Electroreduction of CO2 into Ethanol over an Active Catalyst: Copper Supported on Titania


Abstract
:
1. Introduction
2. Results and Discussion
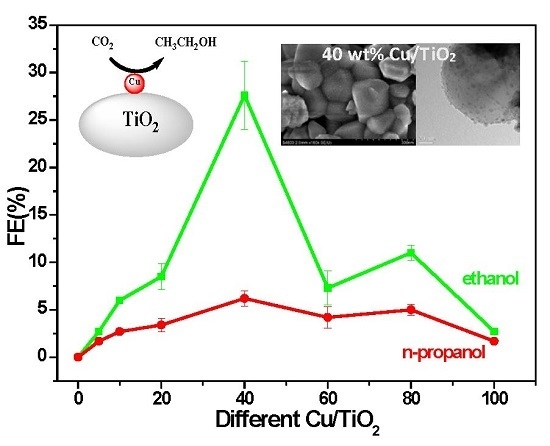
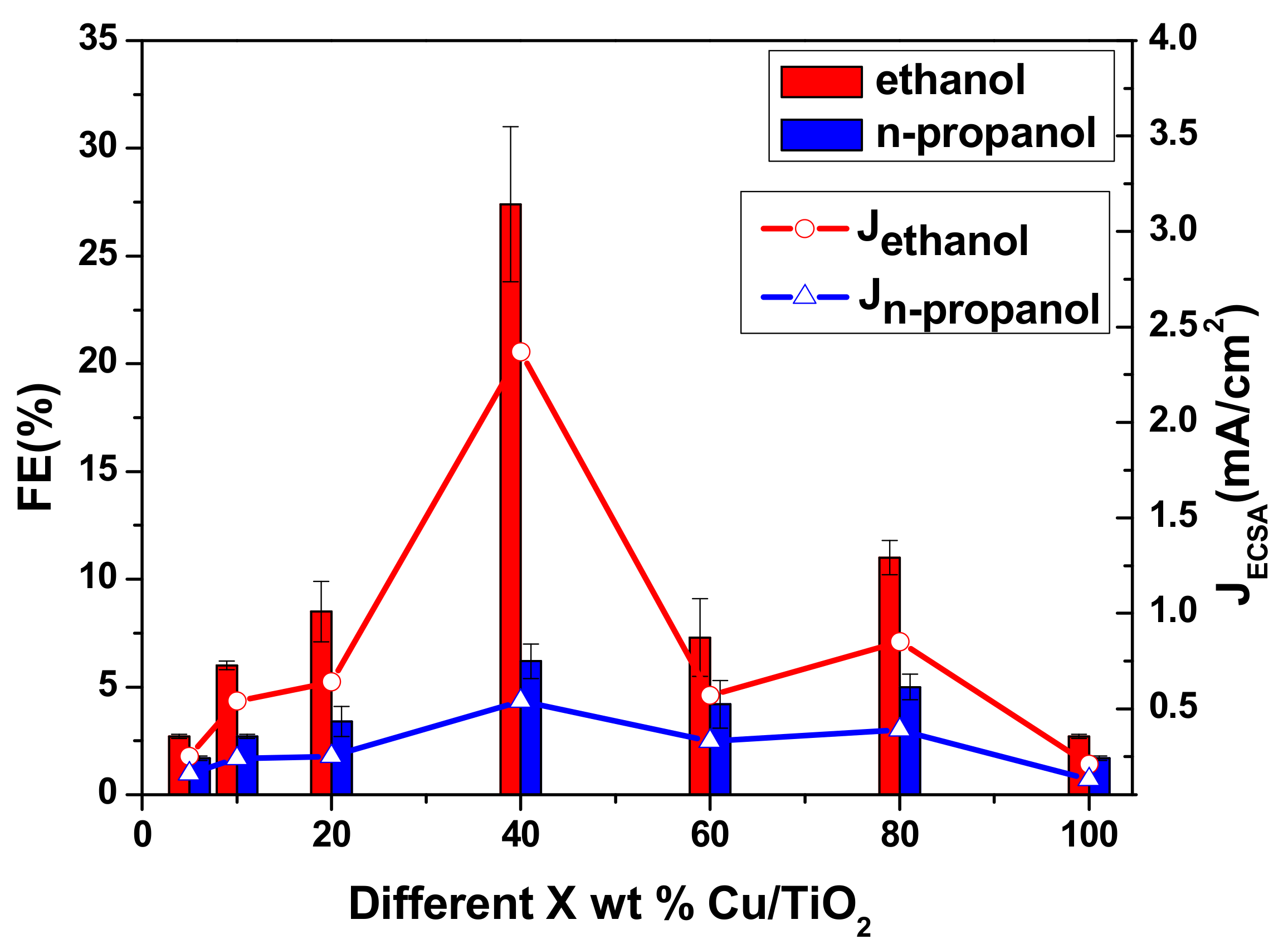
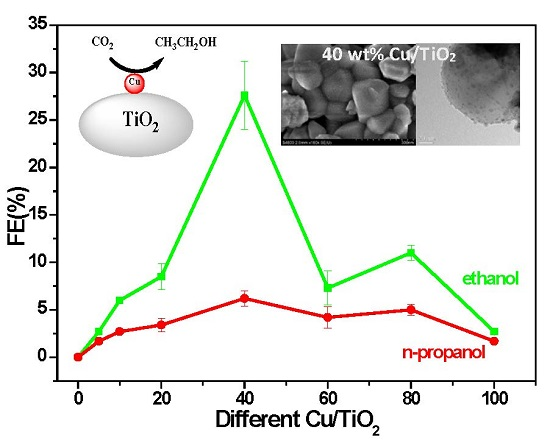
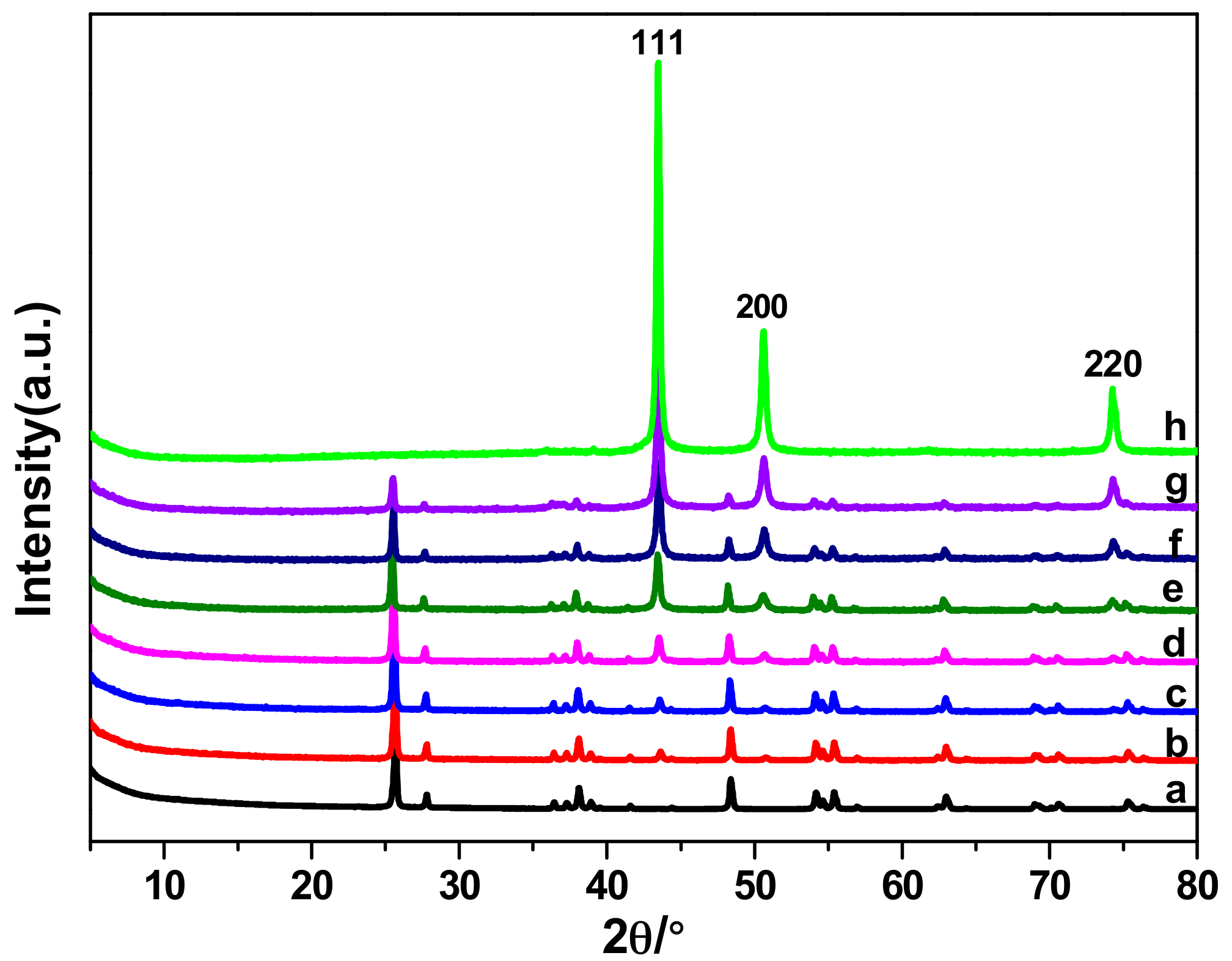
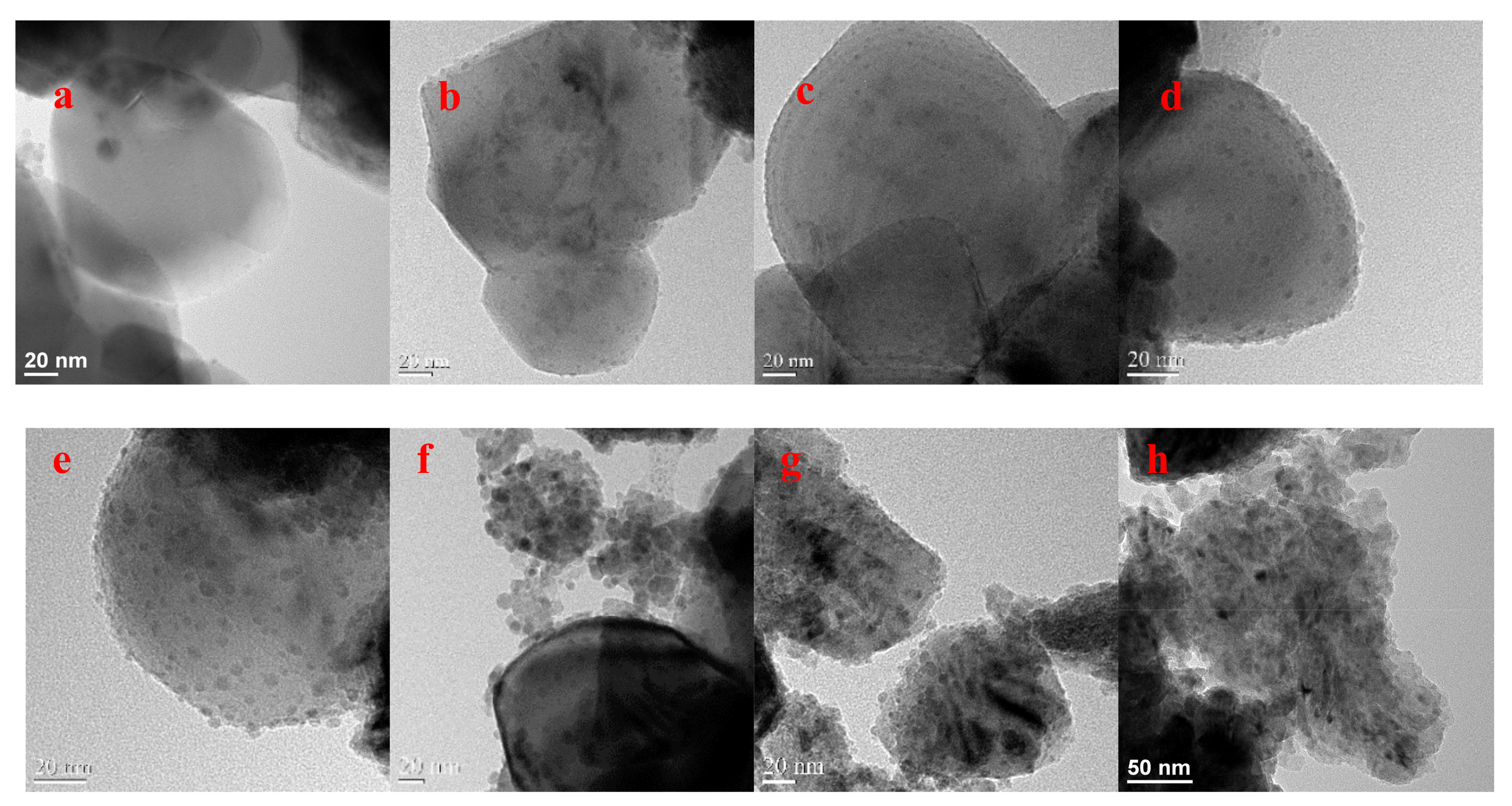
2.1. Performance Comparison of Different Cu-Supported TiO2 Catalysts
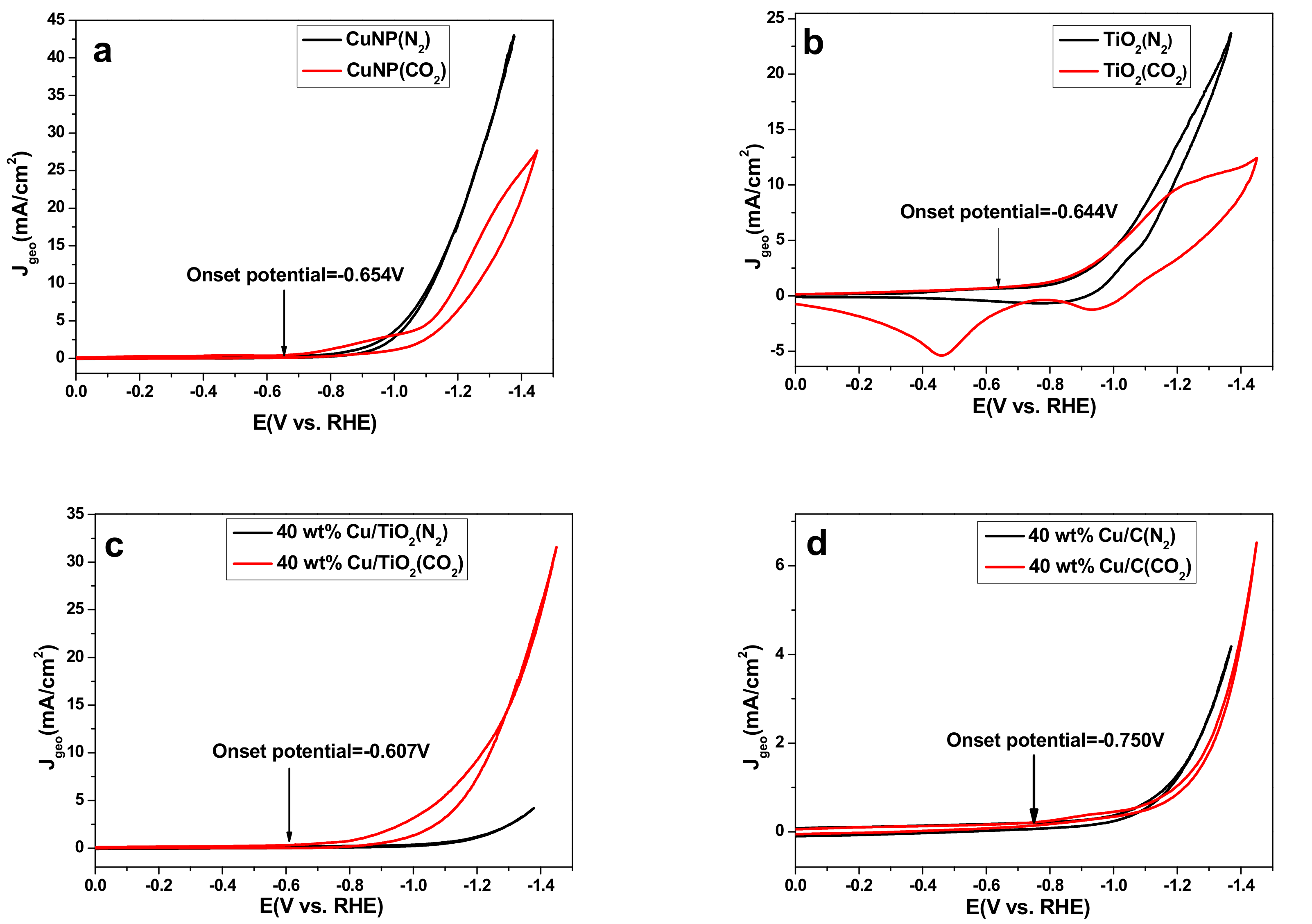
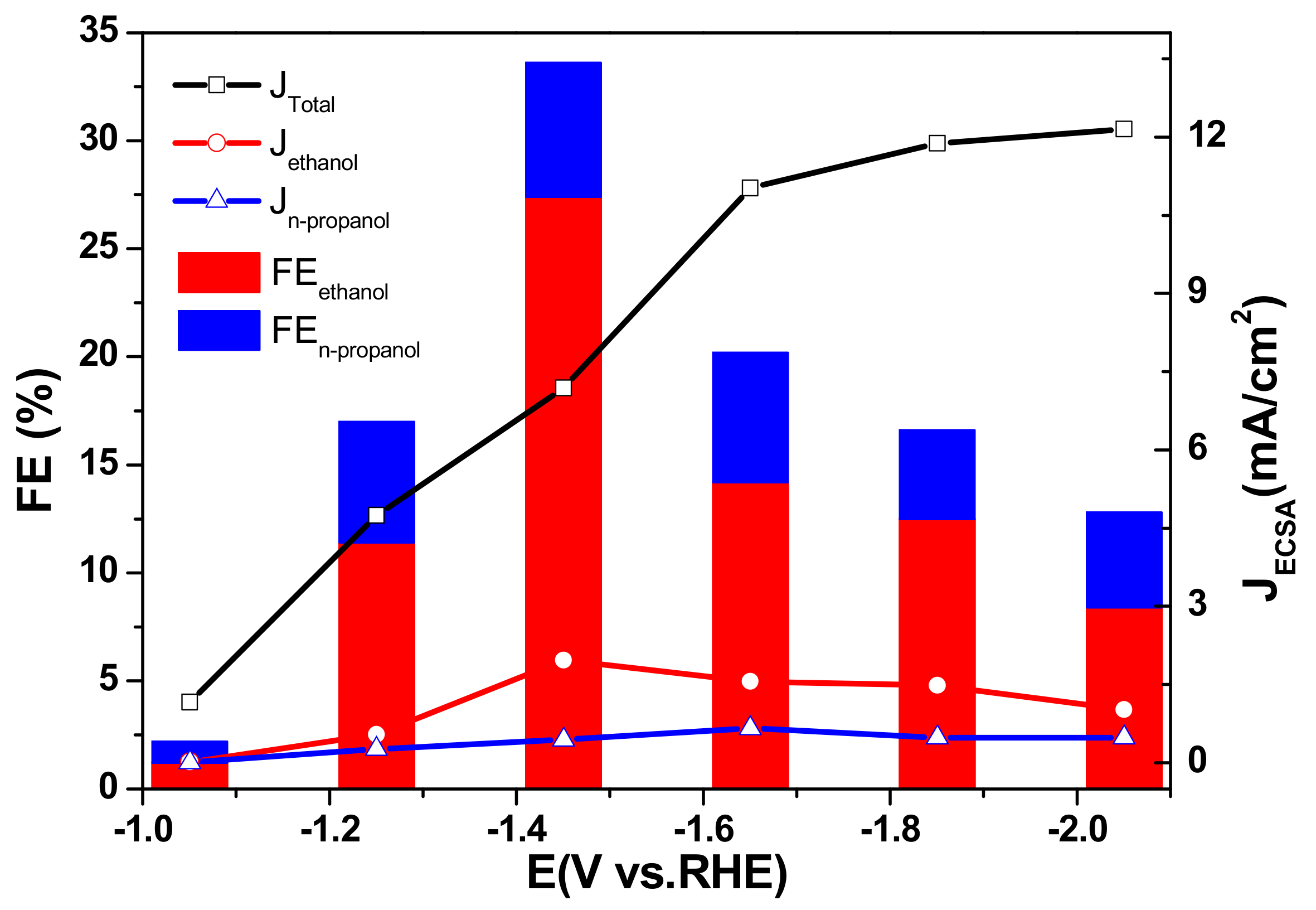
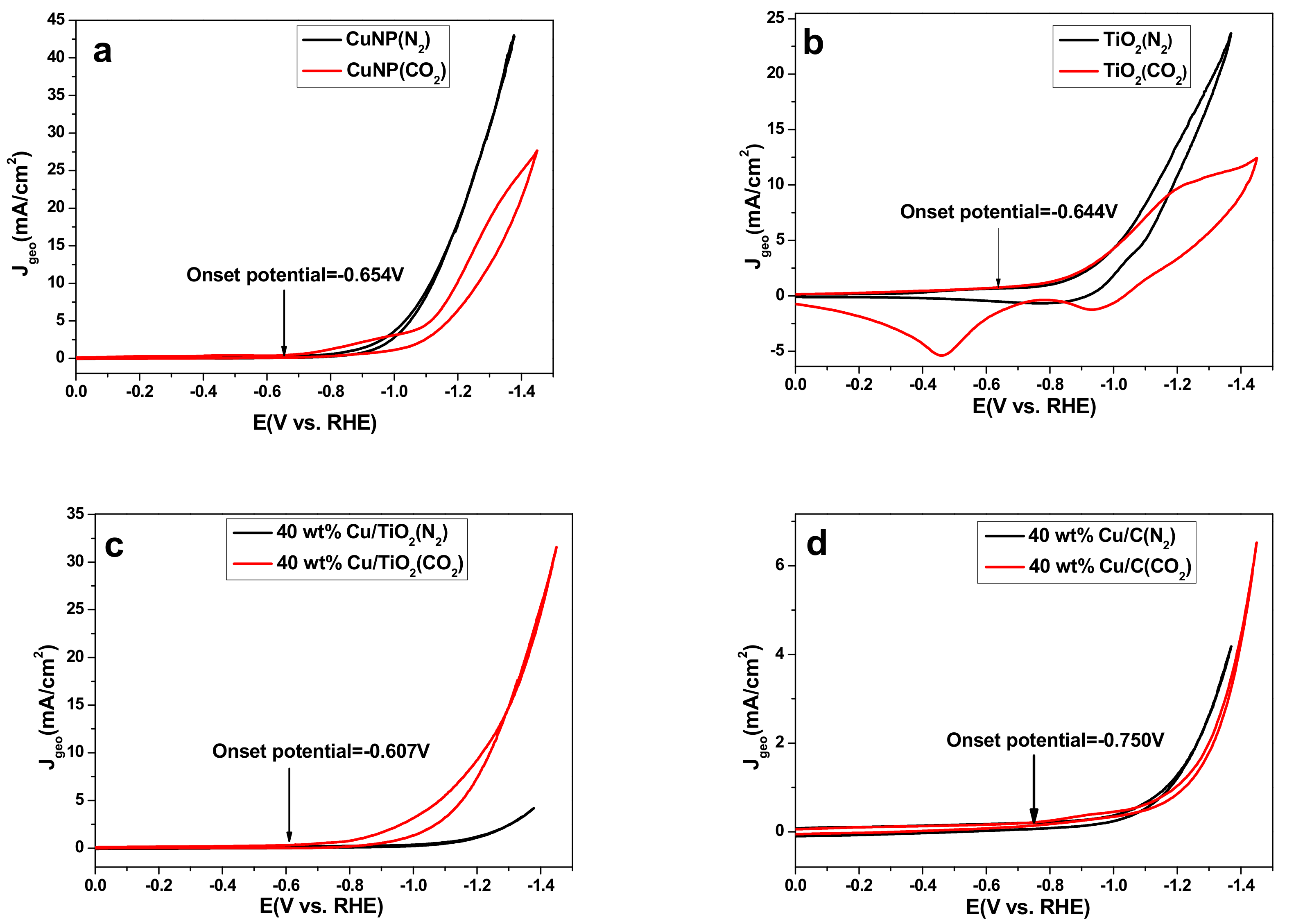
2.2. Catalyst Activity in a Standard Three-Electrode Cell
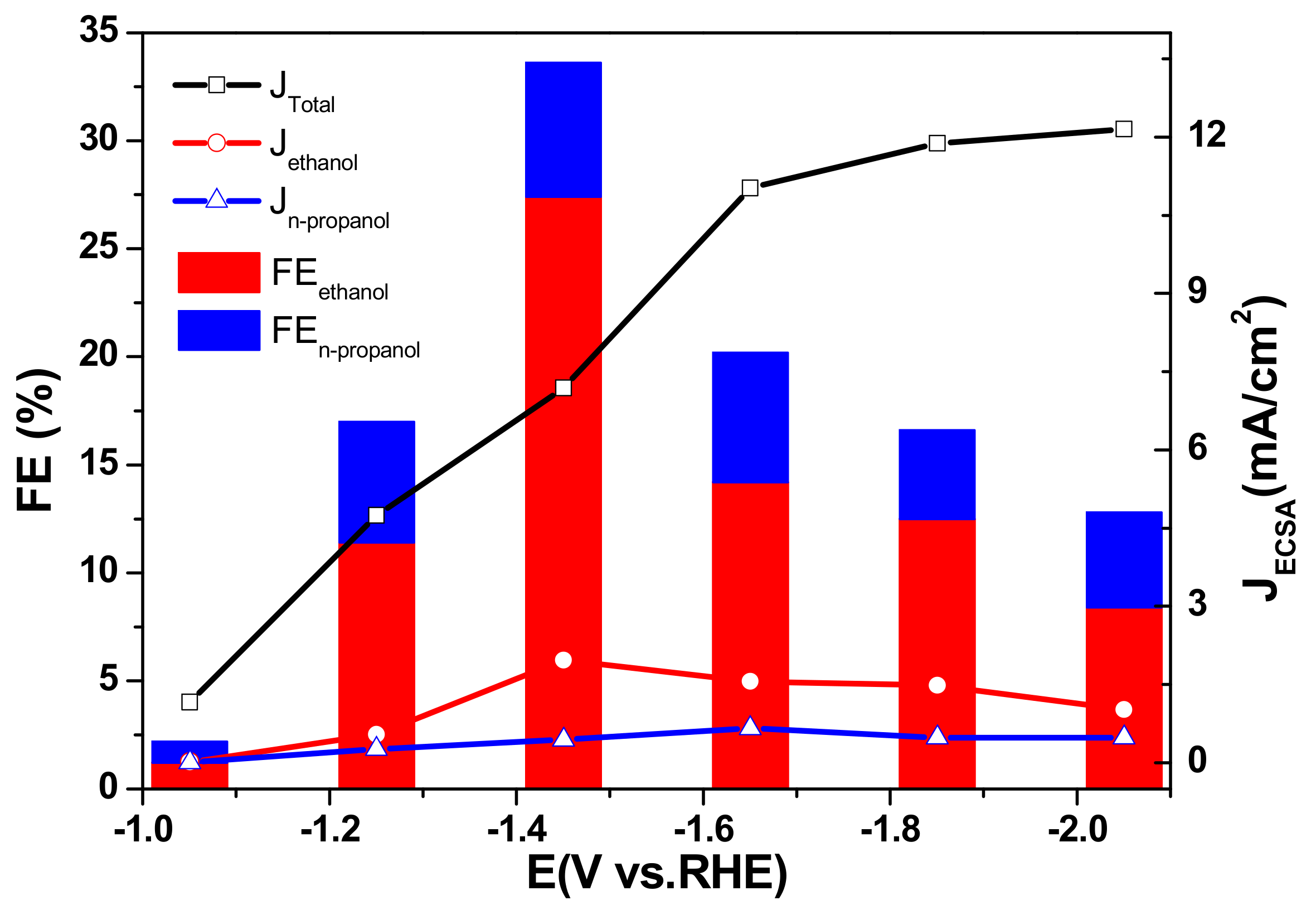
2.3. The Electrocatalytic Activity of Various Cu/TiO2
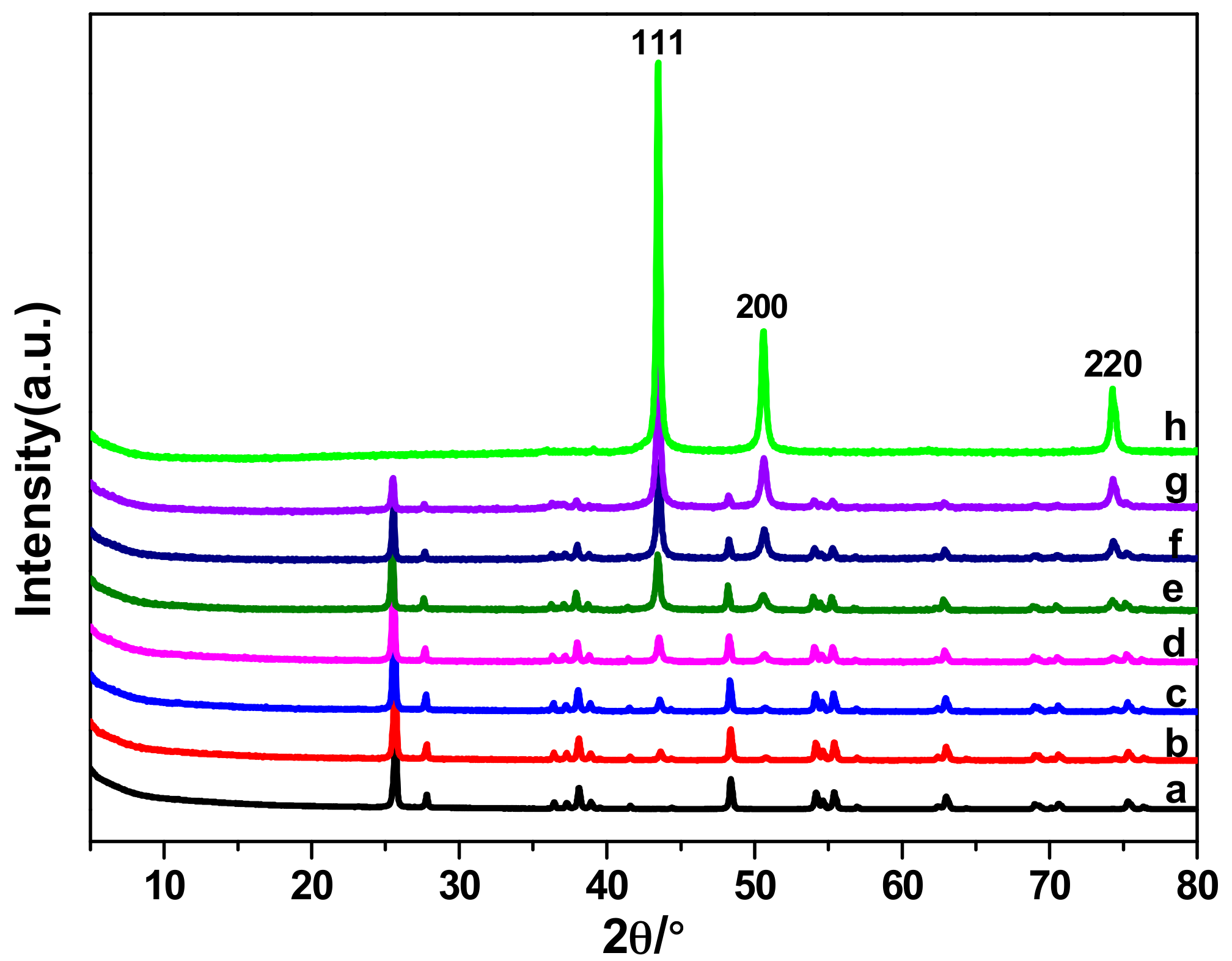
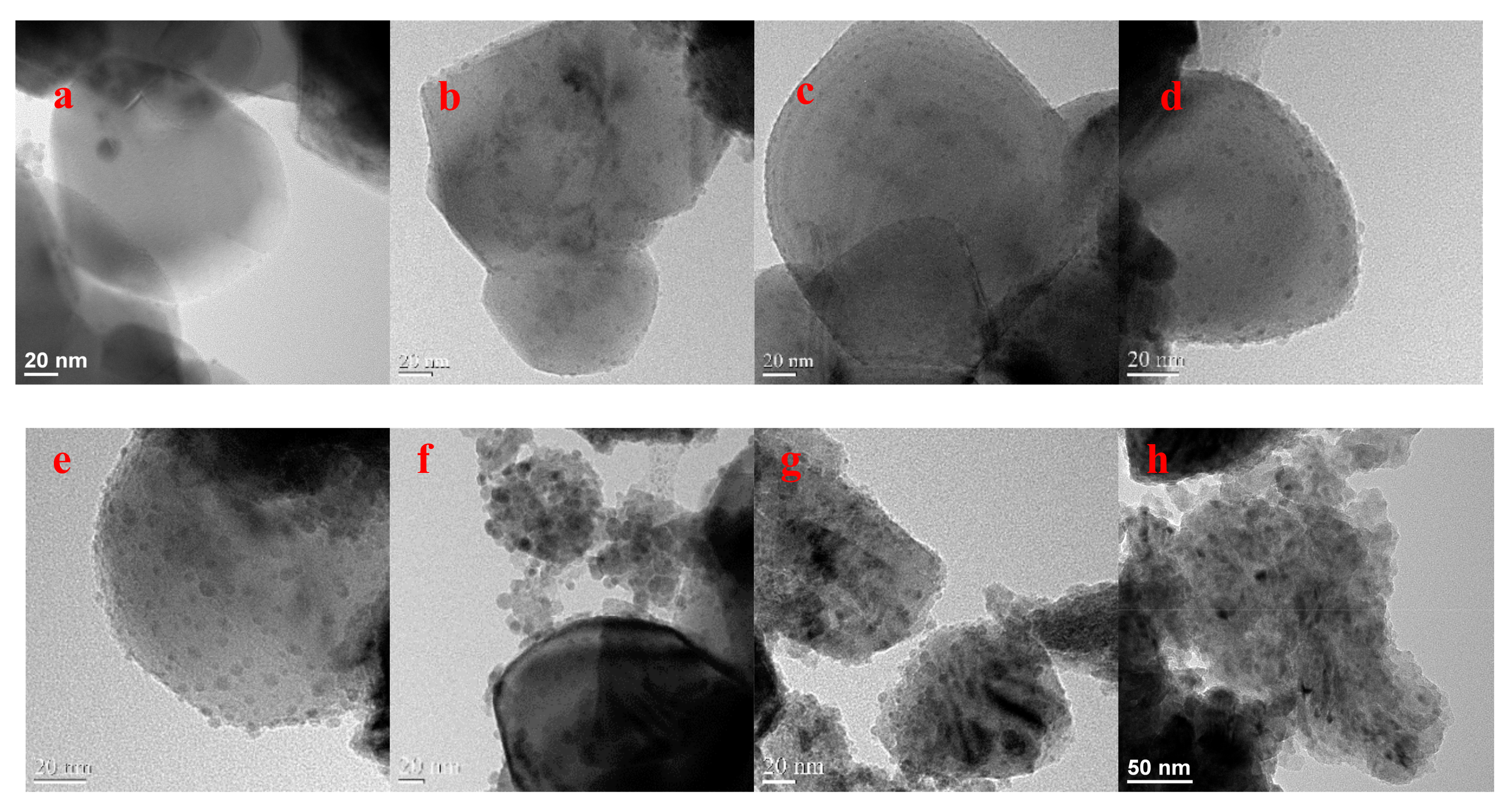
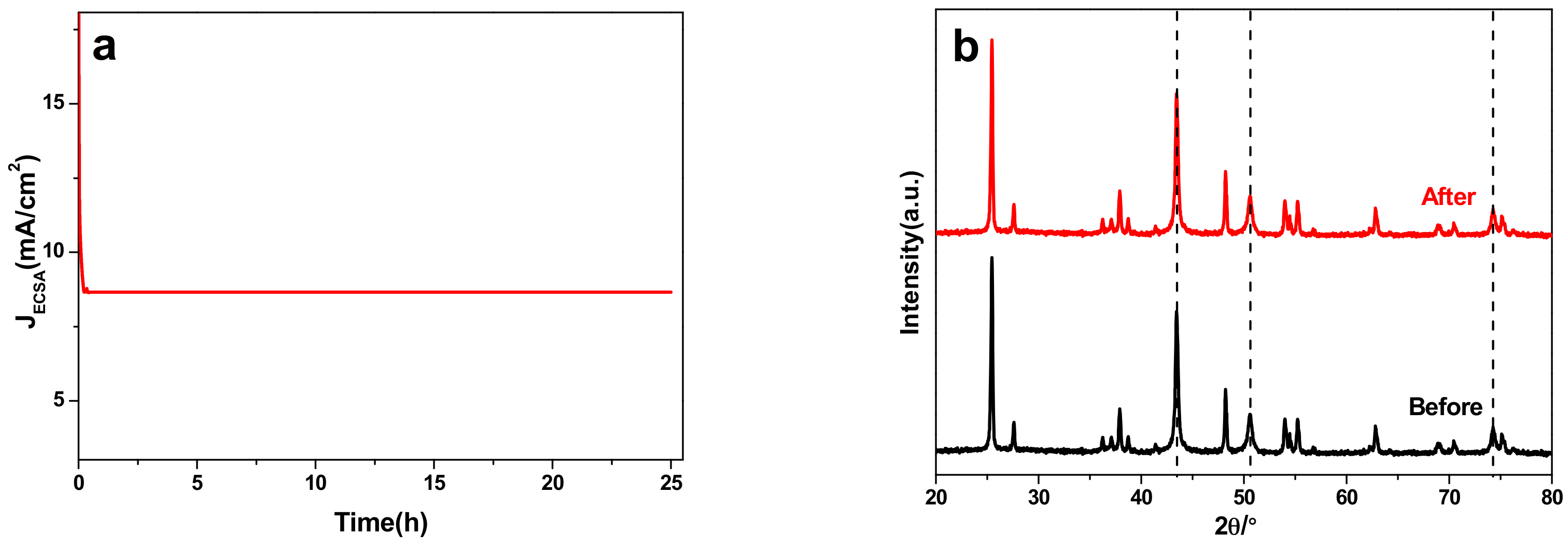
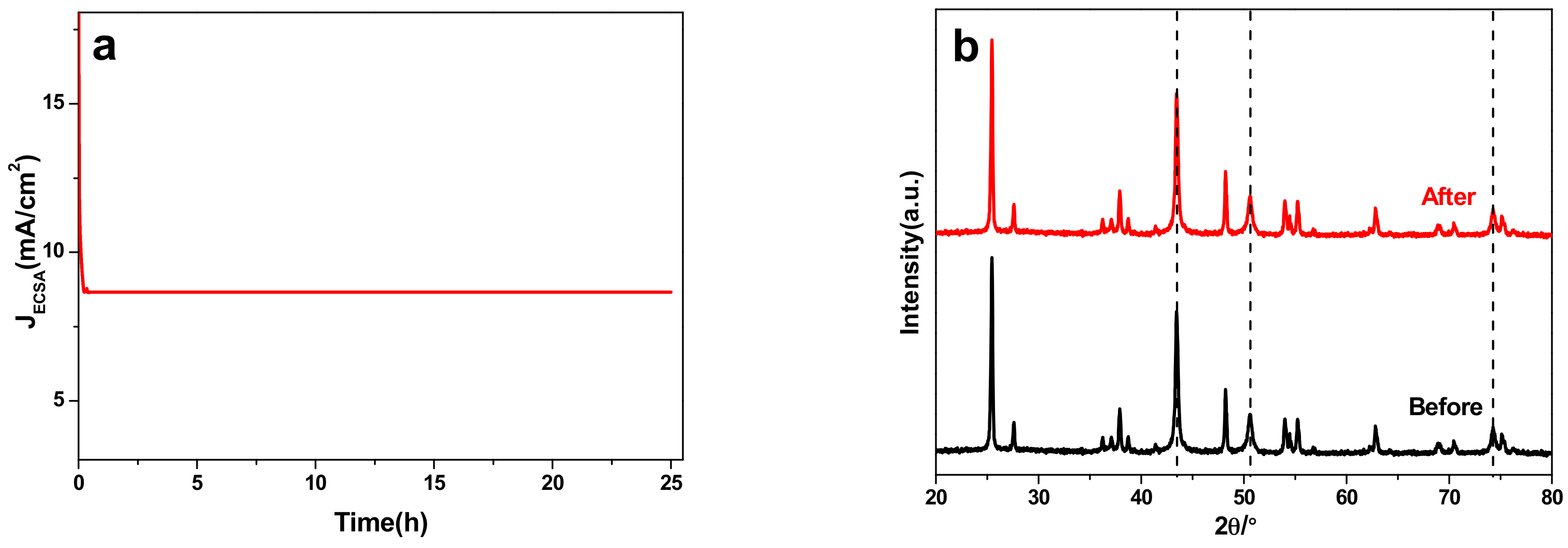
2.4. Electrocatalytic Characterization
3. Materials and Methods
3.1. Materials
3.2. Material Characterization and Product Measurement
3.3. Material Synthesis
3.4. Electrode Preparation and Electrochemical Test
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Figueroa, J.D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R.D. Advances in CO2 capture technology—The U.S. Department of Energy’s Carbon Sequestration Program. Int. J. Greenh. Gas Control. 2008, 2, 9–20. [Google Scholar] [CrossRef]
- Shakun, J.D.; Clark, P.U.; He, F.; Marcott, S.A.; Mix, A.C.; Liu, Z.Y.; Bliesner, B.O.; Schmittner, A.; Bard, E. Global warming preceded by increasing carbon dioxide concentrations during the last deglaciation. Nature 2012, 484, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic Chemical Carbon Cycle for a Sustainable Future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y. Electrochemical CO2 reduction on metal electrodes. In Modern Aspects of Electrochemistry, 3rd ed.; Vayenas, C., Ed.; Faculty of Engineering, Chiba University: Chiba, Japan, 2008; Volume 42, pp. 89–189. [Google Scholar]
- Shi, G.D.; Yu, L.; Ba, X.; Zhang, X.S.; Zhou, J.Q.; Yu, Y. Copper nanoparticle interspersed MoS2 nanoflowers with enhanced efficiency for CO2 electrochemical reduction to fuel. Dalton Trans. 2017. [Google Scholar] [CrossRef]
- Li, Q.; Fu, J.J.; Zhu, W.L.; Chen, Z.Z.; Shen, B.; Wu, L.H.; Xi, Z.; Wang, T.Y.; Lu, G.; Zhu, J.J.; et al. Tuning Sn-Catalysis for Electrochemical Reduction of CO2 to CO via the Core/Shell Cu/SnO2 Structure. J. Am. Chem. Soc. 2017, 139, 4290–4293. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kanan, M.W. Controlling H+ vs. CO2 Reduction Selectivity on Pb Electrodes. ACS Catal. 2015, 5, 465–469. [Google Scholar]
- Feng, X.F.; Jiang, K.L.; Fan, S.S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Kanan, M.W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef] [PubMed]
- Gattrell, M.; Gupta, N. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B.R.; Strasser, P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, K.P.; Cave, E.R.; Abramc, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Mori, K.; Yamashita, H.; Anpo, M. Photocatalytic reduction of CO2 with H2O on various titanium oxide photocatalysts. RSC Adv. 2012, 2, 3165–3172. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.T.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Koudelka, M.; Monnier, A.; Augustynski, J. Electrocatalysis of the Cathodic Reduction of Carbon Dioxide on Platinized Titanium Dioxide Film Electrodes. J. Electrochem. Soc. 1984, 131, 745–750. [Google Scholar] [CrossRef]
- Bandi, A. Electrochemical Reduction of Carbon Dioxide on Conductive Metallic Oxides. J. Electrochem. Soc. 1990, 137, 2157–2160. [Google Scholar] [CrossRef]
- Cueto, L.F.; Hirata, G.A.; Sanchez, E.M. Thin-film TiO2 electrode surface characterization upon CO2 reduction processes. J. Sol–Gel Sci. Technol. 2006, 37, 105–109. [Google Scholar] [CrossRef]
- Beck, F.; Gabriel, W. Heterogeneous Redox Catalysis on Ti/TiO2 Cathodes-Reduction of Nitrobenzene. Angew. Chem. Int. Ed. 1985, 24, 771–772. [Google Scholar] [CrossRef]
- Chu, D.; Qin, G.; Yuan, X.; Xu, M.; Zheng, P.; Lu, J. Fixation of CO2 by electrocatalytic reduction and electropolymerization in lonic liquid-H2O solution. ChemSusChem 2008, 1, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyahara, K.; Toyoshima, I. Adsorption of CO2 on TiO2 and Pt/TiO2 Studled by X-ray Photoelectron Spectroscopy and Auger Electron Spectroscopy. J. Phys. Chem. 1984, 88, 3504–3508. [Google Scholar] [CrossRef]
- Markovits, A.; Fahmi, A.; Minot, C. A theoretical study of CO2 adsorption on TiO2. J. Mol. Struct.-Theochem. 1996, 371, 219–235. [Google Scholar] [CrossRef]
- Huang, S.Y.; Ganesan, P.; Popov, B.N. Titania supported platinum catalyst with high electrocatalytic activity and stability for polymer electrolyte membrane fuel cell. Appl. Catal. B Environ. 2011, 102, 71–77. [Google Scholar] [CrossRef]
- Ma, S.C.; Lan, Y.C.; Perez, G.M.J.; Moniri, S.; Kenis, P.J.A. Silver Supported on Titania as an Active Catalyst for Electrochemical Carbon Dioxide Reduction. ChemSusChem 2014, 7, 866–874. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Cu loading (wt %) | |
|---|---|---|
| Intended | Actual a | |
| 5 wt % Cu/TiO2 | 5 | 4.68 |
| 10 wt % Cu/TiO2 | 10 | 8.52 |
| 20 wt % Cu/TiO2 | 20 | 18.39 |
| 40 wt % Cu/TiO2 | 40 | 37.12 |
| 60 wt % Cu/TiO2 | 60 | 53.06 |
| 80 wt % Cu/TiO2 | 80 | 73.02 |
| Cu/TiO2 Amount (mg/cm2) | FEethanol (%) b | FEn-propanol (%) b |
|---|---|---|
| 5 | 2.1 | 1.2 |
| 4 | 4.6 | 1.8 |
| 3 | 7.2 | 3.2 |
| 2 | 19.0 | 6.0 |
| 1 | 27.4 | 6.2 |
| 0.5 | 6.3 | 2.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, J.; Liu, L.; Guo, R.-R.; Zeng, S.; Wang, H.; Lu, J.-X. Electroreduction of CO2 into Ethanol over an Active Catalyst: Copper Supported on Titania. Catalysts 2017, 7, 220. https://doi.org/10.3390/catal7070220
Yuan J, Liu L, Guo R-R, Zeng S, Wang H, Lu J-X. Electroreduction of CO2 into Ethanol over an Active Catalyst: Copper Supported on Titania. Catalysts. 2017; 7(7):220. https://doi.org/10.3390/catal7070220
Chicago/Turabian StyleYuan, Jing, Li Liu, Rong-Rong Guo, Sheng Zeng, Huan Wang, and Jia-Xing Lu. 2017. "Electroreduction of CO2 into Ethanol over an Active Catalyst: Copper Supported on Titania" Catalysts 7, no. 7: 220. https://doi.org/10.3390/catal7070220
APA StyleYuan, J., Liu, L., Guo, R.-R., Zeng, S., Wang, H., & Lu, J.-X. (2017). Electroreduction of CO2 into Ethanol over an Active Catalyst: Copper Supported on Titania. Catalysts, 7(7), 220. https://doi.org/10.3390/catal7070220