
Efficient Hydrogenolysis of Guaiacol over Highly Dispersed Ni/MCM-41 Catalyst Combined with HZSM-5


Abstract
:1. Introduction
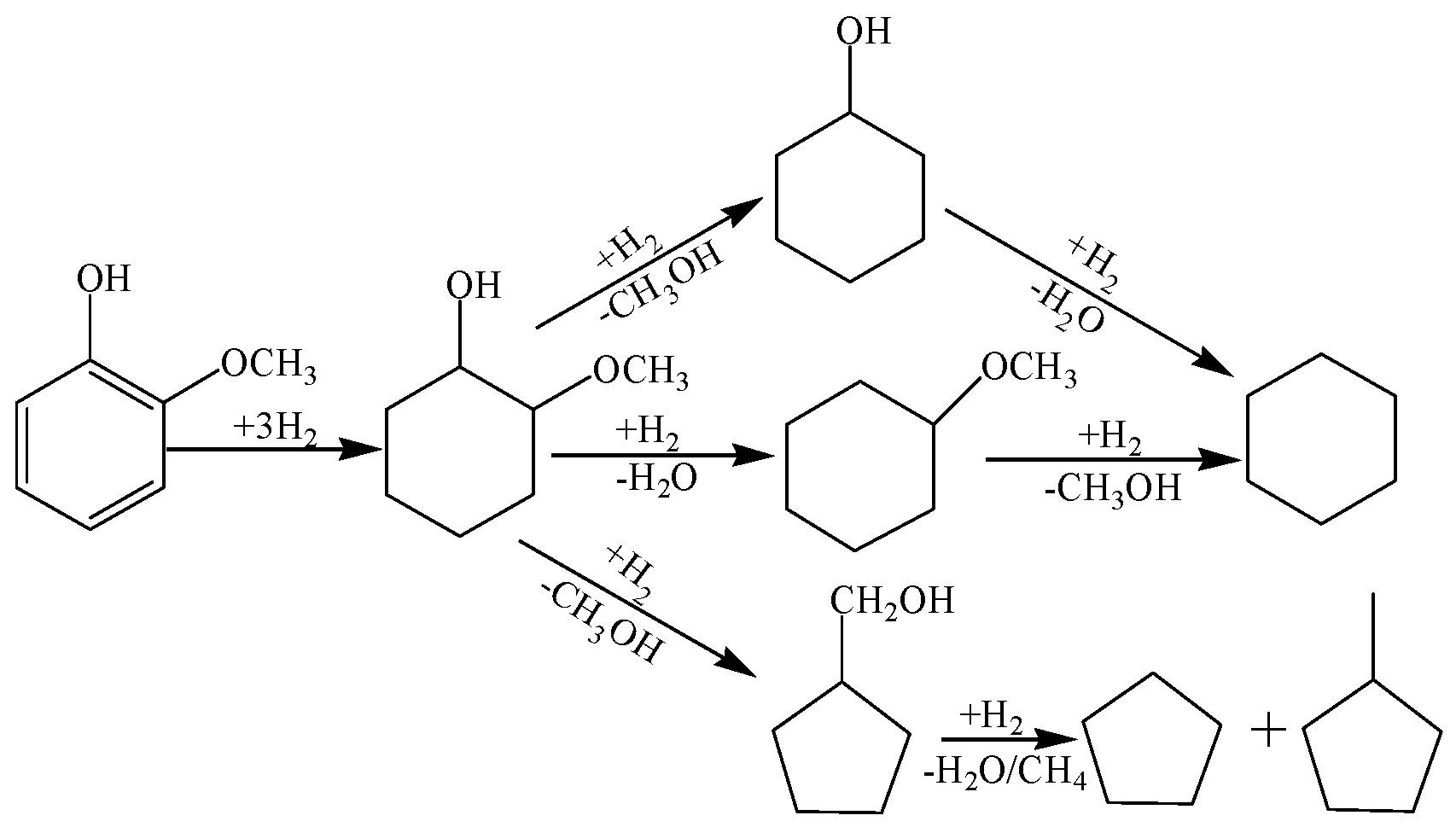
2. Results and Discussion
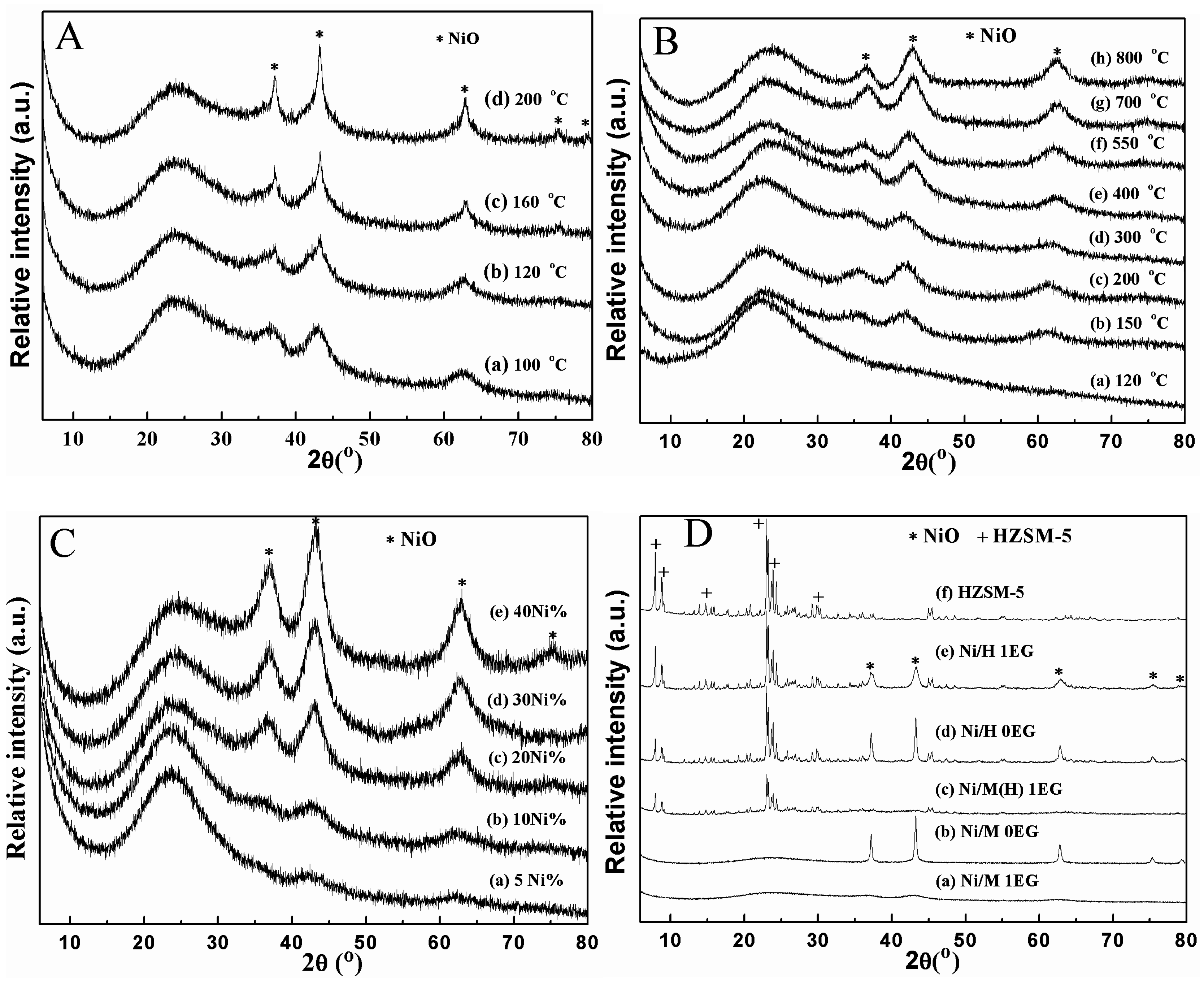
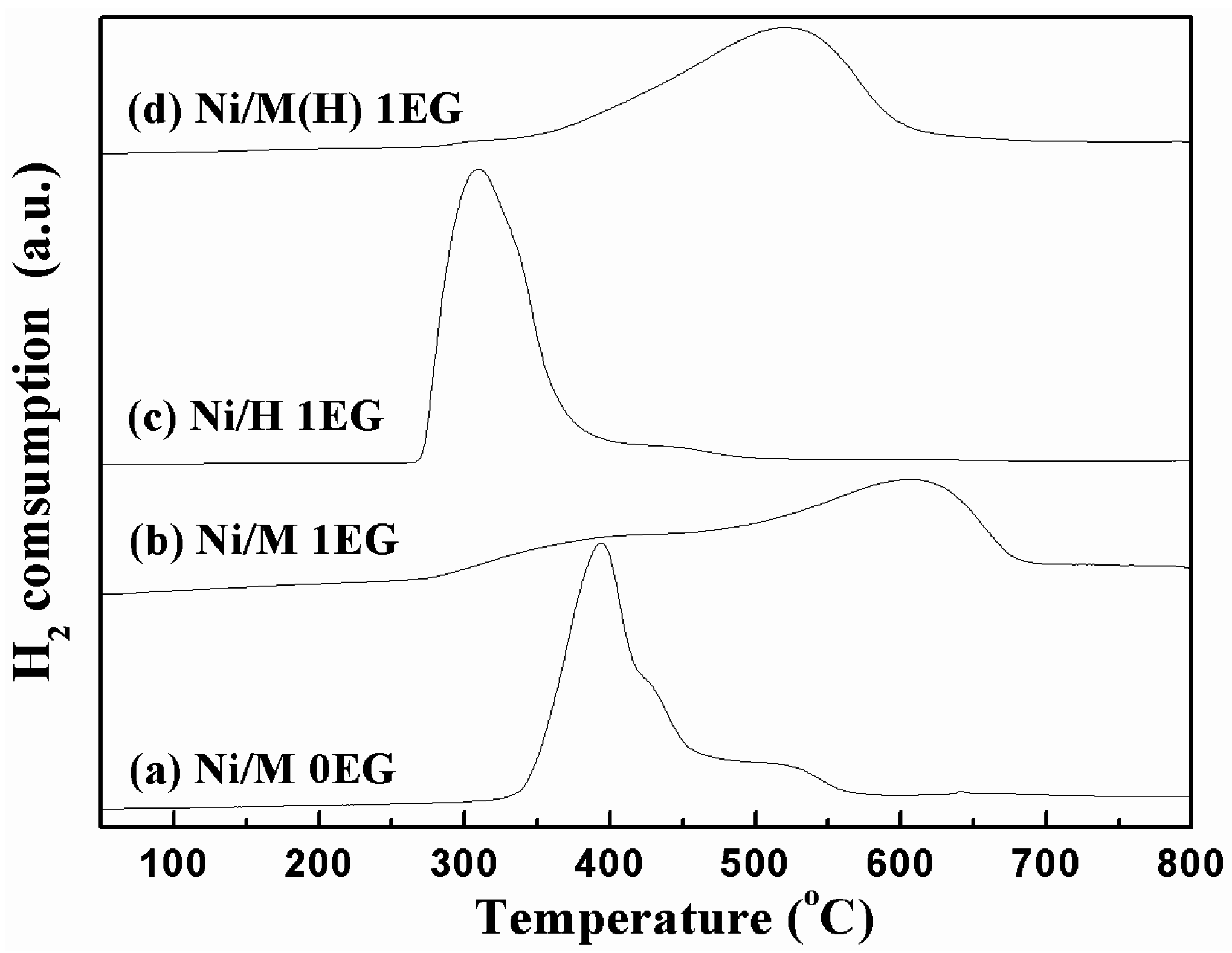
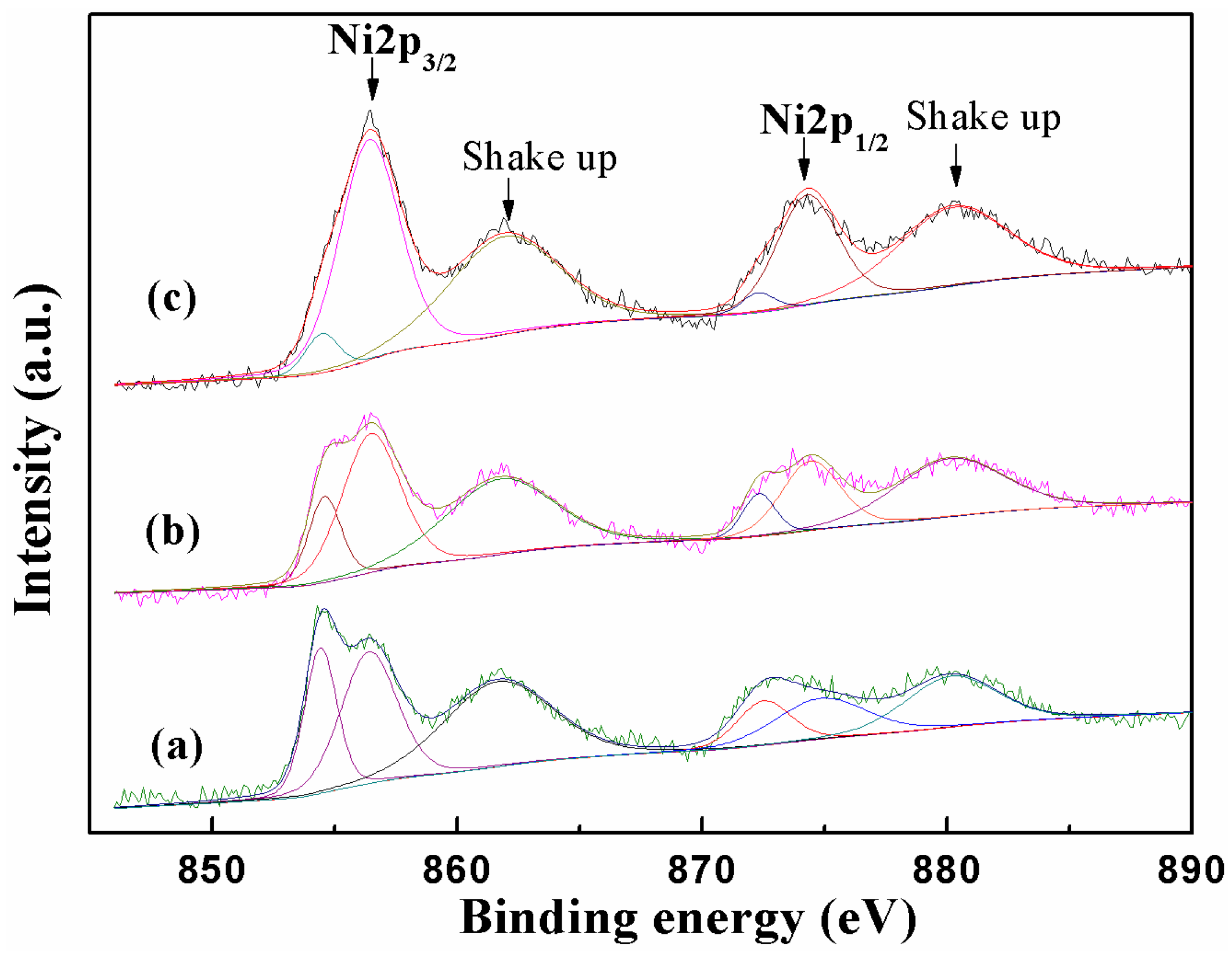
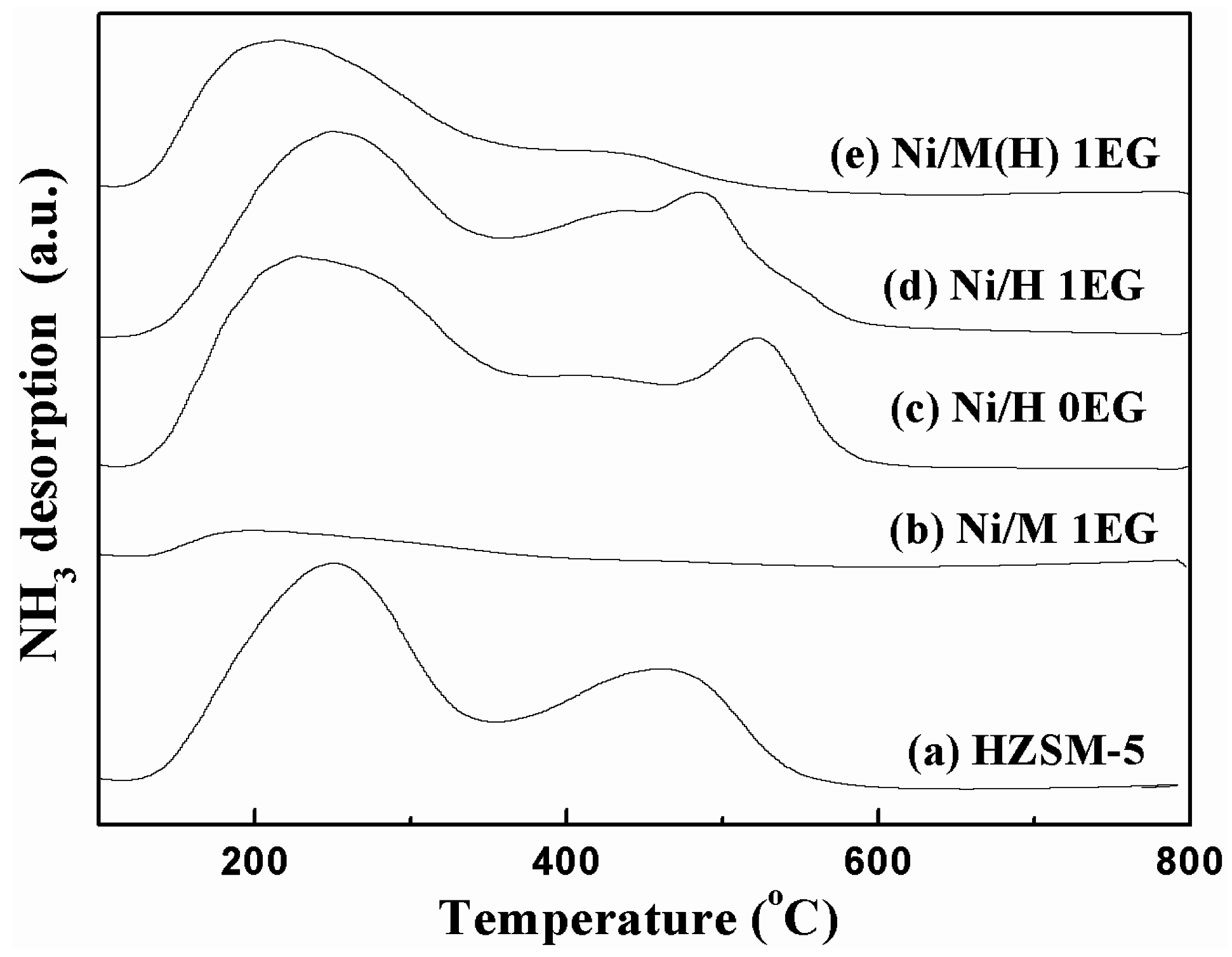
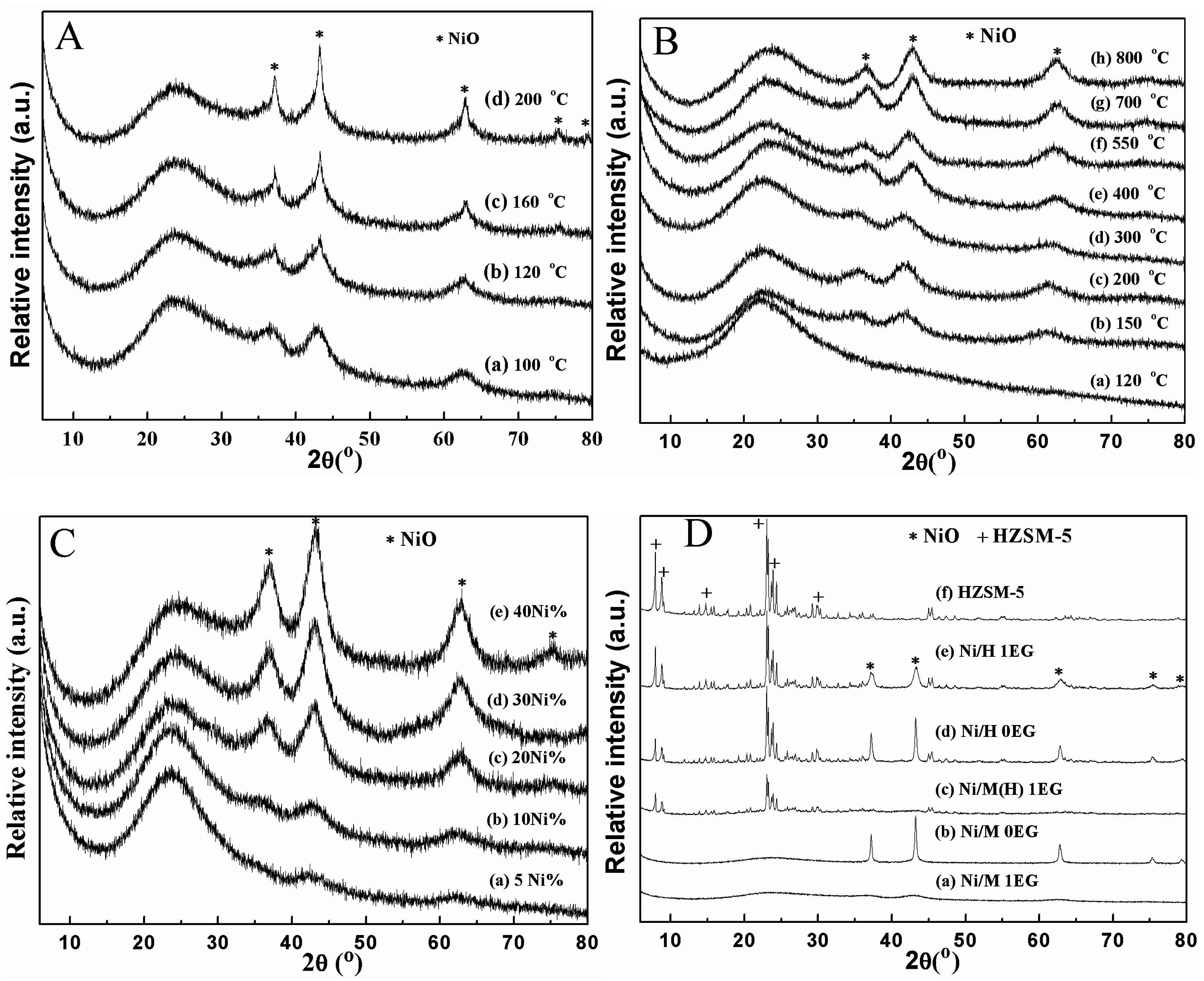
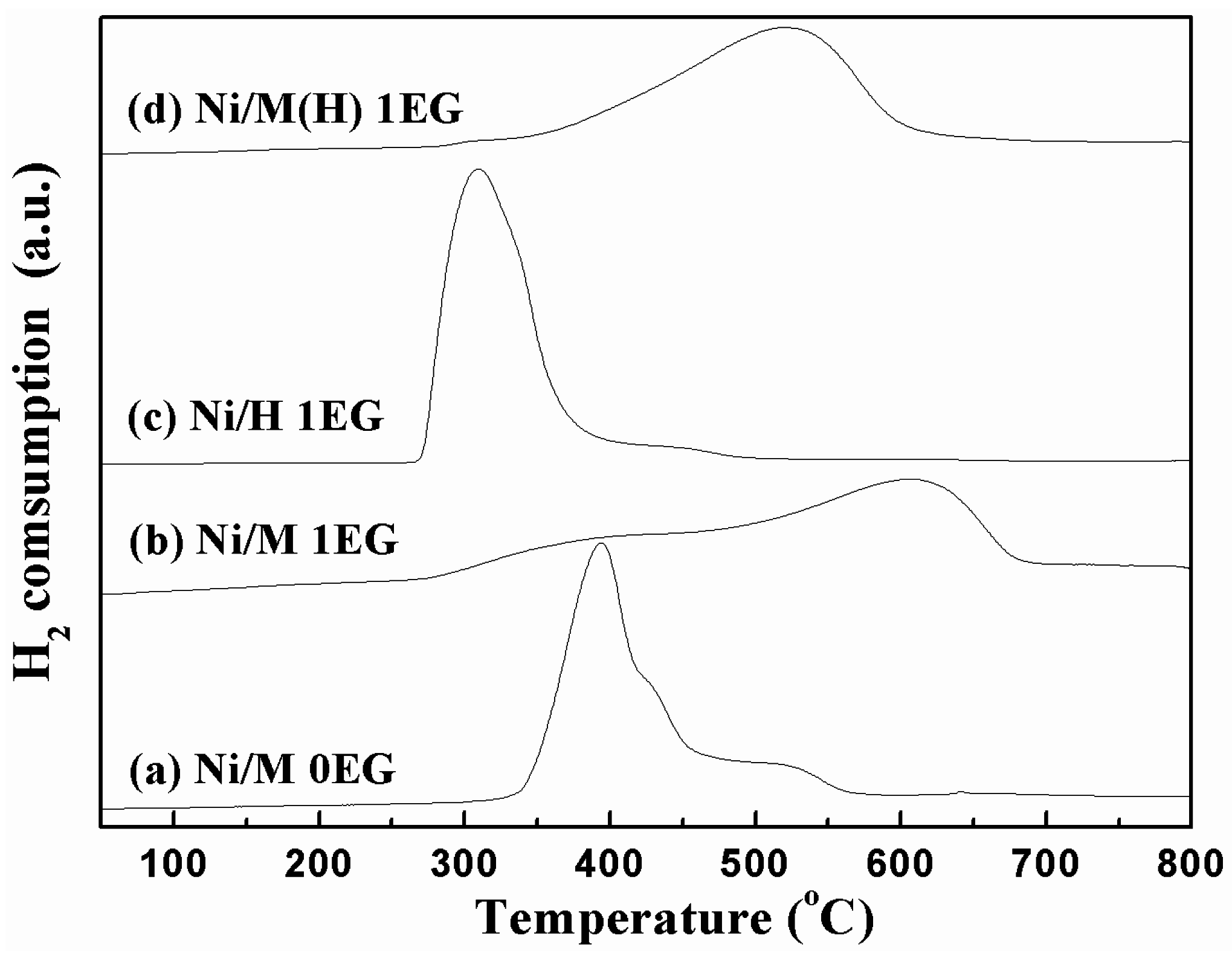
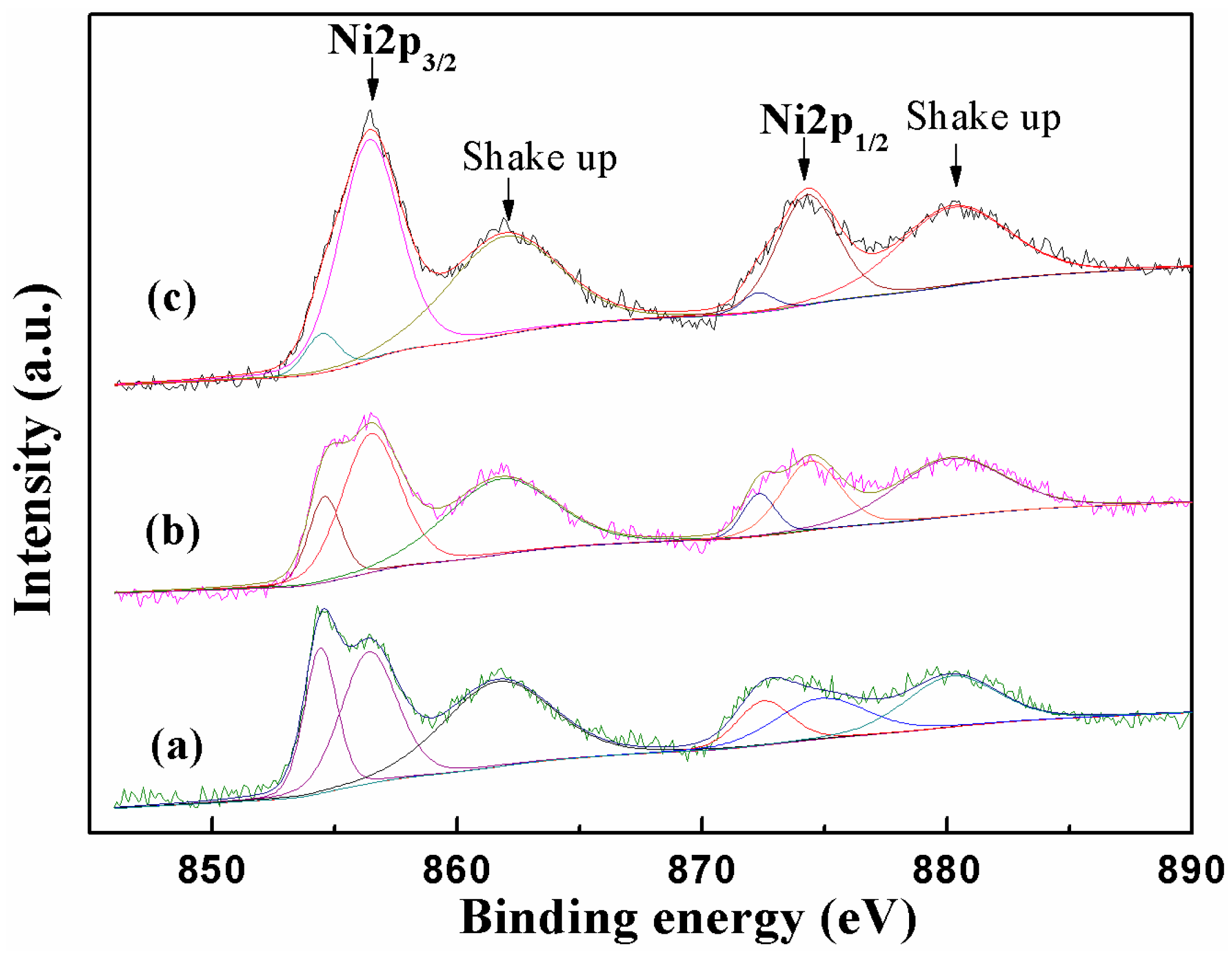
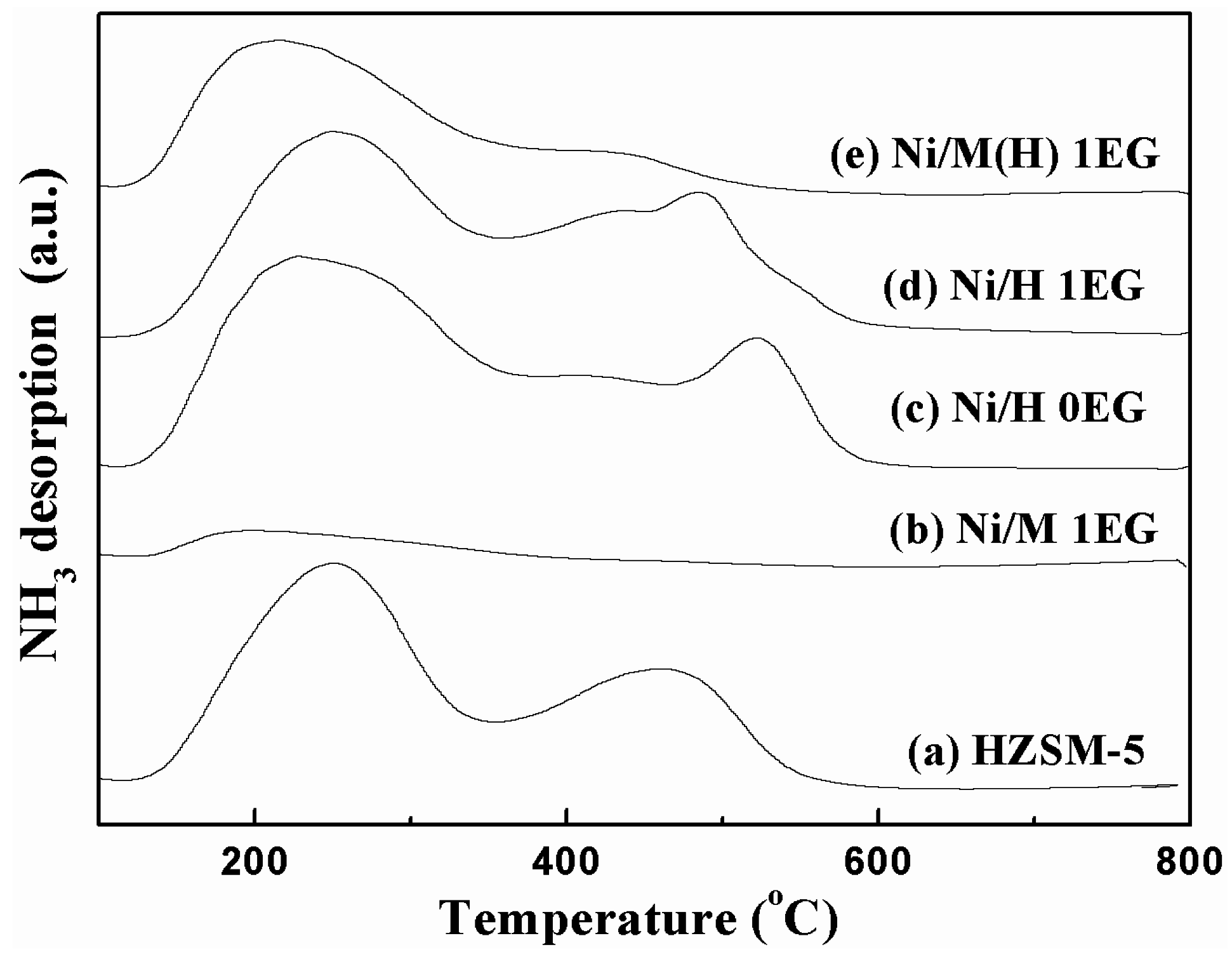
2.1. Catalyst Characterization
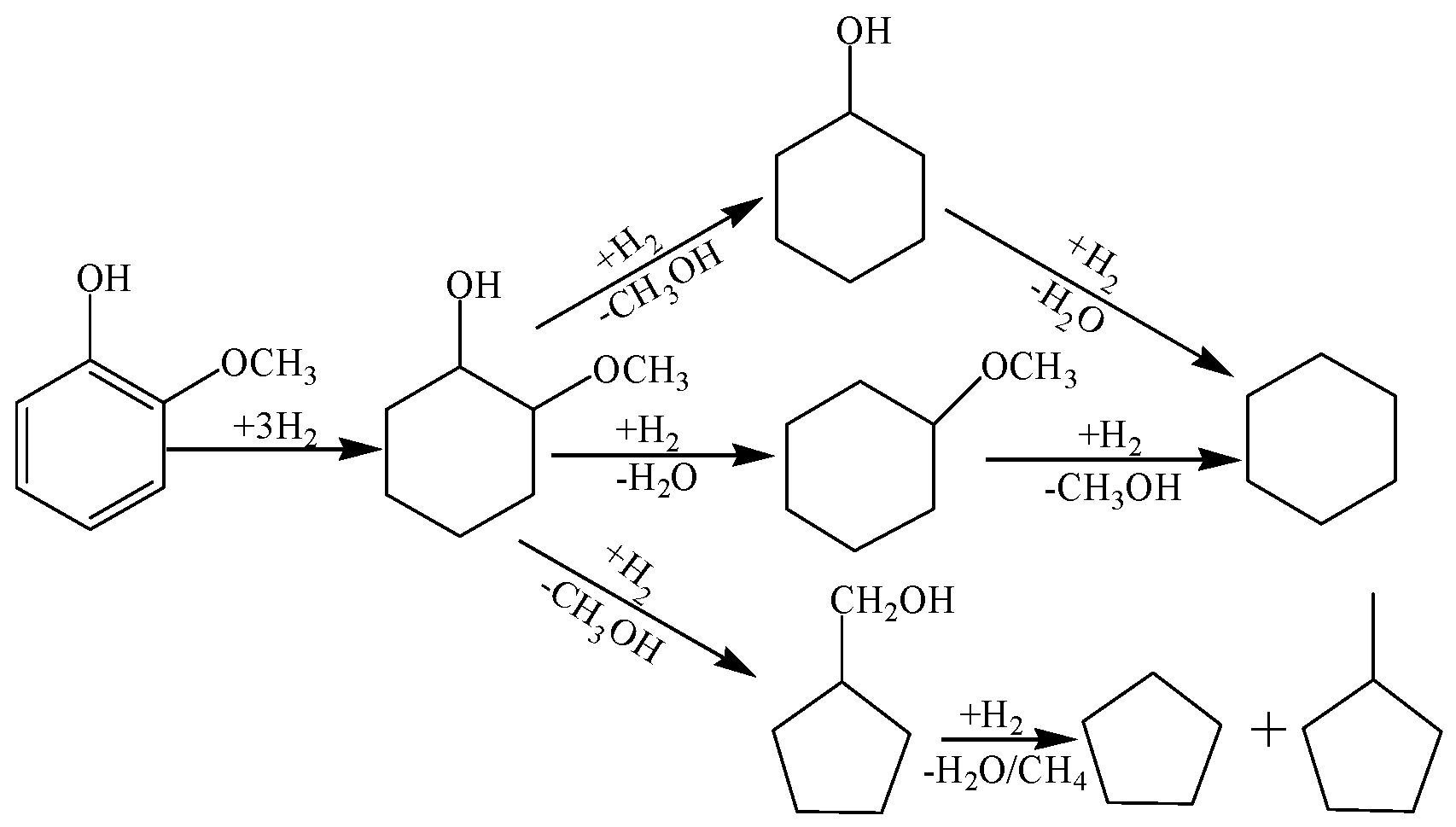
2.2. Guaiacol Hydrogenolysis Activity of Prepared Catalysts
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalyst Tests
3.3. Catalyst Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Dillen, A.J.; Terörde, R.M.; Lensveld, D.J.; Geus, J.W.; de Jong, K.P. Synthesis of supported catalysts by impregnation and drying using aqueous chelated metal complexes. J. Catal. 2003, 216, 257–264. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Lopez, L.; Velasco, J.; Montes, V.; Marinas, A.; Cabrera, S.; Boutonnet, M.; Jaras, S. Synthesis of Ethanol from Syngas over Rh/MCM-41 Catalyst: Effect of Water on Product Selectivity. Catalysts 2015, 5, 1737–1755. [Google Scholar] [CrossRef]
- Nares, R.; Ramirez, J.; Gutierrez-Alejandre, A.; Cuevas, R. Characterization and Hydrogenation Activity of Ni/Si(Al)-MCM-41. Catalysts Prepared by Deposition-Precipitation. Ind. Eng. Chem. Res. 2009, 48, 1154–1162. [Google Scholar] [CrossRef]
- Lensveld, D.J.; Mesu, J.G.; van Dillen, A.J.; de Jong, K.P. Synthesis and characterization of MCM-41 supported nickel oxide catalysts. Microporous Mesoporous Mater. 2001, 44–45, 401–407. [Google Scholar] [CrossRef]
- Yonemitsu, M.; Tanaka, Y.; Iwamoto, M. Metal Ion-Planted MCM-41. 1. Planting of Manganese (II) Ion into MCM-41 by a Newly Developed Template-Ion Exchange Method. Chem. Mater. 1997, 9, 2679–2681. [Google Scholar] [CrossRef]
- Chanquia, C.M.; Winkler, E.L.; Causa, M.T.; Eimer, G.A. Evolution of Copper Nanospecies in the Synthesis Stages of MCM-41-Type Mesoporous Molecular Sieves. J. Phys. Chem. C 2012, 116, 5376–5382. [Google Scholar] [CrossRef]
- Lu, J.L.; Elam, J.W.; Stair, P.C. Synthesis and Stabilization of Supported Metal Catalysts by Atomic Layer Deposition. Acc. Chem. Res. 2013, 46, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Maki-Arvela, P.; Murzin, D.Y. Effect of catalyst synthesis parameters on the metal particle size. Appl. Catal. A 2012, 451, 251–281. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Yang, G.H.; Sun, S.L.; Tsubaki, N. Effects of impregnation solvent on Co/SiO2 catalyst for Fischer-Tropsch synthesis: A highly active and stable catalyst with bimodal sized cobalt particles. Appl. Catal. A 2007, 321, 79–85. [Google Scholar] [CrossRef]
- Li, Y.; Shen, W.J. Morphology-dependent nanocatalysts: Rod-shaped oxides. Chem. Soc. Rev. 2014, 43, 1543–1574. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zeng, C.Y.; Lin, Q.H.; Lu, P.; Niu, W.Q.; Tsubaki, N. Citric acid assisted one-step synthesis of highly dispersed metallic Co/SiO2 without further reduction: As-prepared Co/SiO2 catalysts for Fischer-Tropsch synthesis. Catal. Toady 2014, 228, 206–211. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Wang, S.R.; Zhu, L.J.; Ge, X.L.; Li, X.B.; Luo, Z.Y. The influence of copper particle dispersion in Cu/SiO2 catalysts on the hydrogenation synthesis of ethylene glycol. Catal. Lett. 2010, 135, 275–281. [Google Scholar] [CrossRef]
- Qiu, S.B.; Zhang, X.; Liu, Q.Y.; Wang, T.J.; Zhang, Q.; Ma, L.L. A simple method to prepare highly active and dispersed Ni/MCM-41 catalysts by co-impregnation. Catal. Commun. 2013, 42, 73–78. [Google Scholar] [CrossRef]
- Qiu, S.B.; Weng, Y.J.; Li, Y.P.; Ma, L.L.; Zhang, Q.; Wang, T.J. Promotion of Ni/MCM-41 Catalyst for Hydrogenation of Naphthalene by co-Impregnation with Polyols. Chin. J. Chem. Phys. 2014, 27, 433–438. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Bi, P.Y.; Wang, J.C.; Jiang, P.W.; Wu, X.P.; Xue, H.; Liu, J.X.; Zhou, X.G.; Li, Q.X. Production of jet and diesel biofuels from renewable lignocellulosic biomass. Appl. Energy 2015, 150, 128–137. [Google Scholar] [CrossRef]
- De, S.; Saha, B.; Luque, R. Hydrodeoxygenation processes: Advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels. Bioresour. Technol. 2015, 178, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Zhao, L.; Qi, Y. Enhancing the productivity of microalgae cultivated in wastewater toward biofuel production: A critical review. Appl. Energy 2015, 137, 282–291. [Google Scholar] [CrossRef]
- Bi, P.Y.; Wang, J.C.; Zhang, Y.J.; Jiang, P.W.; Wu, X.P.; Liu, J.X.; Xue, H.; Wang, T.J.; Li, Q.X. From lignin to cycloparaffins and aromatics: Directional synthesis of jet and diesel fuel range biofuels using biomass. Bioresour. Technol. 2015, 183, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hellinger, M.; Baier, S.; Mortensen, P.M.; Kleist, W.; Jensen, A.D.; Grunwaldt, J.D. Continuous Catalytic Hydrodeoxygenation of Guaiacol over Pt/SiO2 and Pt/H-MFI-90. Catalysts 2015, 5, 1152–1166. [Google Scholar] [CrossRef] [Green Version]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Wang, H.M.; Male, J.; Wang, Y. Recent Advances in Hydrotreating of Pyrolysis Bio-Oil and Its Oxygen-Containing Model Compounds. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Hua, D.R.; Wu, Y.L.; Chen, Y.; Li, J.; Yang, M.D.; Lu, X.N. Co-Pyrolysis Behaviors of the Cotton Straw/PP Mixtures and Catalysis Hydrodeoxygenation of Co-Pyrolysis Products over Ni-Mo/Al2O3 Catalyst. Catalysts 2015, 5, 2085–2097. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, T.; Li, B.; Wang, T.J.; Zhang, X.H.; Zhang, Q.; Ma, L.L. Highly Selective Sorbitol Hydrogenolysis to Liquid Alkanes over Ni/HZSM-5 Catalysts Modified with Pure Silica MCM-41. ChemCatChem 2012, 4, 1084–1087. [Google Scholar] [CrossRef]
- Weng, Y.J.; Qiu, S.B.; Ma, L.L.; Liu, Q.Y.; Ding, M.Y.; Zhang, Q.; Zhang, Q.; Wang, T.J. Jet-Fuel Range Hydrocarbons from Biomass-Derived Sorbitol over Ni-HZSM-5/SBA-15 Catalyst. Catalysts 2015, 5, 2147–2160. [Google Scholar] [CrossRef]
- Song, W.J.; Liu, Y.S.; Barath, E.; Zhao, C.; Lercher, J.A. Synergy effect of Ni and acid sites for C–O bond cleavage of phenol, catechol, and guaiacol. Green Chem. 2015, 17, 1204–1218. [Google Scholar] [CrossRef]
- Hellinger, M.; Carvalho, H.W.P.; Baier, S.; Wang, D.; Kleist, W.; Grunwaldt, J.D. Catalytic hydrodeoxygenation of guaiacol over platinum supported on metal oxides and zeolites. Appl. Catal. A 2015, 490, 181–192. [Google Scholar] [CrossRef]
- Lv, X.Y.; Chen, J.F.; Tan, Y.S.; Zhang, Y. A highly dispersed nickel supported catalyst for dry reforming of methane. Catal. Commun. 2012, 20, 6–11. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, T.J.; Li, B.; Jiang, T.; Ma, L.L.; Zhang, X.H.; Liu, Q.Y. Aqueous phase reforming of sorbitol to bio-gasoline over Ni/HZSM-5 catalysts. Appl. Energy 2012, 97, 509–513. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Monteverdi, S.; Mercy, M.; Nowak, I.; Ziolek, M.; Bettahar, M.M. Nickel containing MCM-41 and AlMCM-41 mesoporous molecular sieves: Characteristics and activity in the hydrogenation of benzene. Appl. Catal. A 2004, 268, 241–253. [Google Scholar] [CrossRef]
- Li, X.B.; Wang, S.R.; Cai, Q.J.; Zhu, L.J.; Yin, Q.Q.; Luo, Z.Y. Effects of Preparation Method on the Performance of Ni/Al2O3 Catalysts for Hydrogen Production by Bio-Oil Steam Reforming. Appl. Biochem. Biotech. 2012, 168, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Moya, S.F.; Martins, R.L.; Schmal, M. Monodispersed and nanostructrured Ni/SiO2 catalyst and its activity for non oxidative methane activation. Appl. Catal. A 2011, 396, 159–169. [Google Scholar] [CrossRef]
- Dutta, D.; Borah, B.J.; Saikia, L.; Pathak, M.G.; Sengupta, P.; Dutta, D.K. Synthesis and catalytic activity of Ni-acid activated montmorillonite nanoparticles. Appl. Clay Sci. 2011, 53, 650–656. [Google Scholar] [CrossRef]
- Ren, H.P.; Hao, Q.Q.; Wang, W.; Song, Y.H.; Cheng, J.; Liu, Z.W.; Liu, Z.T.; Lu, J.; Hao, Z.P. High-performance Ni-SiO2 for pressurized carbon dioxide reforming of methane. Int. J. Hydrog. Energy 2014, 39, 11592–11605. [Google Scholar] [CrossRef]
- Kirumakki, S.R.; Shpeizer, B.G.; Sagar, G.V.; Chary, K.V.R.; Clearfield, A. Hydrogenation of naphthalene over NiO/SiO2-Al2O3 catalysts: Structure-activity correlation. J. Catal. 2006, 242, 319–331. [Google Scholar] [CrossRef]
- Fang, K.G.; Ren, J.; Sun, Y.H. Effect of nickel precursors on the performance of Ni/AlMCM-41 catalysts for n-dodecane hydroconversion. J. Mol. Catal. A Chem. 2005, 229, 51–58. [Google Scholar] [CrossRef]
- Chao, K.J.; Chiou, B.H.; Cho, C.C.; Jeng, S.Y. Temperature-programmed desorption studies on ZSM-5 zeolites. Zeolites 1984, 4, 2–4. [Google Scholar] [CrossRef]
- Lobree, L.J.; Hwang, I.C.; Reimer, J.A.; Bell, A.T. Investigations of the State of Fe in H-ZSM-5. J. Catal. 1999, 186, 242–253. [Google Scholar] [CrossRef]
- Yang, Y.X.; Ochoa-Hernandez, C.; O’Shea, V.A.D.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Effect of metal–support interaction on the selective hydrodeoxygenation of anisole to aromatics over Ni-based catalysts. Appl. Catal. B 2014, 145, 91–100. [Google Scholar] [CrossRef]
- Li, X.M.; Han, D.Z.; Wang, H.; Liu, G.B.; Wang, B.; Li, Z.; Wu, J.H. Propene oligomerization to high-quality liquid fuels over Ni/HZSM-5. Fuel 2015, 144, 9–14. [Google Scholar] [CrossRef]
- Van der Borght, K.; Galvita, V.V.; Marin, G.B. Reprint of “Ethanol to higher hydrocarbons over Ni, Ga, Fe-modified ZSM-5: Effect of metal content”. Appl. Catal. A 2015, 504, 621–630. [Google Scholar] [CrossRef]
- Yu, M.J.; Park, S.H.; Jeon, J.K.; Ryu, C.; Sohn, J.M.; Kim, S.C.; Park, Y.K. Hydrodeoxygenation of Guaiacol Over Pt/Al-SBA-15 Catalysts. J. Nanosci. Nanotechnol. 2015, 15, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Lee, D.W.; Eom, H.J.; Lee, K.Y. The catalytic activity of Pd/WOx/gamma-Al2O3 for hydrodeoxygenation of guaiacol. Appl. Catal. B 2014, 150, 438–445. [Google Scholar] [CrossRef]
- Nimmanwudipong, T.; Runnebaum, R.C.; Block, D.E.; Gates, B.C. Catalytic Conversion of Guaiacol Catalyzed by Platinum Supported on Alumina: Reaction Network Including Hydrodeoxygenation Reactions. Energy Fuel 2011, 25, 3417–3427. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples a | Mole Ratio Ni:EG | SBET (m2/g) b | Pore Volume (cm3/g) b | Average Pore Diameter (nm) b | Mean Particle Size Diameter (nm) c |
|---|---|---|---|---|---|
| MCM-41 | - | 1063 | 1.12 | 2.8 | - |
| Ni/M 0EG | 1:0 | 807 | 0.67 | 2.7 | 45.0 |
| Ni/M 1EG | 1:1 | 785 | 0.70 | 2.8 | 3.3 |
| HZSM-5 | - | 420 | 0.21 | 3.7 | - |
| Ni/H 0EG | 1:0 | 231 | 0.09 | 3.7 | 42.9 |
| Ni/H 1EG | 1:1 | 216 | 0.06 | 3.6 | 13.3 |
| Ni/M(H) 1EG | 1:1 | 510 | 0.42 | 2.9 | <3.0 |
| Catalyst | Peak Position (°C) | Acid Amount (umol/g) | |||
|---|---|---|---|---|---|
| T1 | T2 | Total | Weak | Strong | |
| HZSM-5 | 250 | 467 | 622 | 414 | 208 |
| Ni/H 0EG | 253 | 508 | 550 | 372 | 178 |
| Ni/H 1EG | 252 | 485 | 492 | 328 | 164 |
| Ni/M 1EG | 192 | - | 87 | 87 | 0 |
| Ni/M(H) 1EG | 212 | 412 | 350 | 309 | 41 |
| Catalyst | Temp. °C | XGUA % | SMethoxycyclohexanol % | SCyclohexanol % | SMethoxycyclohexane % | SCyclohexane b % |
|---|---|---|---|---|---|---|
| Ni/M 0EG | 150 | 15.5 | 96.3 | 1.8 | 0 | 0.4 |
| Ni/M 0EG | 200 | 92.6 | 91.1 | 5.1 | 0.5 | 1.3 |
| Ni/M 1EG | 100 | 12.3 | 94.4 | 5.6 | 0 | 0 |
| Ni/M 1EG | 120 | 34.8 | 93.0 | 6.4 | 0.4 | 0.2 |
| Ni/M 1EG | 150 | 97.4 | 93.9 | 5.3 | 0.5 | 0.2 |
| Ni/M 1EG | 200 | 100 | 89.7 | 5.9 | 1.1 | 2.1 |
| Ni/H 1EG | 200 | 14.4 | 89.5 | 2.7 | 0.7 | 3.5 |
| 5% Pd/C | 100 | 18.1 | 97.0 | 1.8 | 0.9 | 0.3 |
| 5% Pd/C | 120 | 61.7 | 97.4 | 2.3 | 0.3 | 0 |
| 5% Pd/C | 150 | 100 | 97.8 | 1.4 | 0.7 | 0 |
| 5% Ru/C | 100 | 83.4 | 82.8 | 16.1 | 0.1 | 0 |
| 5% Ru/C | 120 | 92.3 | 82.3 | 15.8 | 1.0 | 0 |
| 5% Ru/C | 150 | 100 | 71.3 | 25.8 | 0.9 | 0 |
| Ni/M 1EG +H | 150 | 87.3 | 93.6 | 4.4 | 0.3 | 0.9 |
| Ni/M 1EG +H | 200 | 100 | 66.5 | 29.2 | 2.1 | 2.2 |
| Ni/M (H) 1EG | 120 | 45.1 | 90.6 | 8.2 | 0.6 | 0.3 |
| Ni/M (H) 1EG | 150 | 97.9 | 94.1 | 4.5 | 0.5 | 0.5 |
| Ni/M (H) 1EG | 200 | 100 | 75.4 | 1.9 | 2.1 | 15.9 |
| Catalyst | Ni/M 0EG | Ni/M 1EG | Ni/H 1EG b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | 220 | 240 | 250 | 220 | 240 | 250 | 220 | 250 | 280 |
| SMethoxycyclohexanol | 81.2 | 56.9 | 29.1 | 78.3 | 29.7 | 0.2 | 74.4 | 50.9 | 0.1 |
| SCyclohexanol | 6.1 | 5.6 | 3.8 | 6.5 | 4.6 | 0 | 3.7 | 1.3 | 0 |
| SMethoxycyclohexane | 1.3 | 0 | 3.9 | 1.9 | 5.0 | 0 | 1.6 | 0.8 | 0 |
| SMethylolcyclopentane | 3.7 | 10.1 | 12.7 | 3.2 | 5.6 | 0 | 0.4 | 0.6 | 0 |
| SCyclohexane | 6.6 | 25.5 | 44.8 | 9.0 | 43.4 | 73.0 | 12.9 | 39.6 | 85.4 |
| SMethylcyclopentane | 0 | 0.3 | 0.7 | 0 | 0.5 | 1.3 | 1.4 | 4.0 | 9.2 |
| SCyclopentane c | 0.1 | 1.2 | 4.6 | 0.5 | 10.7 | 25.2 | 0.5 | 0.3 | 1.1 |
| Catalyst b | Ni/M 1EG+H c | Ni/M(H) 1EG | ||||
|---|---|---|---|---|---|---|
| Temp. (°C) | 220 | 240 | 250 | 220 | 240 | 250 |
| SMethoxycyclohexanol | 26.7 | 3.0 | 0 | 34.0 | 0 | 0.3 |
| SCyclohexanol | 1.5 | 0.3 | 0 | 2.0 | 0 | 0 |
| SMethoxycyclohexane | 4.2 | 1.2 | 0 | 4.7 | 0 | 0 |
| SMethylolcyclopentane | 10.6 | 3.2 | 0 | 7.8 | 0 | 0 |
| SCyclohexane | 52.0 | 82.0 | 88.4 | 45.1 | 84.1 | 84.2 |
| SMethylcyclopentane | 1.9 | 4.6 | 5.8 | 1.5 | 4.8 | 5.4 |
| SCyclopentane d | 2.7 | 4.9 | 4.6 | 4.2 | 10.3 | 8.9 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, S.; Xu, Y.; Weng, Y.; Ma, L.; Wang, T. Efficient Hydrogenolysis of Guaiacol over Highly Dispersed Ni/MCM-41 Catalyst Combined with HZSM-5. Catalysts 2016, 6, 134. https://doi.org/10.3390/catal6090134
Qiu S, Xu Y, Weng Y, Ma L, Wang T. Efficient Hydrogenolysis of Guaiacol over Highly Dispersed Ni/MCM-41 Catalyst Combined with HZSM-5. Catalysts. 2016; 6(9):134. https://doi.org/10.3390/catal6090134
Chicago/Turabian StyleQiu, Songbai, Ying Xu, Yujing Weng, Longlong Ma, and Tiejun Wang. 2016. "Efficient Hydrogenolysis of Guaiacol over Highly Dispersed Ni/MCM-41 Catalyst Combined with HZSM-5" Catalysts 6, no. 9: 134. https://doi.org/10.3390/catal6090134
APA StyleQiu, S., Xu, Y., Weng, Y., Ma, L., & Wang, T. (2016). Efficient Hydrogenolysis of Guaiacol over Highly Dispersed Ni/MCM-41 Catalyst Combined with HZSM-5. Catalysts, 6(9), 134. https://doi.org/10.3390/catal6090134