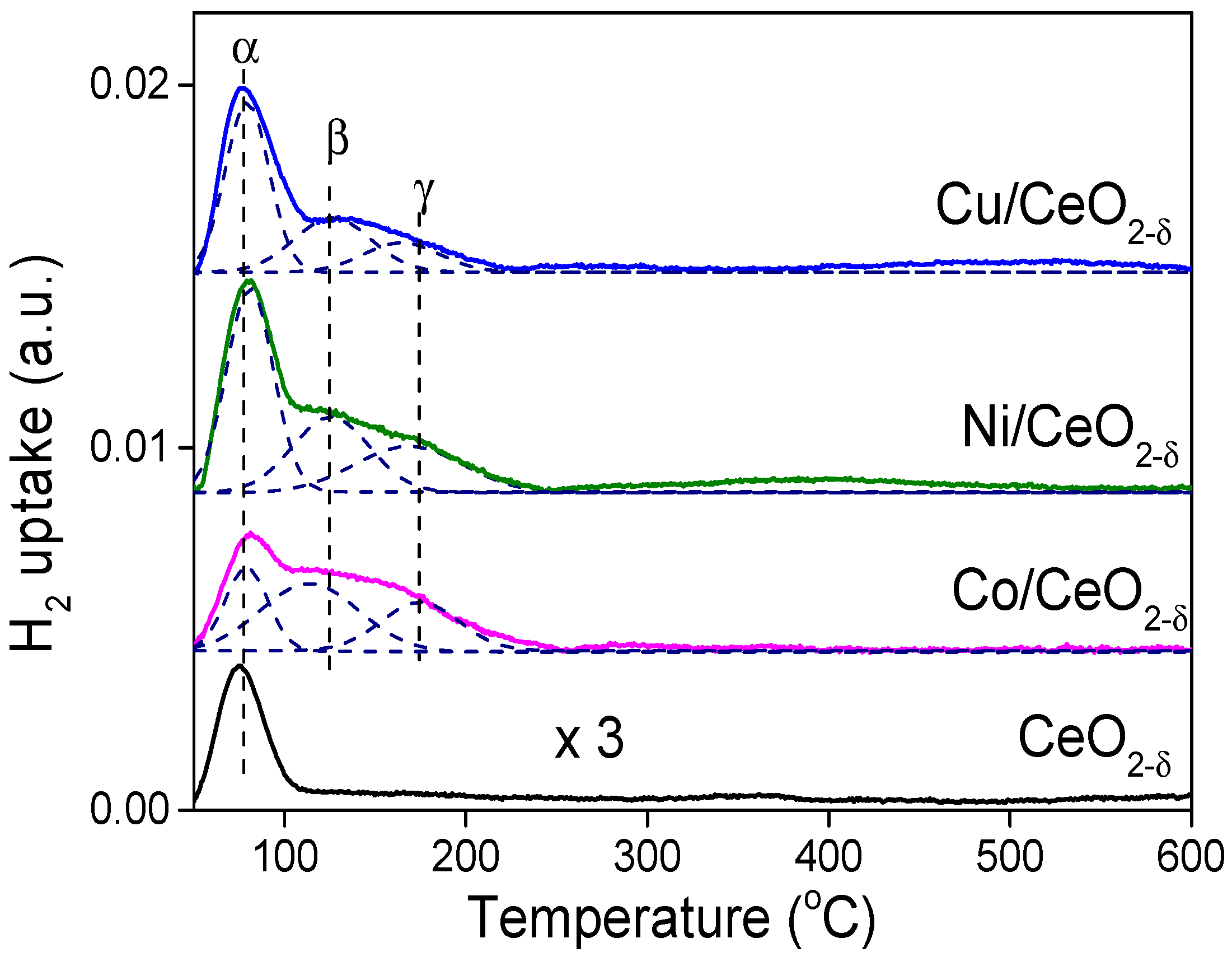
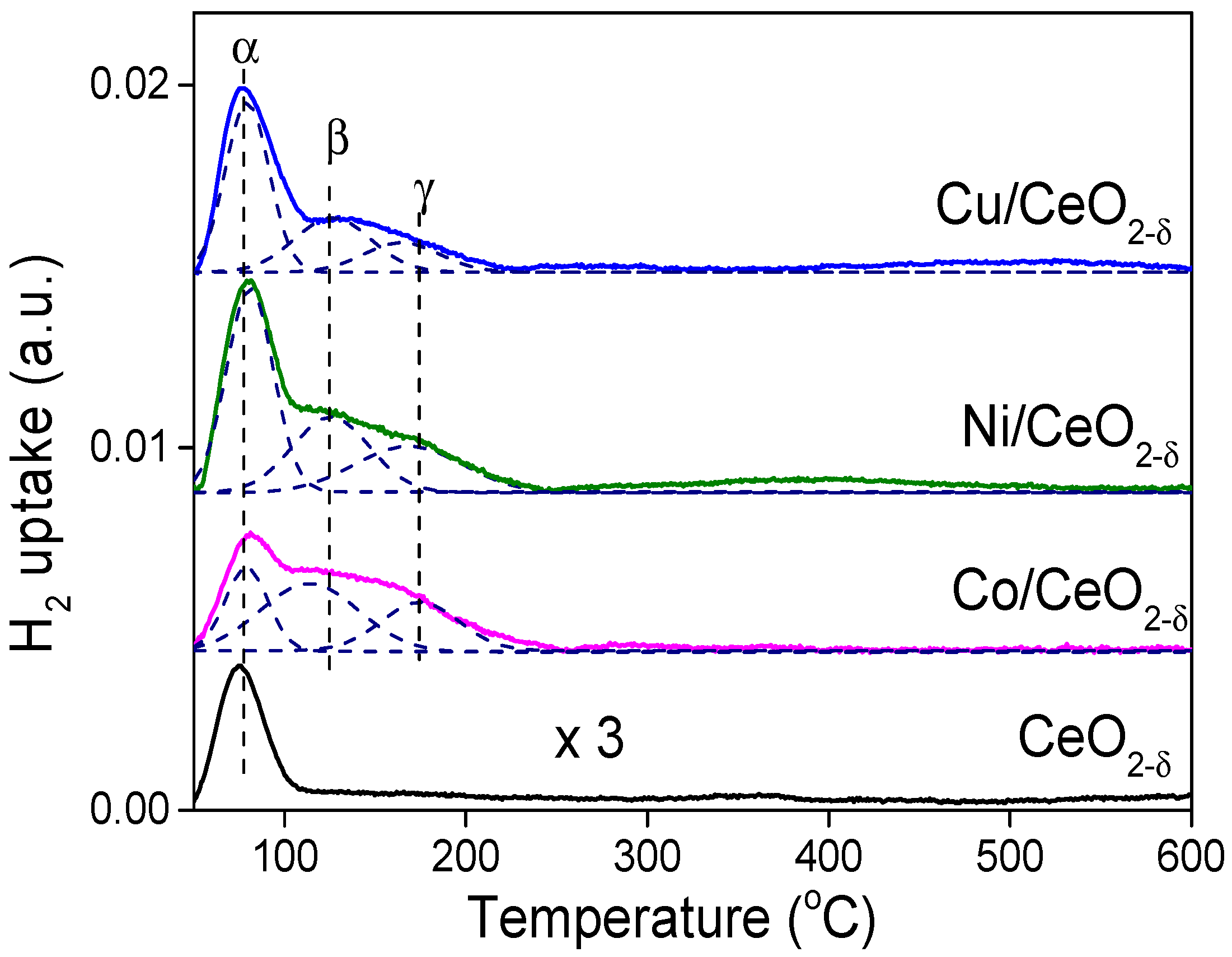
2.4. Hydrogen Activation on the M/CeO2−δ Catalysts
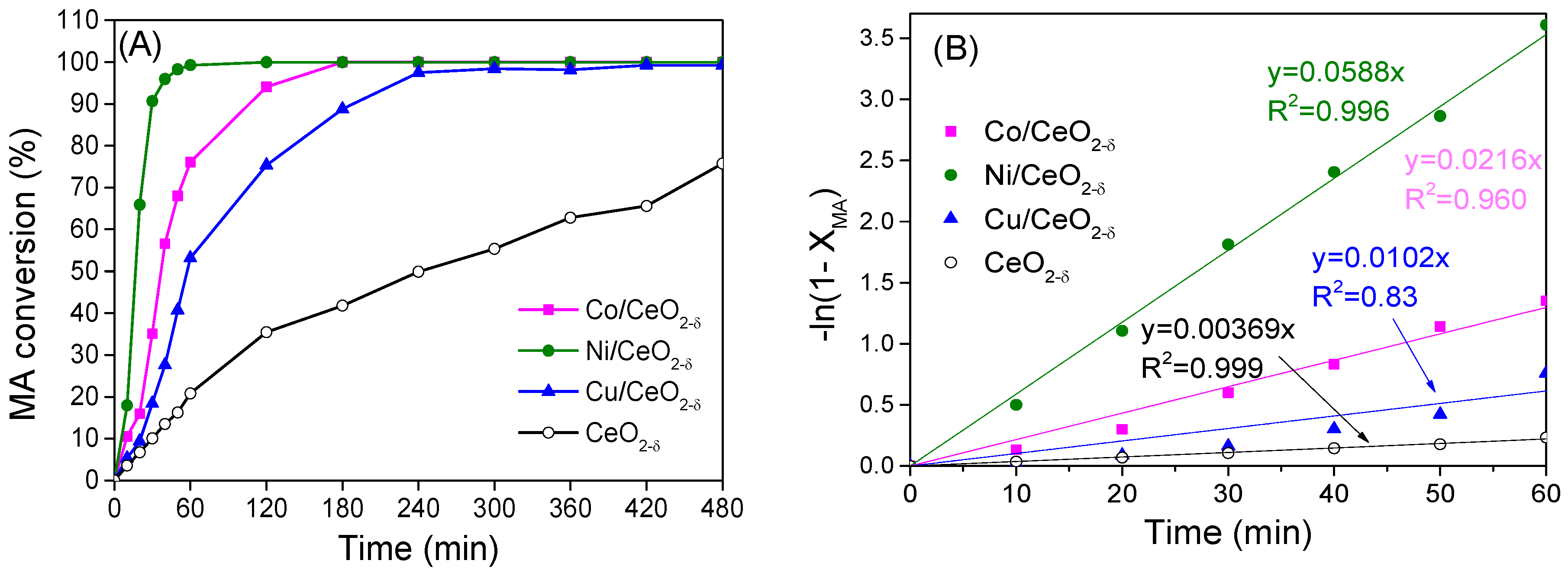
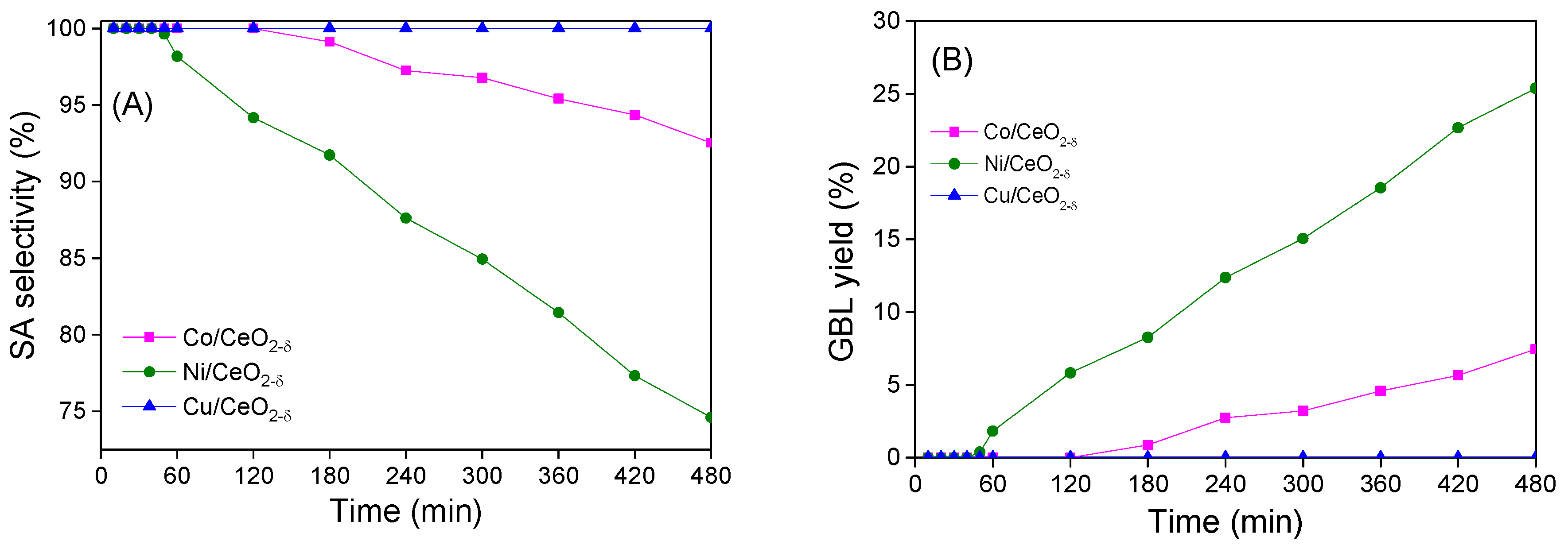
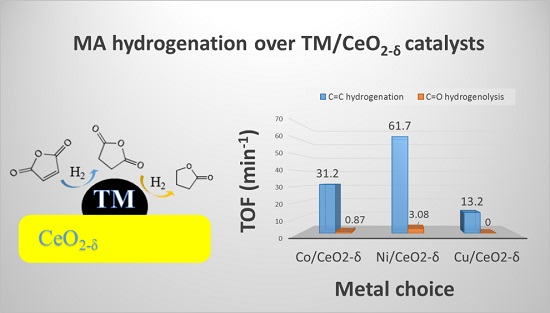
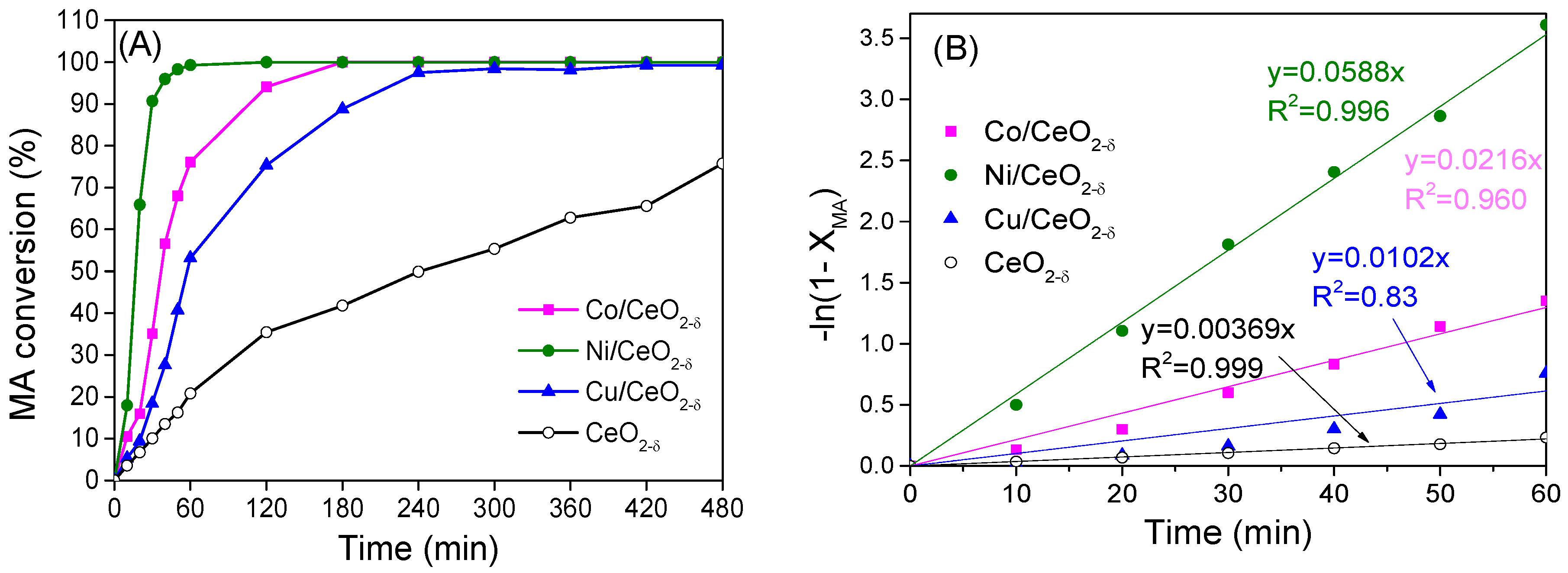
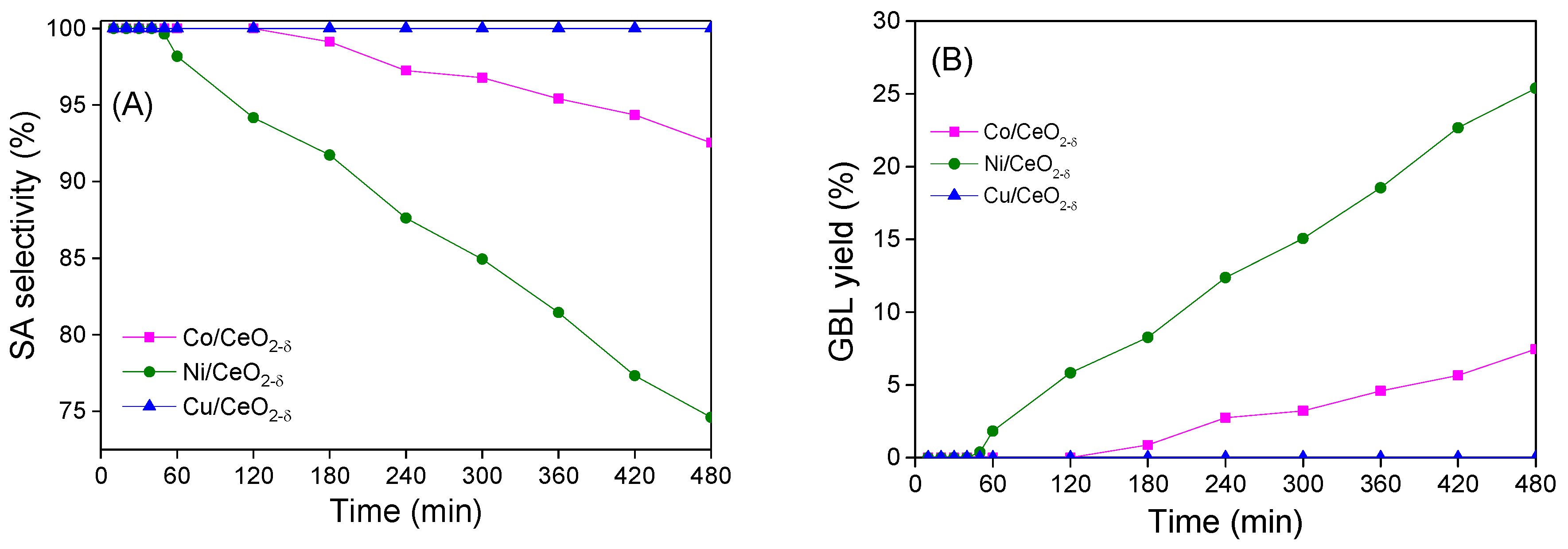
2.5. Catalytic Hydrogenation of Maleic Anhydride
3. Materials and Methods
3.1. Ceria Support Preparation
3.2. M/CeO2−δ (M = Co, Ni and Cu) Catalysts Preparation
3.3. Catalyst Characterizations
3.4. Catalyst Test
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liao, X.; Zhang, Y.; Hill, M.; Xia, X.; Zhao, Y.; Jiang, Z. Highly efficient Ni/CeO2 catalyst for the liquid phase hydrogenation of maleic anhydride. Appl. Catal. A 2014, 488, 256–264. [Google Scholar] [CrossRef]
- Bertone, M.E.; Meyer, C.I.; Regenhardt, S.A.; Sebastian, V.; Garetto, T.F.; Marchi, A.J. Highly selective conversion of maleic anhydride to γ-butyrolactone over Ni-supported catalysts prepared by precipitation–deposition method. Appl. Catal. A Gen. 2015, 503, 135–146. [Google Scholar] [CrossRef]
- Meyer, C.I.; Regenhardt, S.A.; Marchi, A.J.; Garetto, T.F. Gas phase hydrogenation of maleic anhydride at low pressure over silica-supported cobalt and nickel catalysts. Appl. Catal. A 2012, 417–418, 59–65. [Google Scholar] [CrossRef]
- Delhomme, C.; Weuster-Botz, D.; Kühn, F.E. Succinic acid from renewable resources as a C4 building-block chemical—A review of the catalytic possibilities in aqueous media. Green Chem. 2009, 11, 13–26. [Google Scholar] [CrossRef]
- Castiglioni, G.; Vaccari, A.; Fierro, G.; Inversi, M.; Lojacono, M.; Minelli, G.; Pettiti, I.; Porta, P.; Gazzano, M. Structure and reactivity of copper-zinc-cadmium chromite catalysts. Appl. Catal. A 1995, 123, 123–144. [Google Scholar] [CrossRef]
- Regenhardt, S.A.; Meyer, C.I.; Garetto, T.F.; Marchi, A.J. Selective gas phase hydrogenation of maleic anhydride over Ni-supported catalysts: Effect of support on the catalytic performance. Appl. Catal. A 2012, 449, 81–87. [Google Scholar] [CrossRef]
- Castiglioni, G.L.; Ferrari, M.; Guercio, A.; Vaccari, A.; Lancia, R.; Fumagalli, C. Chromium-free catalysts for selective vapor phase hydrogenation of maleic anhydride to γ-butyrolactone. Catal. Today 1996, 27, 181–186. [Google Scholar] [CrossRef]
- Zhang, D.; Yin, H.; Xue, J.; Ge, C.; Jiang, T.; Yu, L.; Shen, Y. Selective hydrogenation of maleic anhydride to tetrahydrofuran over Cu–Zn–M (M = Al, Ti, Zr) catalysts using ethanol as a solvent. Ind. Eng. Chem. Res. 2009, 48, 11220–11224. [Google Scholar] [CrossRef]
- Meyer, C.I.; Marchi, A.J.; Monzon, A.; Garetto, T.F. Deactivation and regeneration of Cu/SiO2 catalyst in the hydrogenation of maleic anhydride. Kinetic modeling. Appl. Catal. A Gen. 2009, 367, 122–129. [Google Scholar] [CrossRef]
- Bertone, M.E.; Regenhardt, S.A.; Meyer, C.I.; Sebastian, V.; Garetto, T.F.; Marchi, A.J. Highly selective Cu-modified Ni/SiO2–Al2O3 catalysts for the conversion of maleic anhydride to γ-butyrolactone in gas phase. Top. Catal. 2015, 59, 159–167. [Google Scholar] [CrossRef]
- Li, J.; Qian, L.-P.; Hu, L.-Y.; Yue, B.; He, H.-Y. Low-temperature hydrogenation of maleic anhydride to succinic anhydride and γ-butyrolactone over pseudo-boehmite derived alumina supported metal (metal=Cu, Co and Ni) catalysts. Chin. Chem. Lett. 2016, 27, 1004–1008. [Google Scholar] [CrossRef]
- Yu, Y.; Guo, Y.; Zhan, W.; Guo, Y.; Wang, Y.; Wang, Y.; Zhang, Z.; Lu, G. Gas-phase hydrogenation of maleic anhydride to γ-butyrolactone at atmospheric pressure over Cu–CeO2–Al2O3 catalyst. J. Mol. Catal. A Chem. 2011, 337, 77–81. [Google Scholar] [CrossRef]
- Lu, W.; Lu, G.; Guo, Y.; Guo, Y.; Wang, Y. Gas-phase hydrogenation of maleic anhydride to butyric acid over Cu/TiO2/γ-Al2O3 catalyst promoted by Pd. Catal. Commun. 2003, 4, 177–181. [Google Scholar] [CrossRef]
- Gao, C.-G.; Zhao, Y.-X.; Zhang, Y.; Liu, D.-S. Synthesis characterization and catalytic evaluation of Ni/ZrO2/SiO2 aerogels catalysts. J. Sol-Gel Sci. Technol. 2007, 44, 145–151. [Google Scholar] [CrossRef]
- Zhang, D.; Yin, H.; Zhang, R.; Xue, J.; Jiang, T. Gas phase hydrogenation of maleic anhydride to γ-butyrolactone by cu–zn–ce catalyst in the presence of n-butanol. Catal. Lett. 2007, 122, 176–182. [Google Scholar] [CrossRef]
- Zhang, R.; Yin, H.; Zhang, D.; Qi, L.; Lu, H.; Shen, Y.; Jiang, T. Gas phase hydrogenation of maleic anhydride to tetrahydrofuran by Cu/ZnO/TiO2 catalysts in the presence of n-butanol. Chem. Eng. J. 2008, 140, 488–496. [Google Scholar] [CrossRef]
- Torres, C.C.; Alderete, J.B.; Mella, C.; Pawelec, B. Maleic anhydride hydrogenation to succinic anhydride over mesoporous Ni/TiO2 catalysts: Effects of Ni loading and temperature. J. Mol. Catal. A Chem. 2016, 423, 441–448. [Google Scholar] [CrossRef]
- Bruix, A.; Rodriguez, J.A.; Ramirez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. A new type of strong metal-support interaction and the production of H2 through the transformation of water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T. Catalyst-support interactions: Electronic perturbations. Nat. Chem. 2012, 4, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Farmer, J.A.; Campbell, C.T. Ceria maintains smaller metal catalyst particles by strong metal-support bonding. Science 2010, 329, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, J.; Lopez-Duran, D.; Liu, Z.; Duchon, T.; Evans, J.; Senanayake, S.D.; Crumlin, E.J.; Matolin, V.; Rodriguez, J.A.; Ganduglia-Pirovano, M.V. In situ and theoretical studies for the dissociation of water on an active Ni/CeO2 catalyst: Importance of strong metal-support interactions for the cleavage of O-H bonds. Angew. Chem. Int. Ed. Engl. 2015, 54, 3917–3921. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Nagai, Y.; Aoki, Y.; Takahashi, N. Investigation into the catalytic reduction of NOx at copper–ceria interface active sites. Appl. Catal. A 2016, 510, 57–63. [Google Scholar] [CrossRef]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.; Wei, M.; Evans, D.G.; Duan, X. Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, A.B.; Scanlon, D.O.; Watson, G.W. Role of lattice distortions in the oxygen storage capacity of divalently doped CeO2. Chem. Mater. 2011, 23, 4464–4468. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Steam reforming of bio-ethanol to produce hydrogen over Co/CeO2 catalysts derived from Ce1−xCoxO2−y precursors. Catalysts 2016, 6, 26. [Google Scholar] [CrossRef]
- Cui, Y.; Dai, W.-L. Support morphology and crystal plane effect of Cu/CeO2 nanomaterial on the physicochemical and catalytic properties for carbonate hydrogenation. Catal. Sci. Technol. 2016, 6, 7752–7762. [Google Scholar] [CrossRef]
- Sohlberg, K.; Pantelides, S.T.; Pennycook, S.J. Interactions of hydrogen with CeO2. J. Am. Chem. Soc. 2001, 123, 6609–6611. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Shen, W.; Li, C. Structural characteristics and redox behaviors of Ce1−xCuxOy solid solutions. Chem. Mater. 2003, 15, 4761–4767. [Google Scholar] [CrossRef]
- Tang, K.; Liu, W.; Li, J.; Guo, J.; Zhang, J.; Wang, S.; Niu, S.; Yang, Y. The effect of exposed facets of ceria to the nickel species in nickel-ceria catalysts and their performance in a NO + CO reaction. ACS Appl. Mater. Interfaces 2015, 7, 26839–26849. [Google Scholar] [CrossRef] [PubMed]
- Greluk, M.; Rotko, M.; Machocki, A. Conversion of ethanol over Co/CeO2 and K-Co/CeO2 catalysts for hydrogen production. Catal. Lett. 2015, 146, 163–173. [Google Scholar] [CrossRef]
- De Lima, S.M.; da Silva, A.M.; da Costa, L.O.O.; Graham, U.M.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Study of catalyst deactivation and reaction mechanism of steam reforming, partial oxidation, and oxidative steam reforming of ethanol over Co/CeO2 catalyst. J. Catal. 2009, 268, 268–281. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, B.; Li, Y.; Xu, Y.; Xin, Q.; Shen, W. Carbon monoxide oxidation over CuO/CeO2 catalysts. Catal. Today 2004, 93–95, 191–198. [Google Scholar] [CrossRef]
- Matte, L.P.; Kilian, A.S.; Luza, L.; Alves, M.C.M.; Morais, J.; Baptista, D.L.; Dupont, J.; Bernardi, F. Influence of the CeO2 support on the reduction properties of Cu/CeO2 and Ni/CeO2 nanoparticles. J. Phys. Chem. C 2015, 119, 26459–26470. [Google Scholar] [CrossRef]
- Spanier, J.E.; Robinson, R.D.; Zhang, F.; Chan, S.-W.; Herman, I.P. Size-dependent properties of CeO2−y nanoparticles as studied by raman scattering. Phys. Rev. B 2001, 64, 245407. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Howe, J.; Meyer, H.M.; Overbury, S.H. Probing defect sites on CeO2 nanocrystals with well-defined surface planes by raman spectroscopy and O2 adsorption. Langmuir ACS J. Surf. Coll. 2010, 26, 16595–16606. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, C.; Wang, F.; He, S.; Chen, H.; Zhao, Y.; Wei, M.; Evans, D.G.; Duan, X. Enhanced low-temperature activity of CO2 methanation over highly-dispersed Ni/TiO2 catalyst. Catal. Sci. Technol. 2013, 3, 2627. [Google Scholar] [CrossRef]
- Znak, L.; Zieliński, J. Effects of support on hydrogen adsorption/desorption on nickel. Appl. Catal. A 2008, 334, 268–276. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Pallassana, V.; Neurock, M.; Coulston, G.W. Theoretical density functional analysis of maleic anhydride chemisorption on Pd(111), Re(0001), and bimetallic PdML/Re(0001) and PdML/Mo(110) pseudomorphic overlayers. J. Phys. Chem. B 1999, 103, 8973–8983. [Google Scholar] [CrossRef]
- Zhang, S.; Li, J.; Xia, Z.; Wu, C.; Zhang, Z.; Ma, Y.; Qu, Y. Towards highly active Pd/CeO2 for alkene hydrogenation by tuning Pd dispersion and surface properties of the catalysts. Nanoscale 2017, 9, 3140–3149. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.P.; Gong, X.Q.; Lu, G. Role of oxygen vacancies in the surface evolution of H at CeO2(111): A charge modification effect. Phys. Chem. Chem. Phys. 2015, 17, 3544–3549. [Google Scholar] [CrossRef] [PubMed]
- Pallassana, V.; Neurock, M. First-principles periodic density functional study of the hydrogenation of maleic anhydride to succinic anhydride over palladium(111). J. Phys. Chem. B 2000, 104, 9449–9459. [Google Scholar] [CrossRef]
- Pallassana, V.; Neurock, M.; Coulston, G. Towards understanding the mechanism for the selective hydrogenation of maleic anhydride to tetrahydrofuran over palladium. Catal. Today 1999, 50, 589–601. [Google Scholar] [CrossRef]
- Claus, P.; Brückner, A.; Mohr, C.; Hofmeister, H. Supported gold nanoparticles from quantum dot to mesoscopic size scale: Effect of electronic and structural properties on catalytic hydrogenation of conjugated functional groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. [Google Scholar] [CrossRef]
- Vile, G.; Bridier, B.; Wichert, J.; Perez-Ramirez, J. Ceria in hydrogenation catalysis: High selectivity in the conversion of alkynes to olefins. Angew. Chem. Int. Ed. Engl. 2012, 51, 8620–8623. [Google Scholar] [CrossRef] [PubMed]
- Ponec, V. On the role of promoters in hydrogenations on metals; α,β-unsaturated aldehydes and ketones. Appl. Catal. A 1997, 149, 27–48. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Hájek, J.; Salmi, T.; Murzin, D.Y. Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts. Appl. Catal. A 2005, 292, 1–49. [Google Scholar] [CrossRef]
- Dandekar, A.; Vannice, M.A. Crotonaldehyde hydrogenation on Pt/TiO2 and Ni/TiO2 SMSI catalysts. J. Catal. 1999, 183, 344–354. [Google Scholar] [CrossRef]
- Herrmann, U.; Emig, G. Kinetics and mechanism in the liquid-phase hydrogenation of maleic anhydride and intermediates. Chem. Eng. Technol. 1998, 21, 285–295. [Google Scholar] [CrossRef]
- Gil Girol, S.; Strunskus, T.; Muhler, M.; Wöll, C. Reactivity of ZnO surfaces toward maleic anhydride. J. Phys. Chem. B 2004, 108, 13736–13745. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a(CeO2) (nm) | a(CeO2−δ) (nm) | Loading (wt %) | Crystallite Size (nm) | |||
|---|---|---|---|---|---|---|---|
| D(CeO2) | D(CeO2−δ) | D(MOx) | D(M) | ||||
| CeO2 | 0.5438 | 0.5438 | - | 12.5 | 12.9 | - | - |
| Co/CeO2−δ | 0.5430 | 0.5442 | 10.3 | 14.3 | 15.2 | 15.7 | 12.8 |
| Ni/CeO2−δ | 0.5424 | 0.5441 | 9.8 | 12.5 | 16.5 | 13.2 | 9.8 |
| Cu/CeO2−δ | 0.5413 | 0.5442 | 9.7 | 13.2 | 15.7 | 16.7 | 13.4 |
| Sample | Hα (μmol/g) | Hβ (μmol/g) | Hγ (μmol/g) | Σ(Hβ + Hγ) (μmol/g) | ΣH (μmol/g) | DM (%) | Rate Coefficient, k (min−1) |
|---|---|---|---|---|---|---|---|
| Co/CeO2−δ | 17.28 | 24.04 | 11.84 | 35.9 | 53.16 | 3.9 | 0.0588 |
| Ni/CeO2−δ | 28.35 | 22.6 | 14.6 | 37.2 | 65.55 | 4.4 | 0.0216 |
| Cu/CeO2−δ | 28.74 | 14.2 | 8.2 | 22.4 | 52.14 | 2.9 | - |
| CeO2−δ | 7.87 | / | / | / | 7.87 | / | 0.0037 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, X.; Zhang, Y.; Guo, J.; Zhao, L.; Hill, M.; Jiang, Z.; Zhao, Y. The Catalytic Hydrogenation of Maleic Anhydride on CeO2−δ-Supported Transition Metal Catalysts. Catalysts 2017, 7, 272. https://doi.org/10.3390/catal7090272
Liao X, Zhang Y, Guo J, Zhao L, Hill M, Jiang Z, Zhao Y. The Catalytic Hydrogenation of Maleic Anhydride on CeO2−δ-Supported Transition Metal Catalysts. Catalysts. 2017; 7(9):272. https://doi.org/10.3390/catal7090272
Chicago/Turabian StyleLiao, Xin, Yin Zhang, Junqiu Guo, Lili Zhao, Martyn Hill, Zheng Jiang, and Yongxiang Zhao. 2017. "The Catalytic Hydrogenation of Maleic Anhydride on CeO2−δ-Supported Transition Metal Catalysts" Catalysts 7, no. 9: 272. https://doi.org/10.3390/catal7090272
APA StyleLiao, X., Zhang, Y., Guo, J., Zhao, L., Hill, M., Jiang, Z., & Zhao, Y. (2017). The Catalytic Hydrogenation of Maleic Anhydride on CeO2−δ-Supported Transition Metal Catalysts. Catalysts, 7(9), 272. https://doi.org/10.3390/catal7090272