
N2O Direct Dissociation over MgxCeyCo1−x−yCo2O4 Composite Spinel Metal Oxide


Abstract
:1. Introduction
2. Results and Discussion
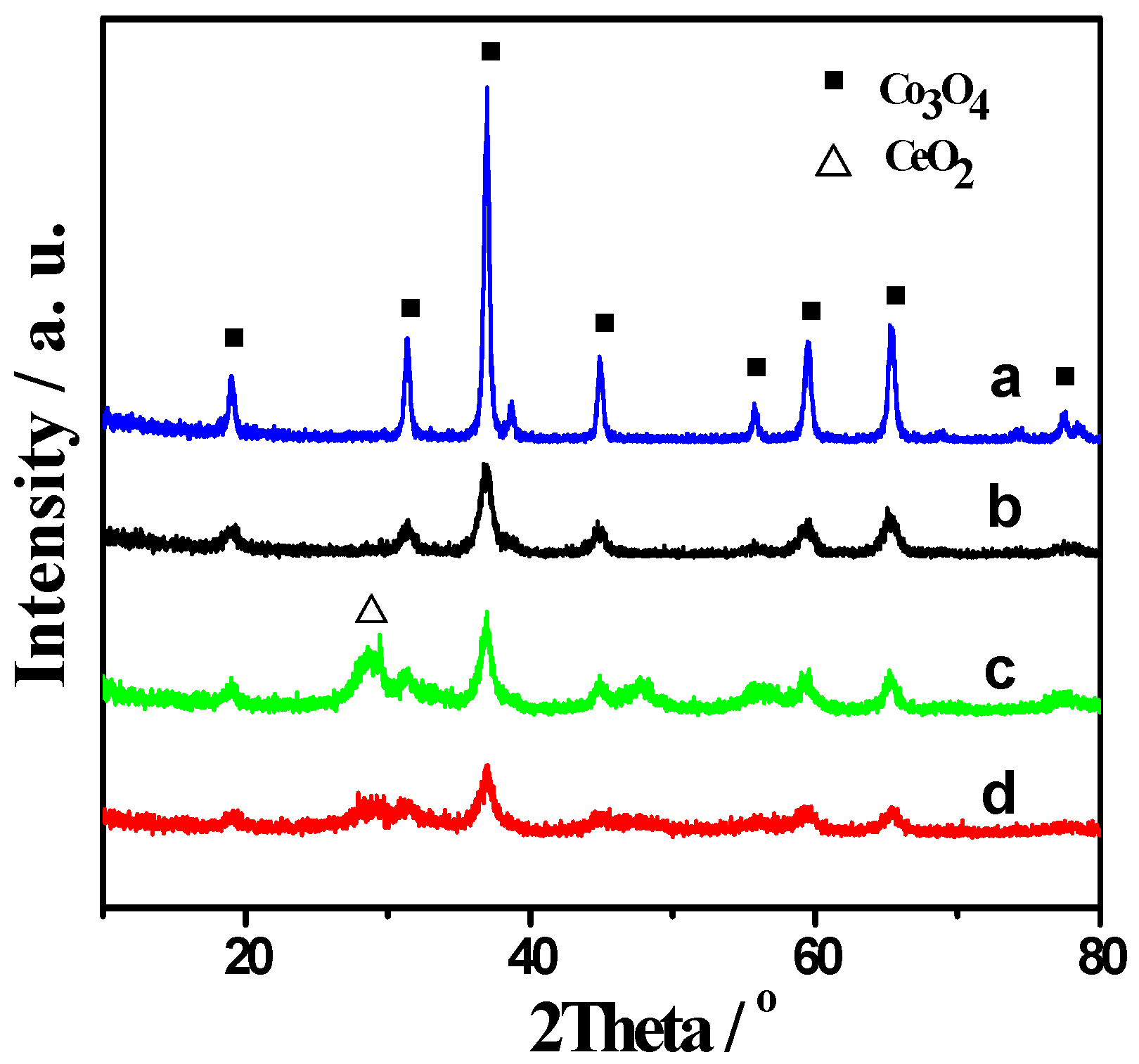
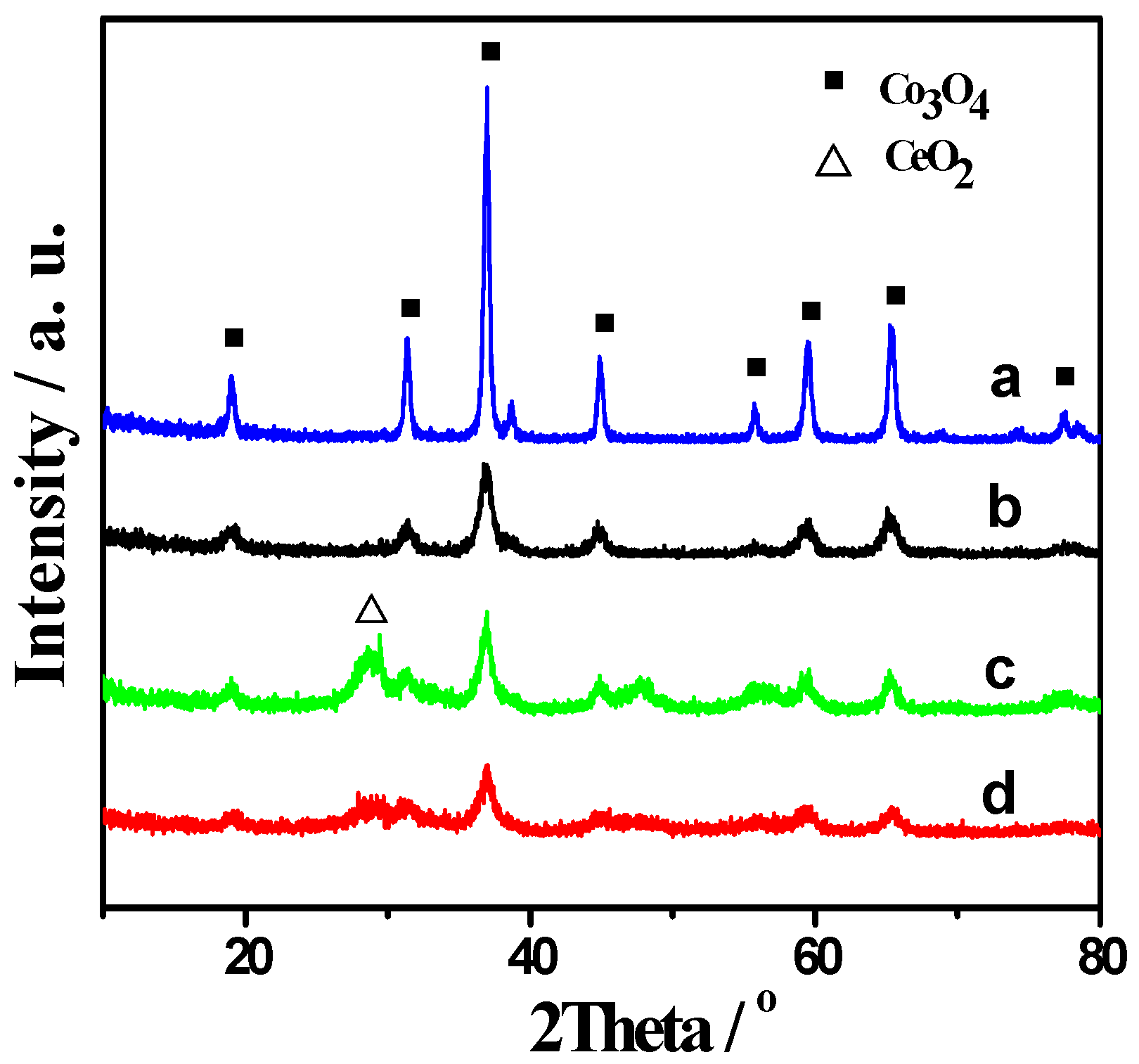
2.1. XRD and BET Results
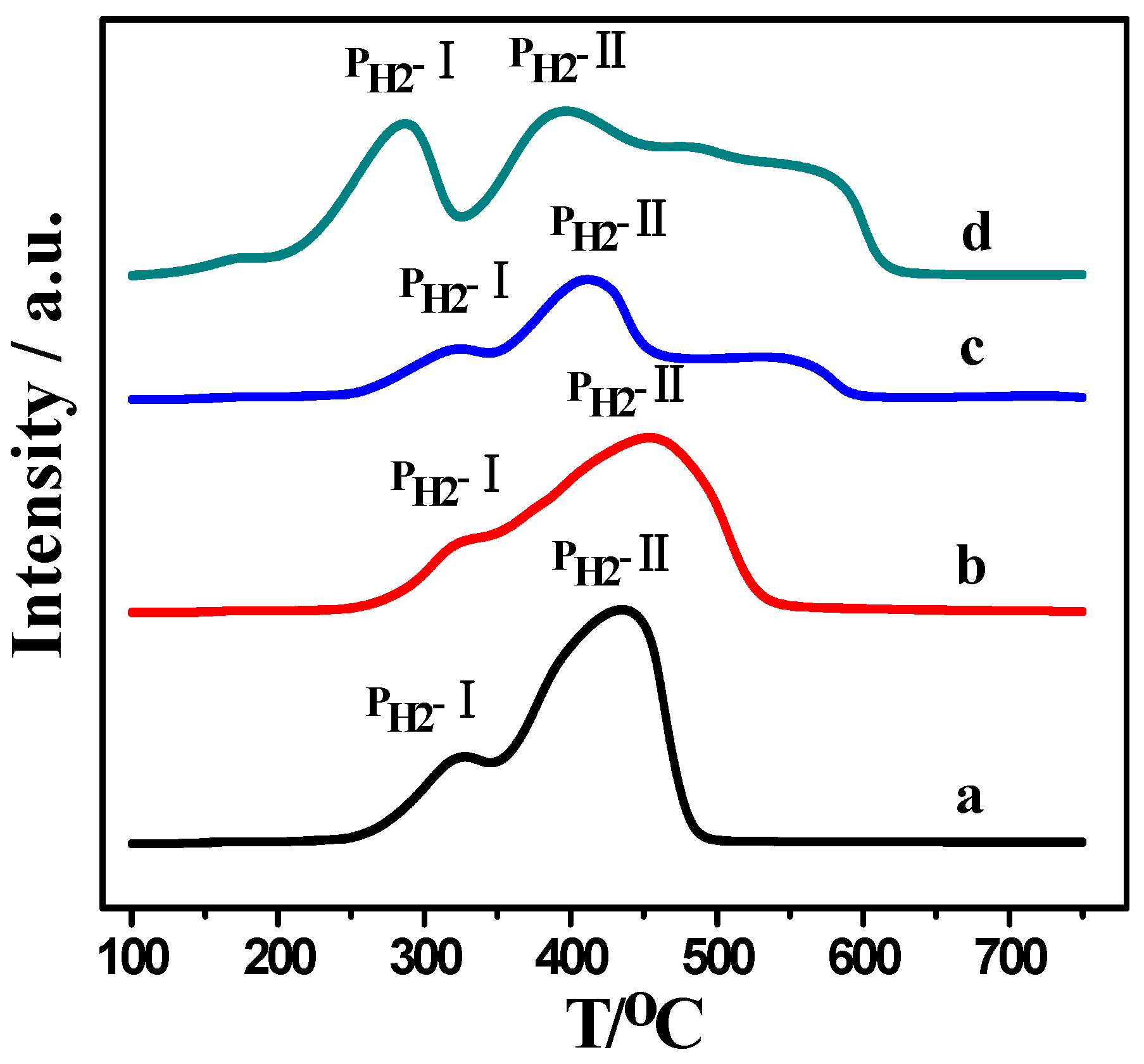
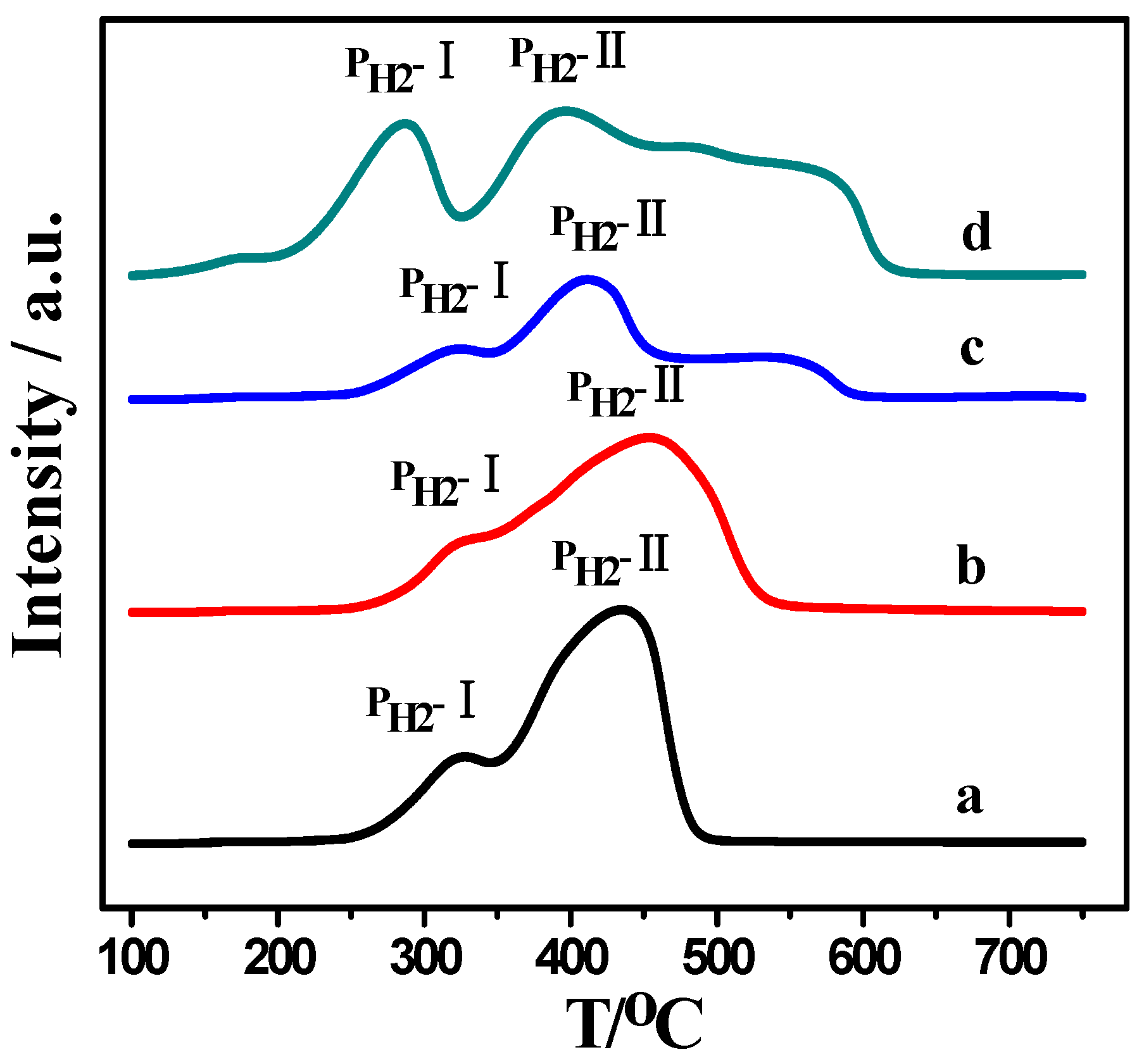
2.2. H2-TPR
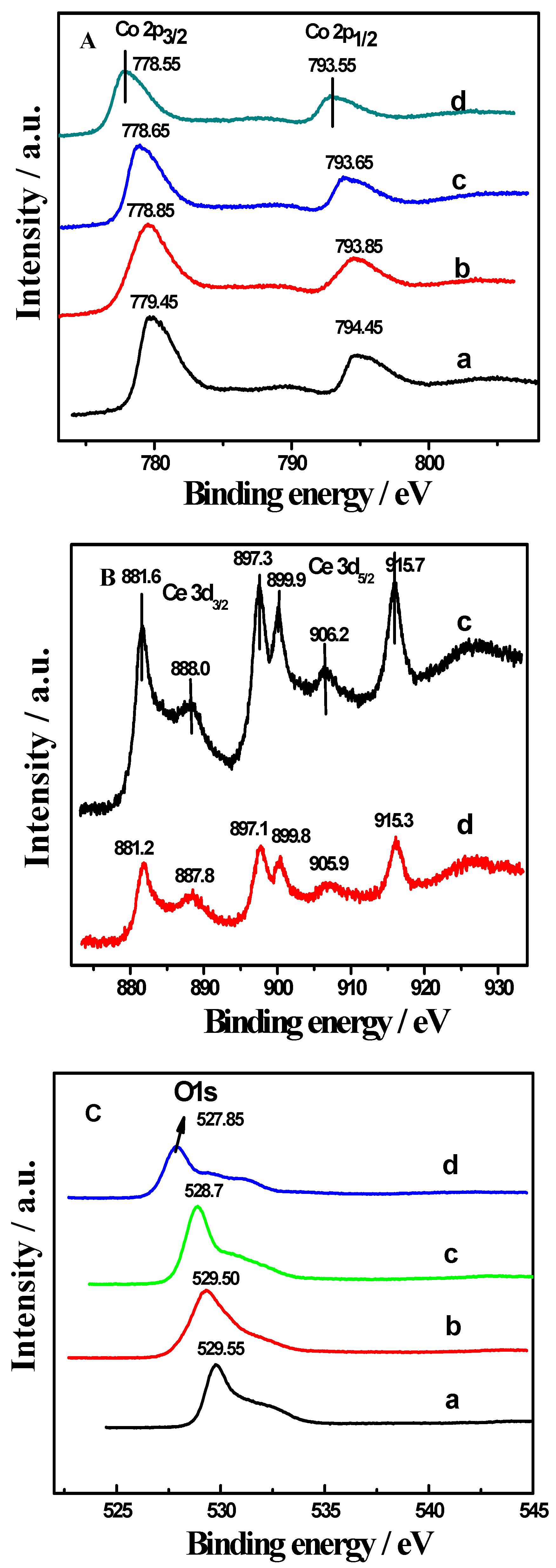
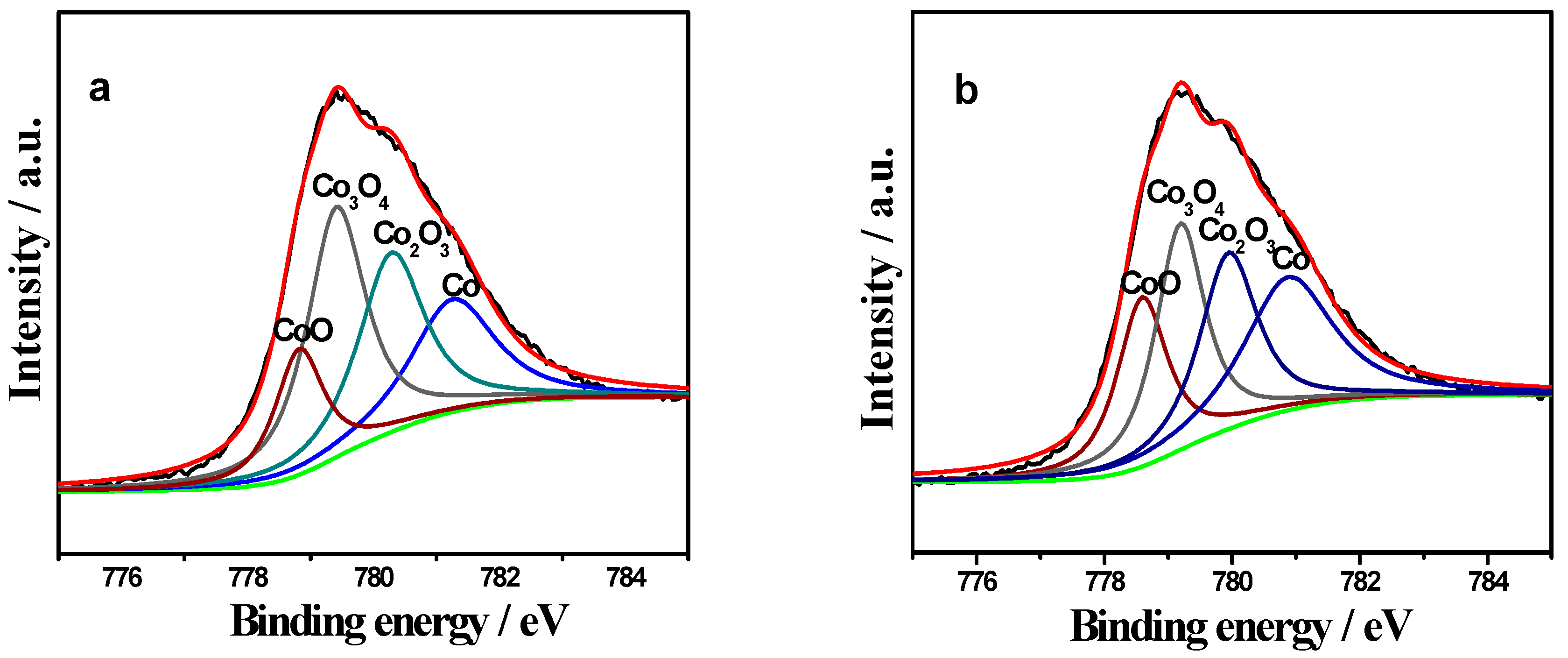
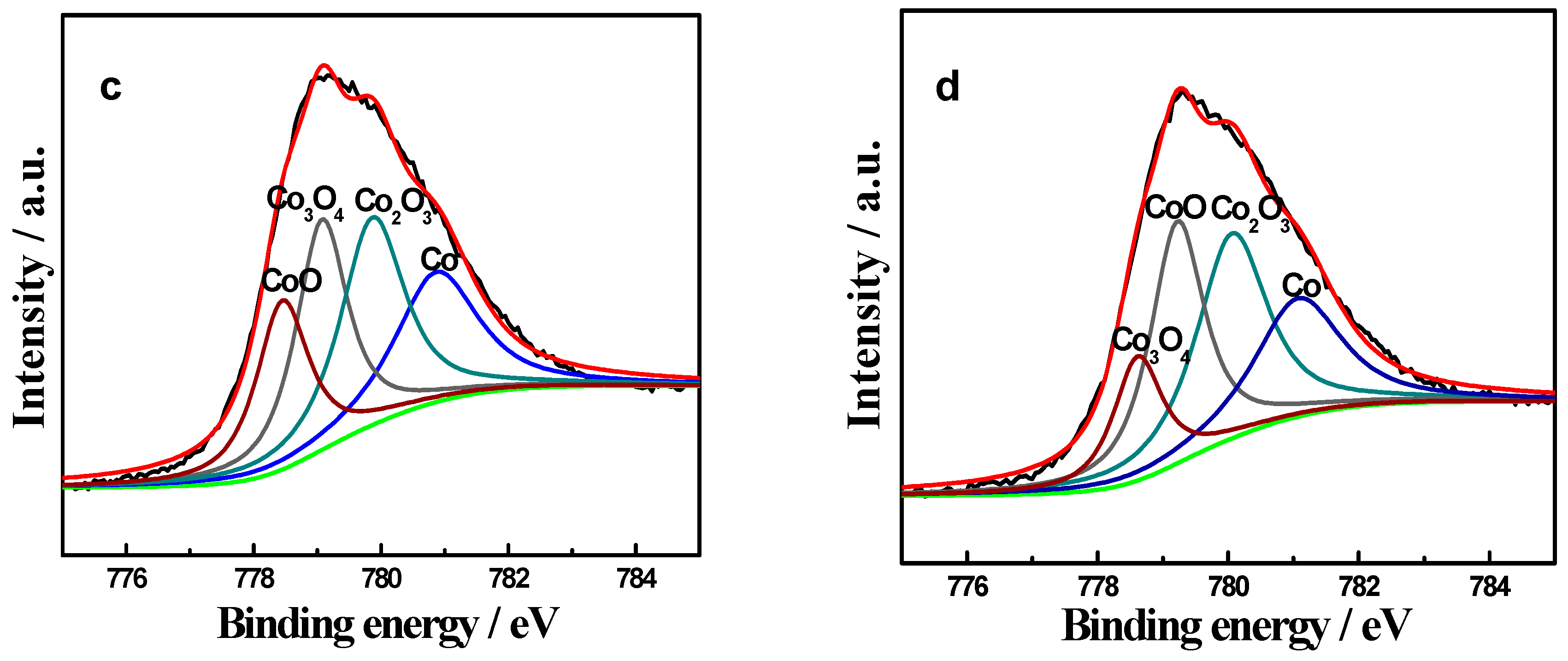
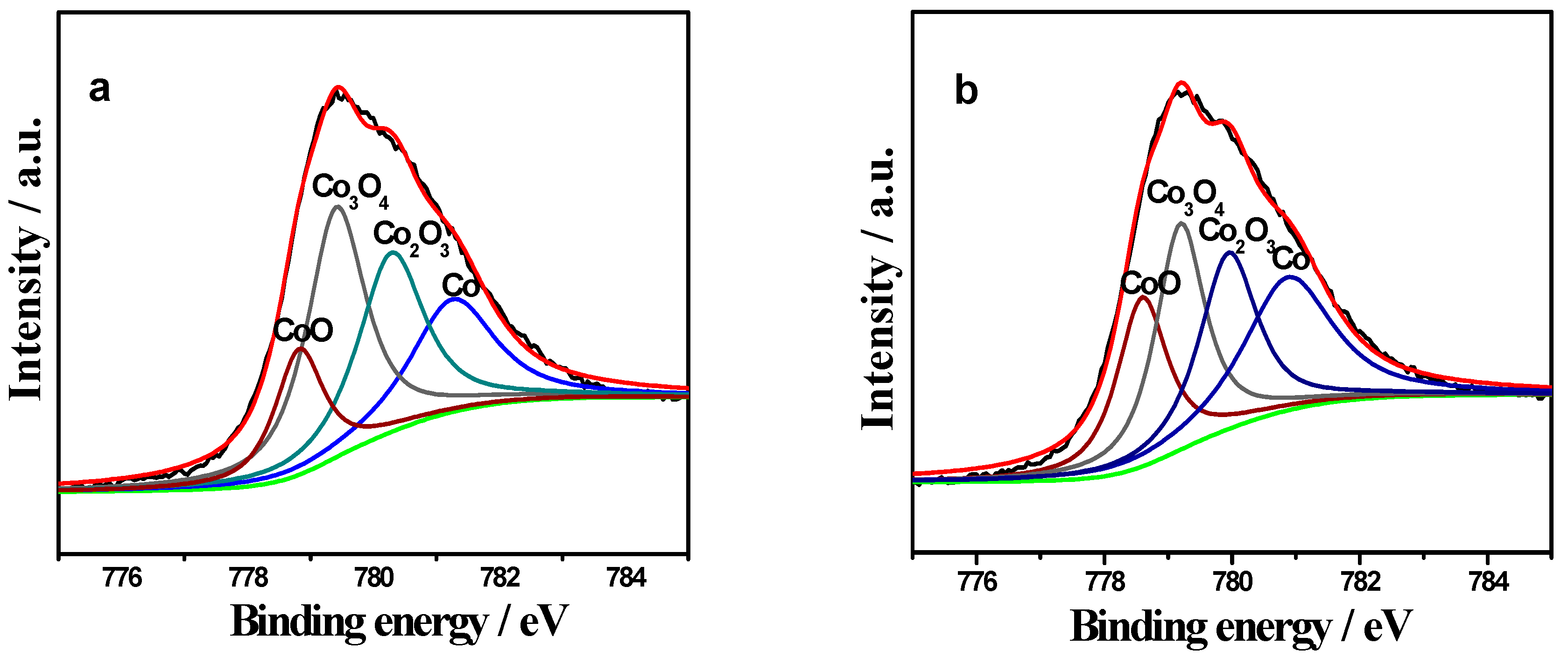
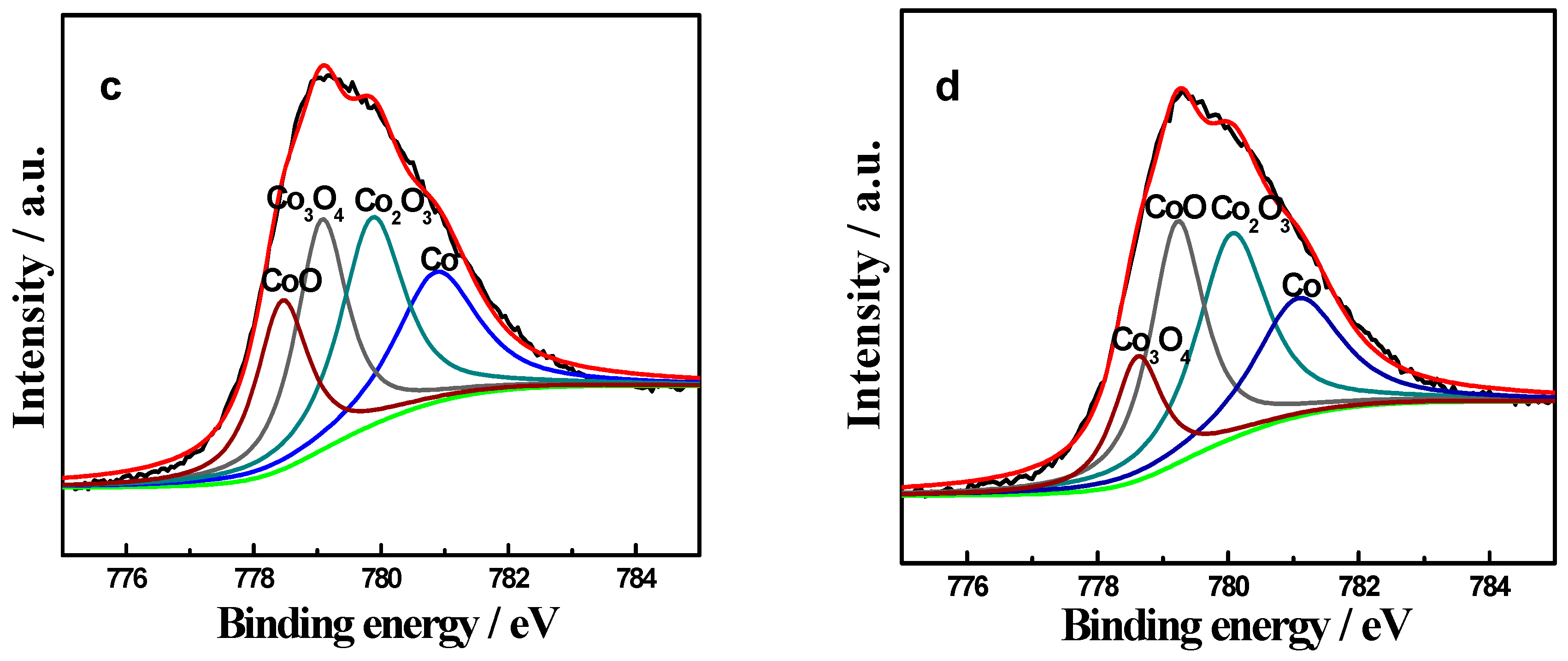
2.3. XPS
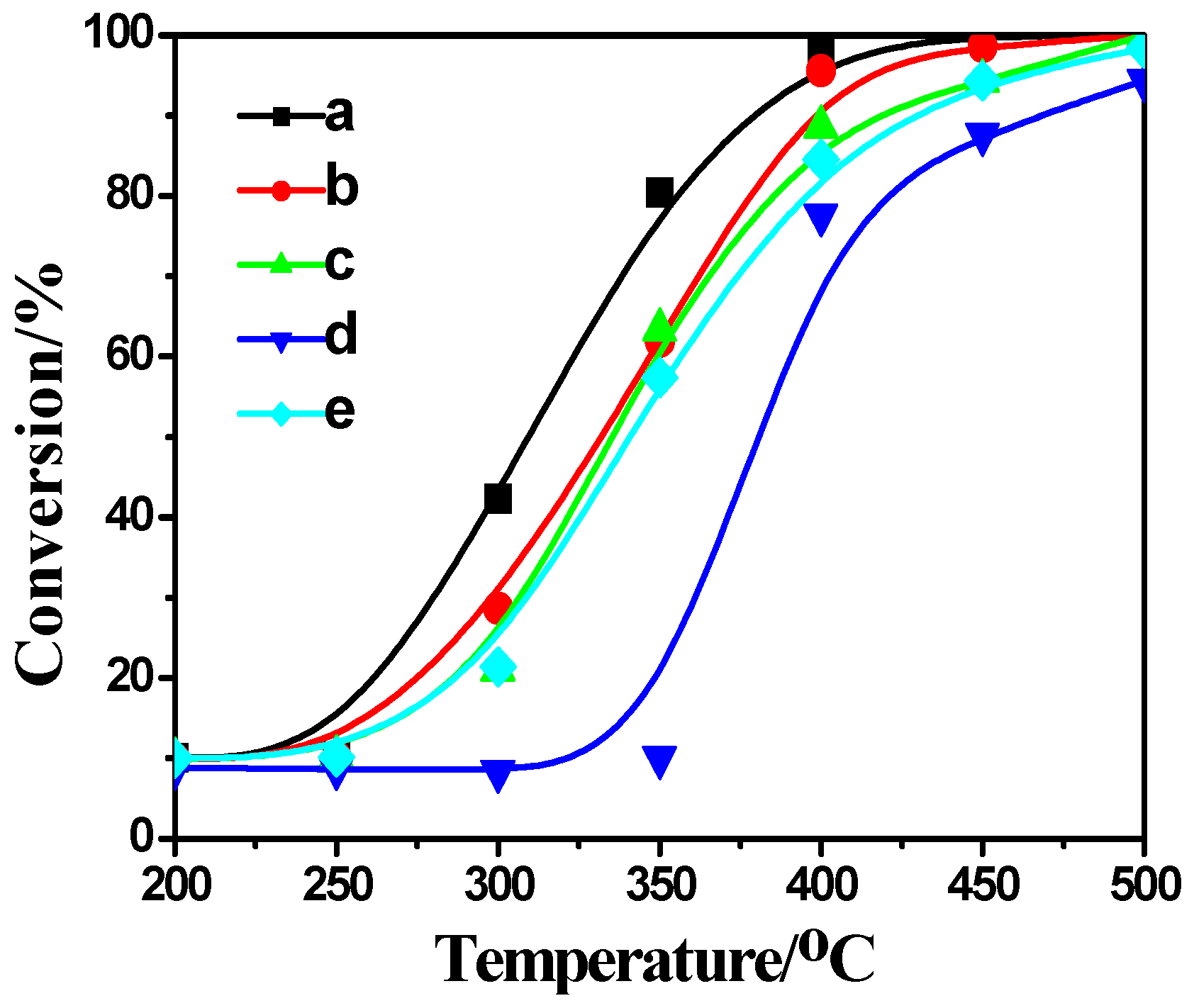
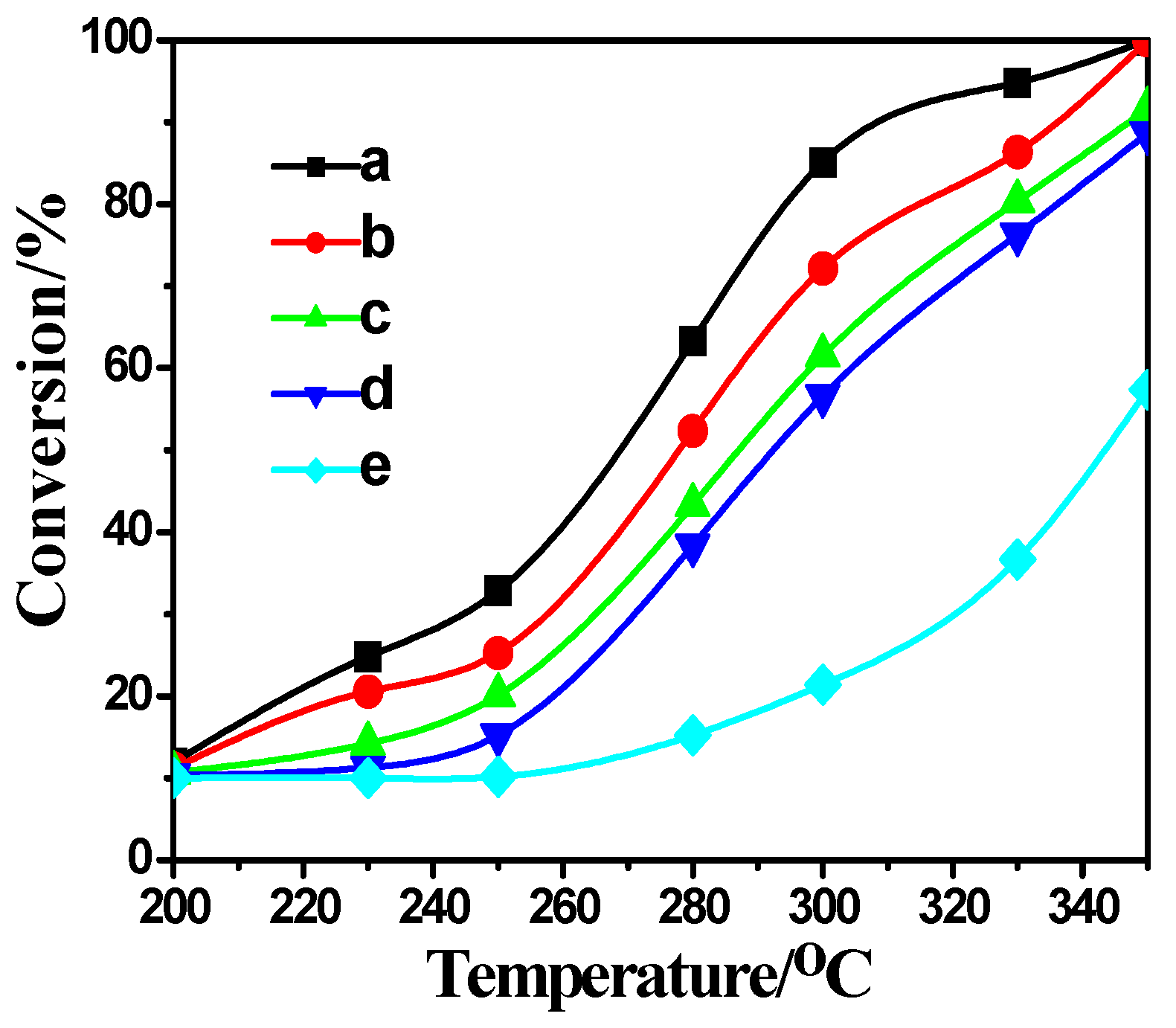
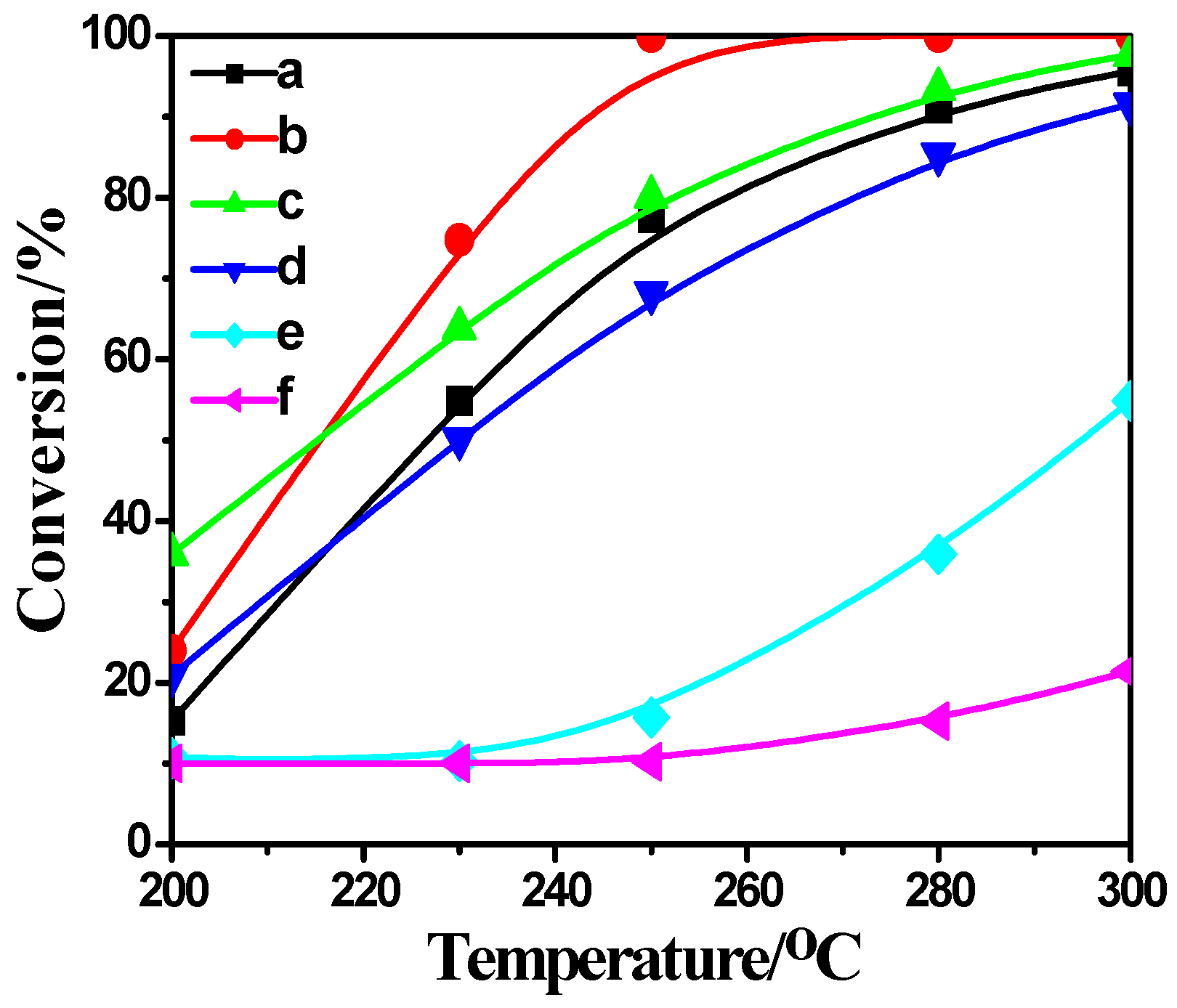
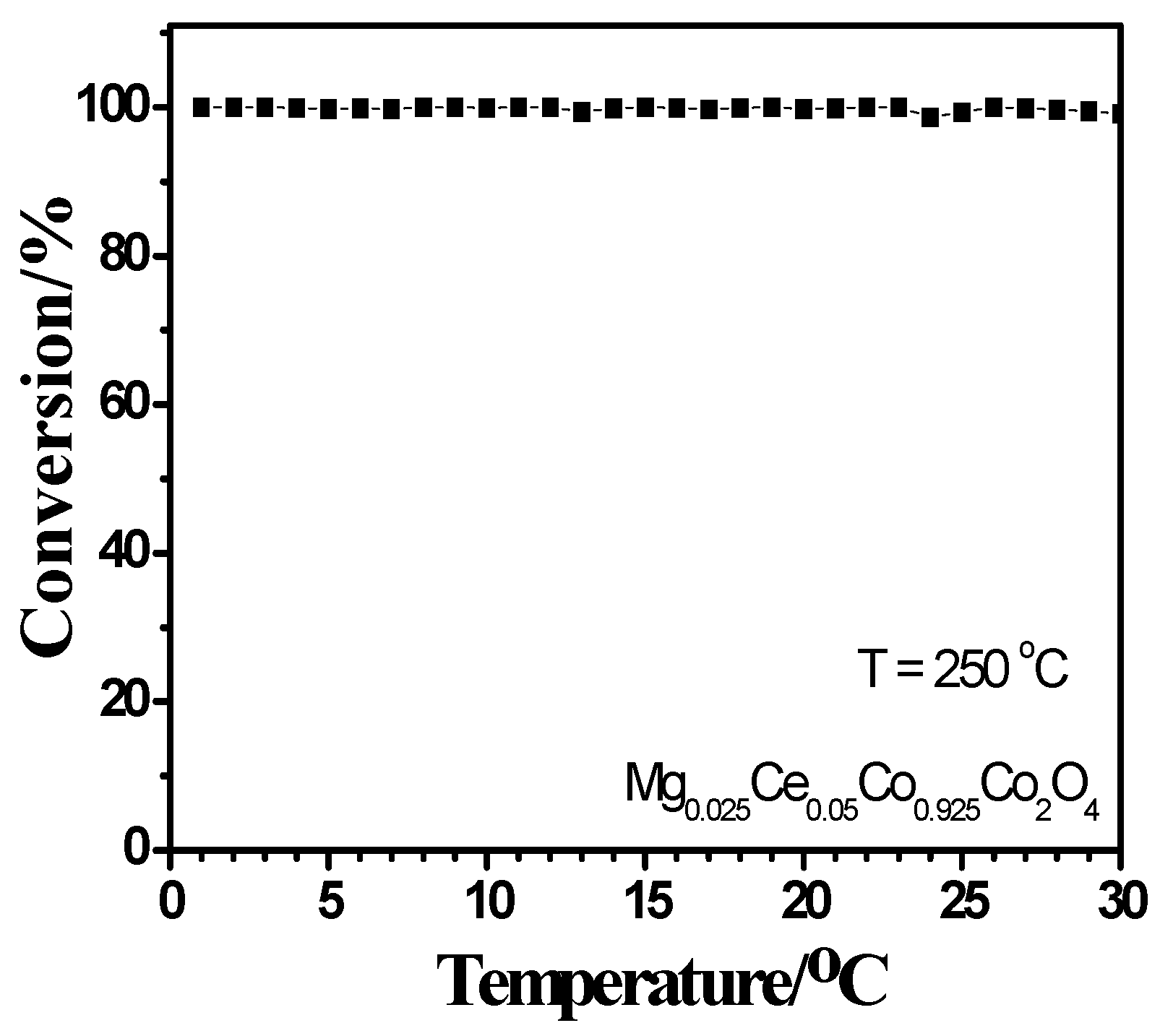
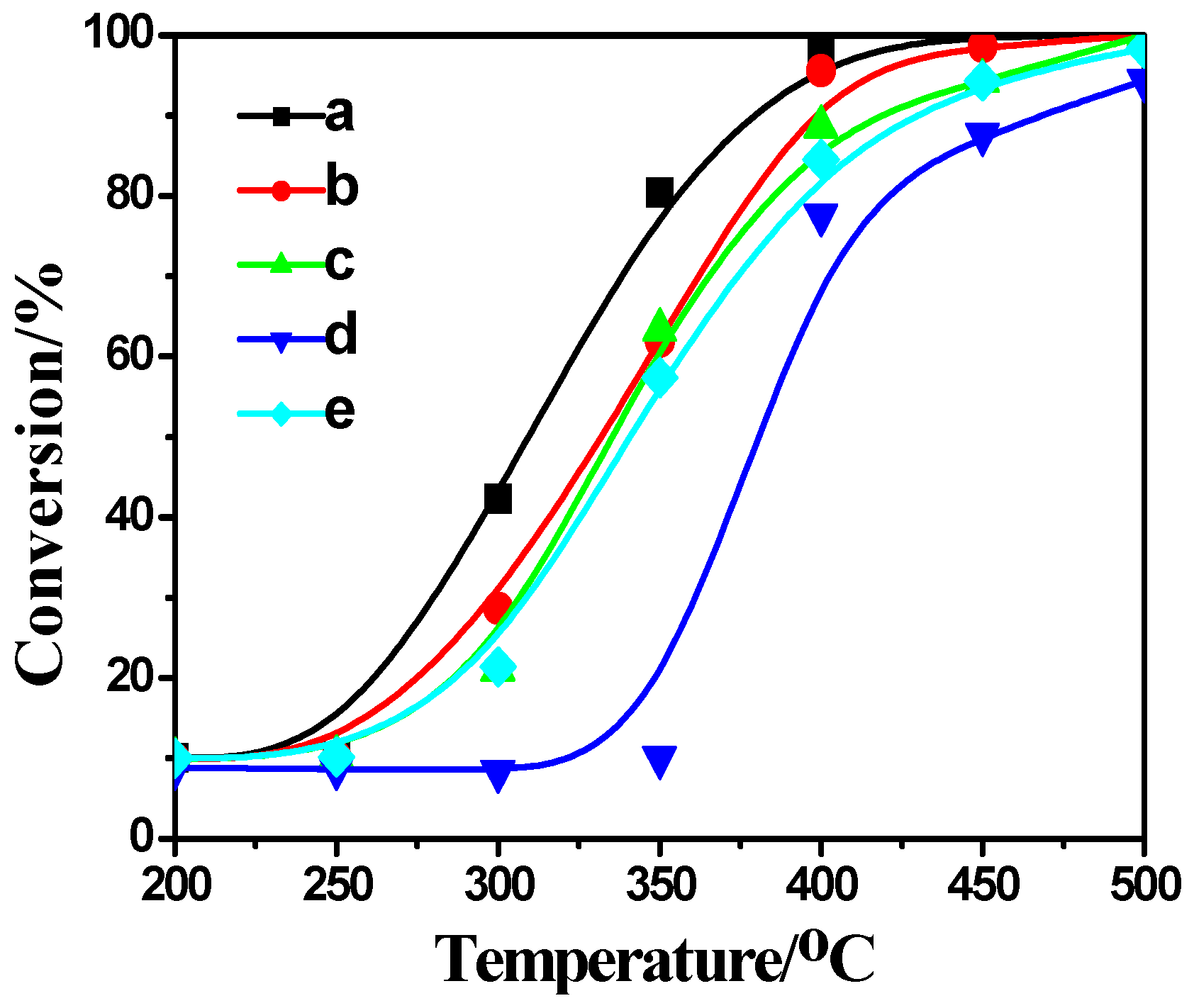
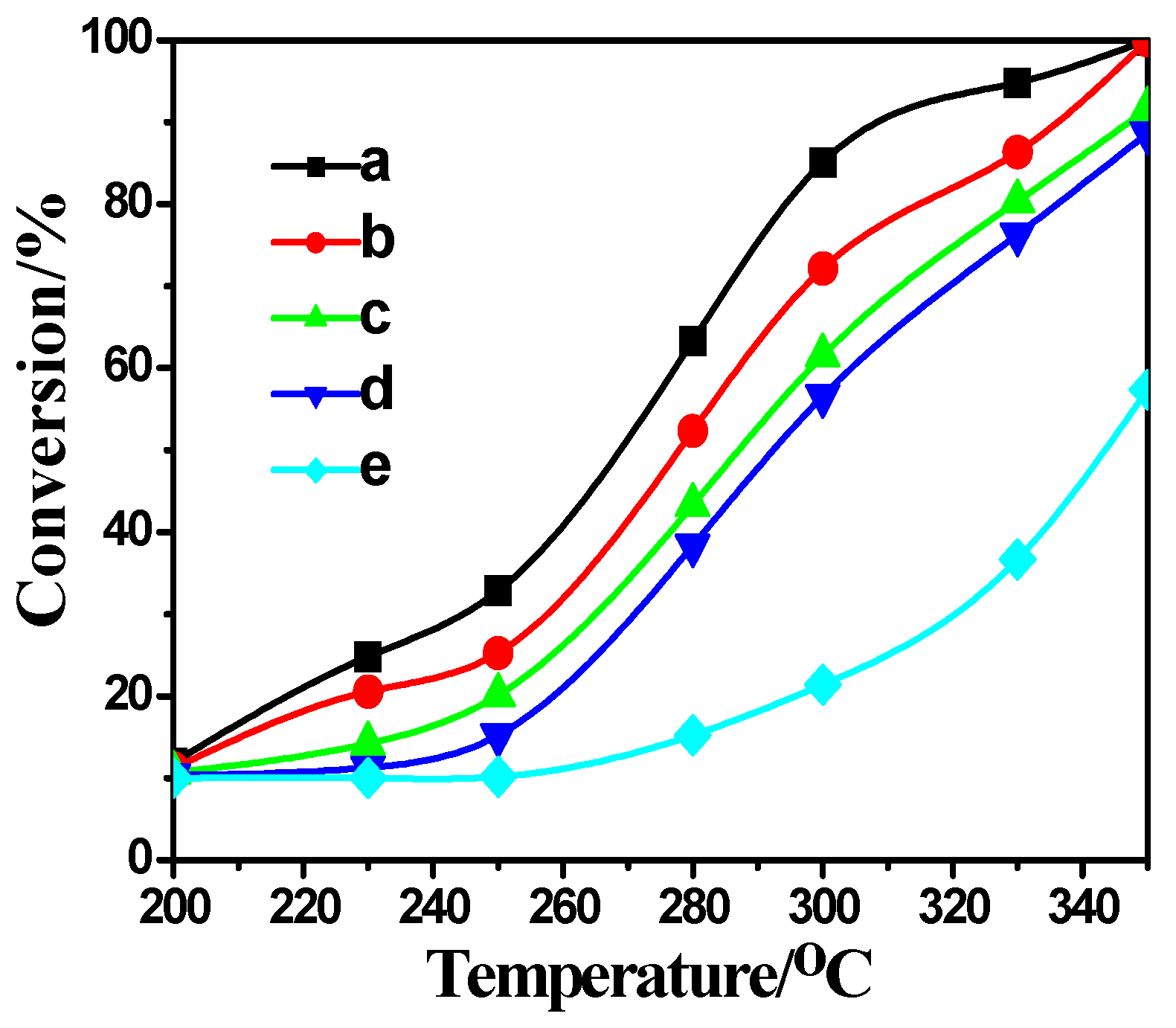
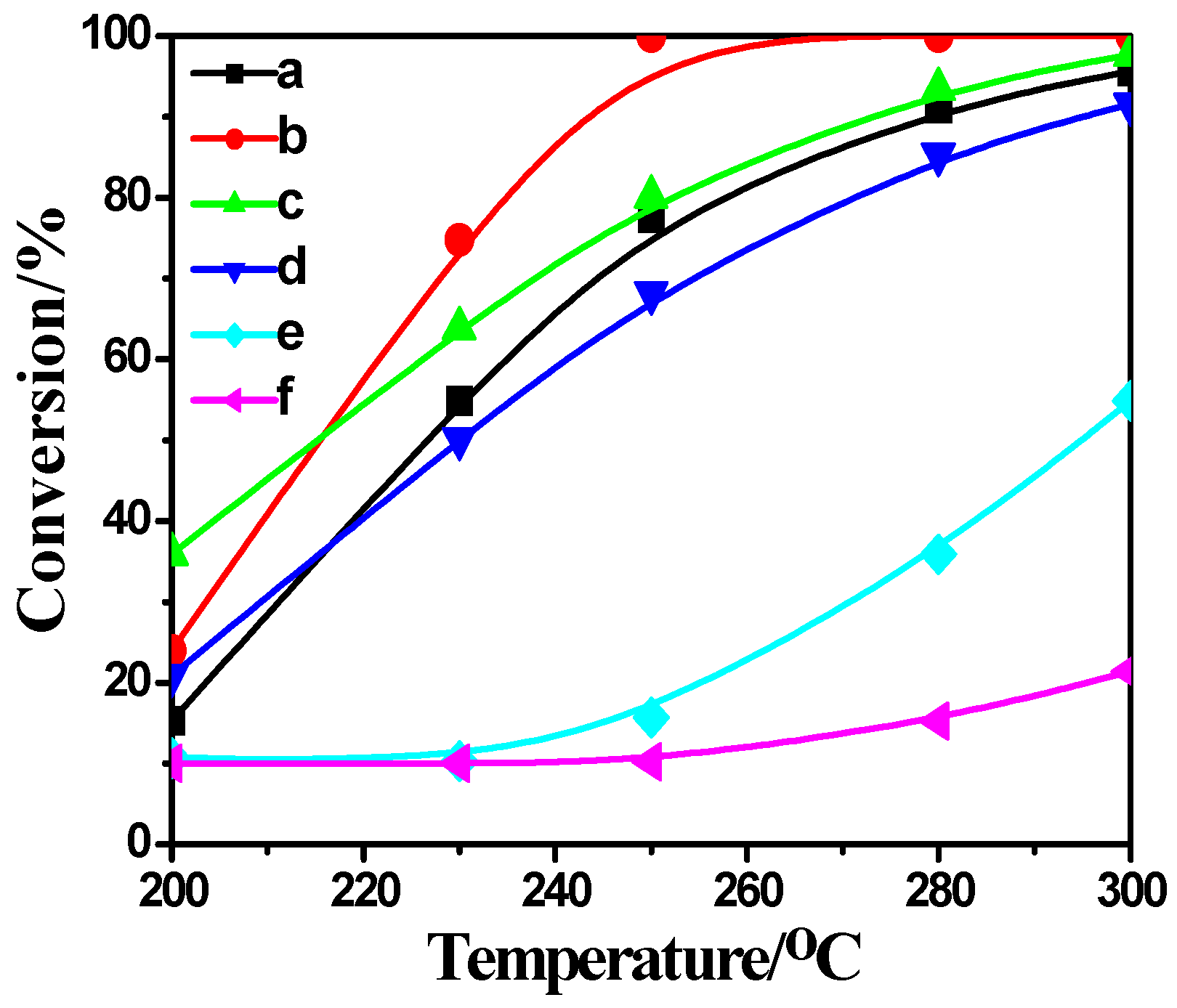
2.4. Activity Measurement
2.5. Correlation between Characterization and Activity Measurement Result
3. Experimental
3.1. Preparation of Catalyst
3.2. Characterization of Catalysts
3.3. Activity Measurement
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trogler, W.C. Physical Properties and Mechanisms of Formation of Niturous Oxide. Coord. Chem. Rev. 1999, 187, 303–327. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, N.; Lei, Z.; Chen, B. Selective Transformation of Various Nitrogen-Containing Exhaust Gases toward N2 over Zeolite Catalysts. Chem. Rev. 2016, 116, 3658–3721. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, N.; Liu, X.; Zhang, R.; Li, Y.; Li, Y.; Sun, X. Study on the Direct Decomposition of Nitrous Oxide over Fe-Beta Zeolites: From Experiment to Theory. Catal. Today 2011, 175, 245–255. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Chen, B.; Li, Y.; Li, Y. Comparative Study on the Direct Decomposition of Nitrous Oxide over M(Fe, Co, Cu)-BEA Zeolites. J. Catal. 2012, 294, 99–112. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S.; Vazzana, F.; Marclla, M.; Tomaselli, M.; Mantegazza, M. Novel Catalysts and Catalytic Technologies for N2O Removal from Industrial Emissions Containing O2, H2O and SO2. Adv. Environ. Res. 2000, 4, 325–338. [Google Scholar] [CrossRef]
- Galle, M.; Agar, D.W.; Watzenberger, O. Thermal N2O Decomposition in Regenerative Heat Exchanger Reactors. Chem. Eng. Sci. 2001, 56, 1587–1595. [Google Scholar] [CrossRef]
- Xia, H.; Sun, K.; Liu, Z.; Feng, Z.; Ying, P.; Li, C. The Promotional Effect of NO on N2O Decomposition over the Bi-Nuclear Fe Sites in Fe/ZSM-5. J. Catal. 2010, 270, 103–109. [Google Scholar] [CrossRef]
- Melian-Cabrera, I.; Mentruit, C.; Pieterse, J.A.Z.; van den Brink, R.W.; Mul, G.; Kapteijn, F.; Moulijn, J.A. Highly Active and Stable Ion-Exchanged Fe-Ferrierite Catalyst for N2O Decomposition under Nitric Acid Tail Gas Conditions. Catal. Commun. 2005, 6, 301–305. [Google Scholar] [CrossRef]
- Kameoka, S.; Suzuki, T.; Yuzaki, K.; Takeda, T.; Tanaka, S.; Ito, S.; Kunimori, K.; Miyadera, T. Selective Catalytic Reduction of N2O with Methane in the Presence of Excess Oxygen over Fe-BEA Zeolite. Chem. Commun. 2000, 9, 745–746. [Google Scholar] [CrossRef]
- Dacquin, J.P.; Dujardin, C.; Granger, P. Surface Reconstruction of Supported Pd on LaCoO3: Consequences on the Catalytic Properties in the Decomposition of N2O. J. Catal. 2008, 253, 37–49. [Google Scholar] [CrossRef]
- Franken, T.; Palkovits, R. Investigation of Potassium Doped Mixed Spinels CuxCo3−xO4 as Catalysts for an Efficient N2O Decomposition in Real Reaction Conditions. Appl. Catal. B 2015, 176–177, 298–305. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Djinović, P.; Tchernychova, E.; Pintar, A. N2O Decomposition over CuO/CeO2 Catalyst: New Insights into Reaction Mechanism and Inhibiting Action of H2O and NO by Operando Techniques. Appl. Catal. B 2016, 197, 146–158. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Catalytic Decomposition of N2O over CeO2 Pomoted Co3O4 Spinel Catalyst. Appl. Catal. B 2007, 75, 167–174. [Google Scholar] [CrossRef]
- Grzybeka, G.; Stelmachowskia, P.; Gudykaa, S.; Indykaa, P.; Sojkaa, Z.; Guillén-Hurtadob, N.; Rico-Pérezb, V.; Bueno-Lópezb, A.; Kotarbaa, A. Strong Dispersion Effect of Cobalt Spinel Active Phase Spread over Ceria for Catalytic N2O Decomposition: The Role of the Interface Periphery. Appl. Catal. B 2016, 180, 622–629. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Maniak, G.; Kaczmarczyk, J.; Zasada, F.; Piskorz, W.; Kotarba, A.; Sojka, Z. Mg and Al Substituted Cobalt Spinels as Catalysts for Low Temperature DeN2O-Evidence for Octahedral Cobalt Active Sites. Appl. Catal. B 2014, 146, 105–111. [Google Scholar] [CrossRef]
- Zhang, R.; Hedjazi, K.; Chen, B.; Li, Y.; Lei, Z.; Liu, N. M(Fe, Co)-BEA Washcoated Honeycomb Cordierite for N2O Direct Decomposition. Catal. Today 2016, 273, 273–285. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Li, Y.; Chen, B. Local Electric Field Effect of TMI (Fe, Co, Cu)-BEA on N2O Direct Dissociation. J. Phys. Chem. C 2014, 118, 10944–10956. [Google Scholar] [CrossRef]
- Liu, N.; Chen, B.; Li, Y.; Zhang, R.; Liang, X.; Lei, Z. Charge Transfer Analysis on the Direct Decomposition of Nitrous Oxide over Fe-BEA Zeolite: An Experimental and Density Functional Study. J. Phys. Chem. C 2011, 115, 12883–12890. [Google Scholar] [CrossRef]
- Watanabe, S.; Ma, X.; Song, C. Characterization of Structural and Surface Properties of Nanocrystalline TiO2-CeO2 Mixed Oxides by XRD, XPS, TPR, and TPD. J. Phys. Chem. C 2009, 113, 14249–14257. [Google Scholar] [CrossRef]
- Li, X.; Hong, H. Co-M (M = La, Ce, Fe, Mn, Cu, Cr) Catalytic Decomposition of Complex Metal Oxides N2O. Acta Phys. Chim. Sin. 2007, 23, 664–670. [Google Scholar]
- Harrison, P.G.; Ball, I.K.; Daniell, W.; Lukinskas, P.; Céspedes, M.; Miró, E.E.; Ulla, M.A. Cobalt Catalysts for the Oxidation of Diesel Soot Particulate. Chem. Eng. J. 2003, 95, 47–55. [Google Scholar] [CrossRef]
- Bahlawane, N.; Rivera, E.F.; Kohse-Höinghaus, K.; Brechling, A.; Kleineberg, U. Characterization and Tests of Planar Co3O4 Model Catalysts Prepared by Chemical Vapor Deposition. Appl. Catal. B 2004, 53, 245–255. [Google Scholar] [CrossRef]
- Karlsen, E.J.; Nygren, M.A.; Pettersson, L.G.M. Theoretical Study on the Decomposition of N2O over Alkaline Earth Metal-Oxides: MgO-BaO. Phys. Chem. A 2002, 106, 41–50. [Google Scholar] [CrossRef]
- He, H. Influence of Alkaline Earth Metals on Cobalt-Cerium Composite Oxide Catalysts for NO Decomposition. Acta Phys. Chim. Sin. 2009, 25, 1033–1039. [Google Scholar]
- Huang, T.J.; Wang, C.H. Roles of Surface and Bulk Lattice Oxygen in Forming CO2 and CO during Methane Reaction over Gadolinia-Doped Ceria. Catal. Lett. 2007, 118, 103–108. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | BET Specific Surface Area (m2·g–1) | Crystal Size (nm) |
|---|---|---|
| Co3O4 | 32.5 | 22.4 |
| Mg0.2Co0.8Co2O4 | 77.8 | 14.6 |
| Ce0.05Co0.95Co2O4 | 106.5 | 11.8 |
| Mg0.025Ce0.05Co0.925Co2O4 | 111.2 | 10.1 |
| Sample | Surface Atomic Score Levels (%) | Co2+/Co3+ | |||
|---|---|---|---|---|---|
| Co | Mg | Ce | O | ||
| Co3O4 | 35.6 | 0 | 0 | 64.4 | 0.65 |
| Mg0.2Co0.8Co2O4 | 32.7 | 2.3 | 0 | 65.1 | 0.76 |
| Ce0.05Co0.95Co2O4 | 17.6 | 0 | 7.4 | 74.9 | 0.91 |
| Mg0.025Ce0.05Co0.925Co2O4 | 17.5 | 2.4 | 3.7 | 76.5 | 1.11 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Chen, P.; Li, Y.; Zhang, R. N2O Direct Dissociation over MgxCeyCo1−x−yCo2O4 Composite Spinel Metal Oxide. Catalysts 2017, 7, 10. https://doi.org/10.3390/catal7010010
Liu N, Chen P, Li Y, Zhang R. N2O Direct Dissociation over MgxCeyCo1−x−yCo2O4 Composite Spinel Metal Oxide. Catalysts. 2017; 7(1):10. https://doi.org/10.3390/catal7010010
Chicago/Turabian StyleLiu, Ning, Ping Chen, Yingxia Li, and Runduo Zhang. 2017. "N2O Direct Dissociation over MgxCeyCo1−x−yCo2O4 Composite Spinel Metal Oxide" Catalysts 7, no. 1: 10. https://doi.org/10.3390/catal7010010
APA StyleLiu, N., Chen, P., Li, Y., & Zhang, R. (2017). N2O Direct Dissociation over MgxCeyCo1−x−yCo2O4 Composite Spinel Metal Oxide. Catalysts, 7(1), 10. https://doi.org/10.3390/catal7010010