
Influence of Cobalt Precursor on Efficient Production of Commercial Fuels over FTS Co/SiC Catalyst


Abstract
:1. Introduction
2. Results and Discussion
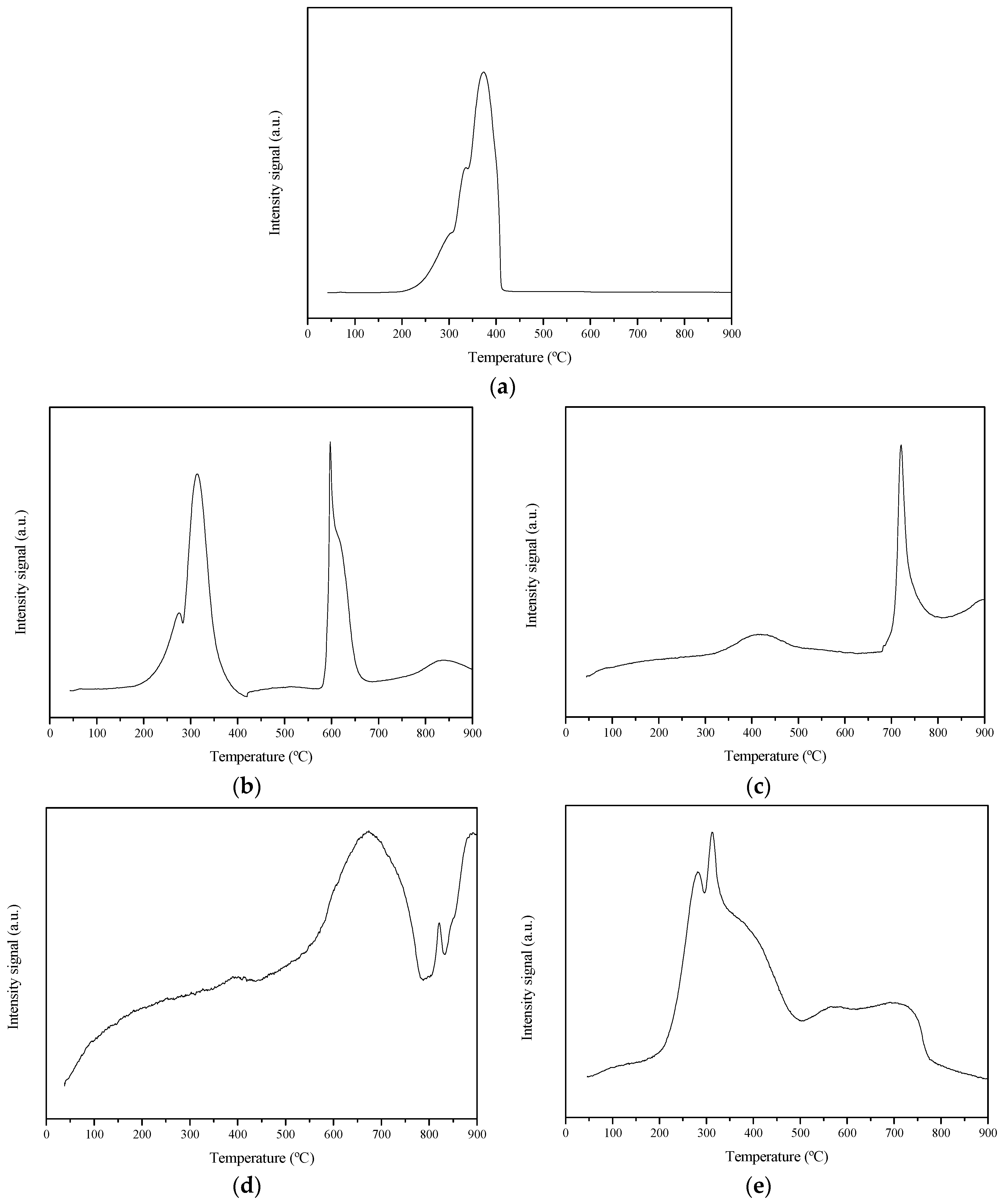
2.1. Thermal Analysis
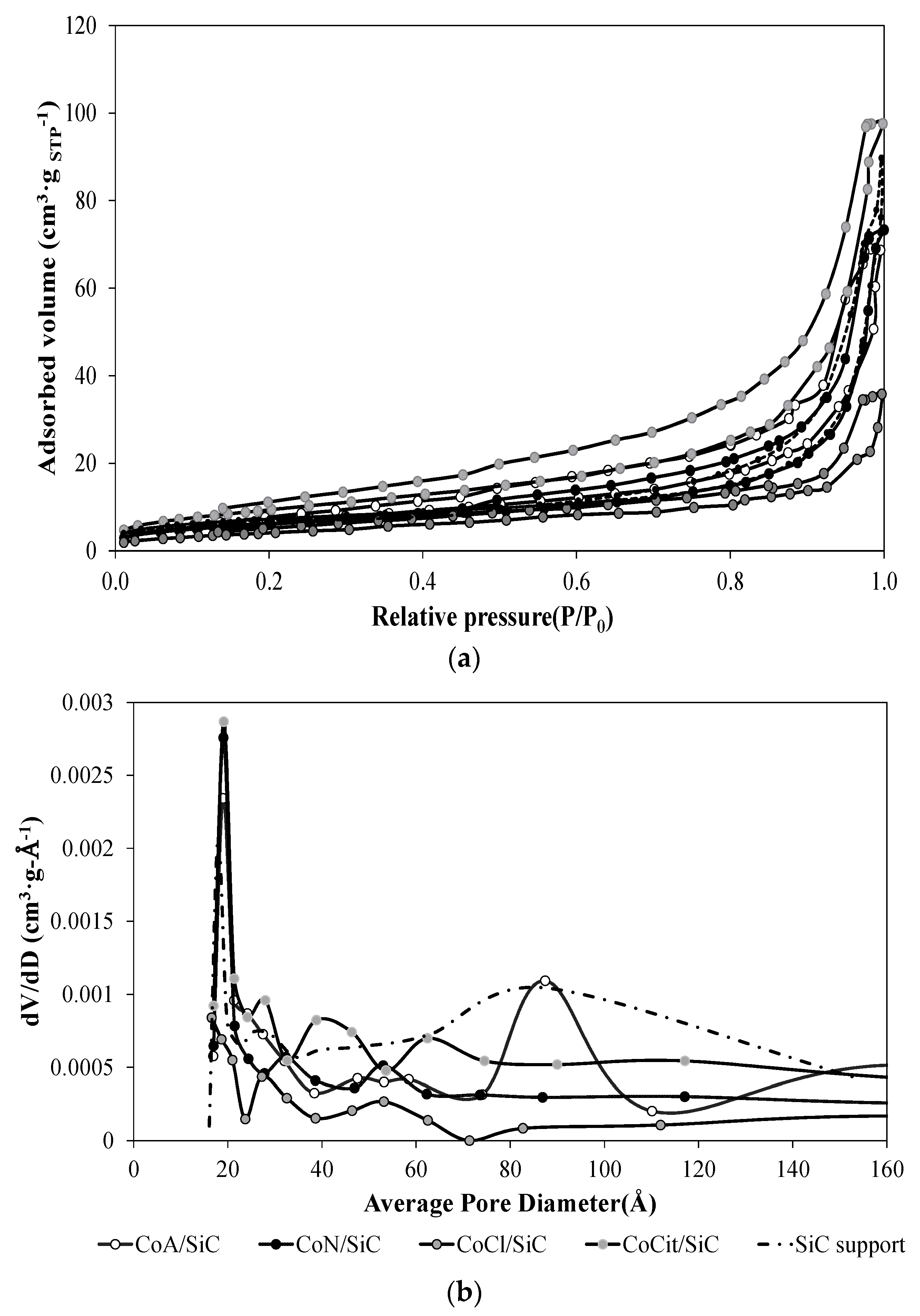
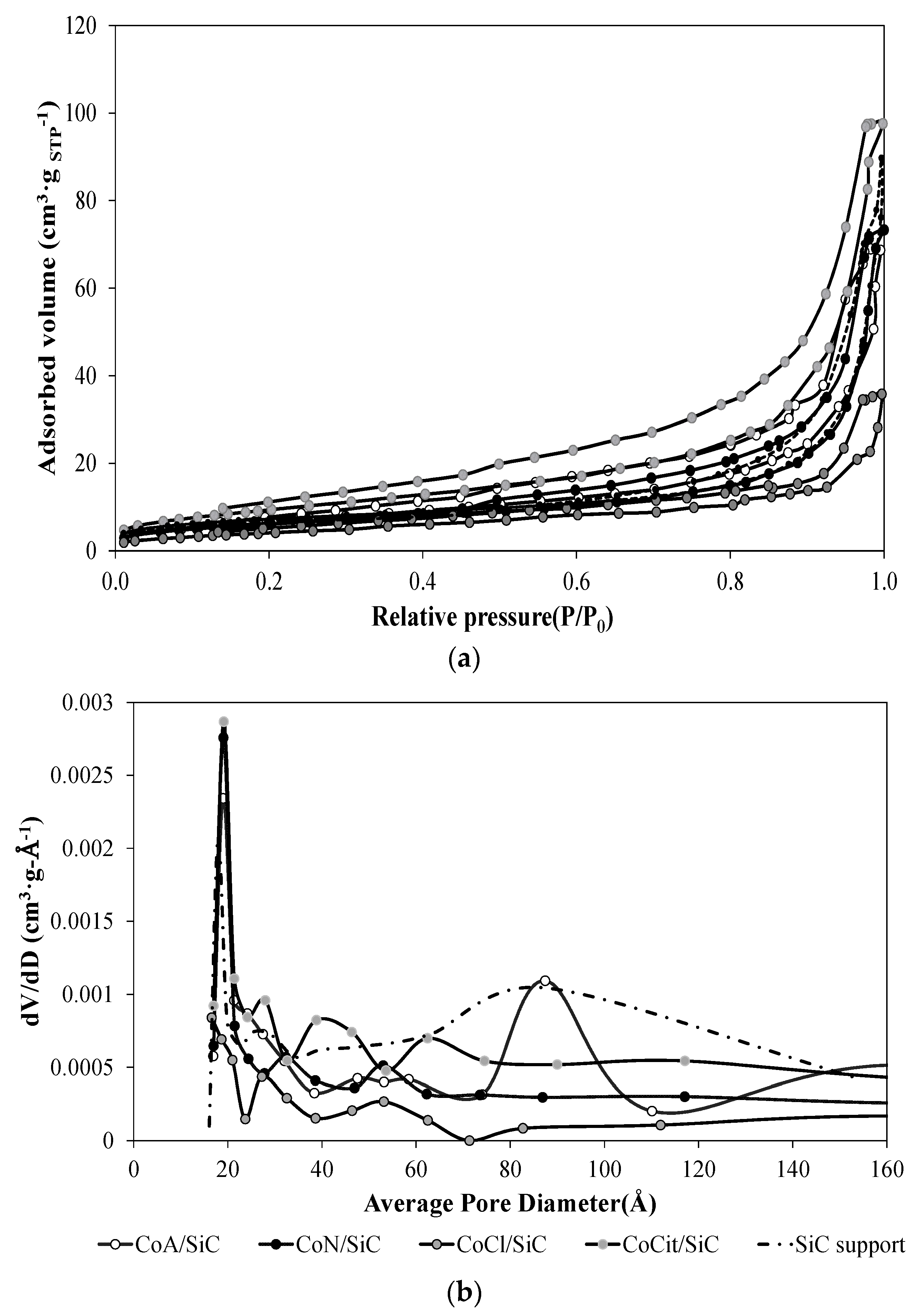
2.2. Nitrogen Adsorption/Desorption Measurements
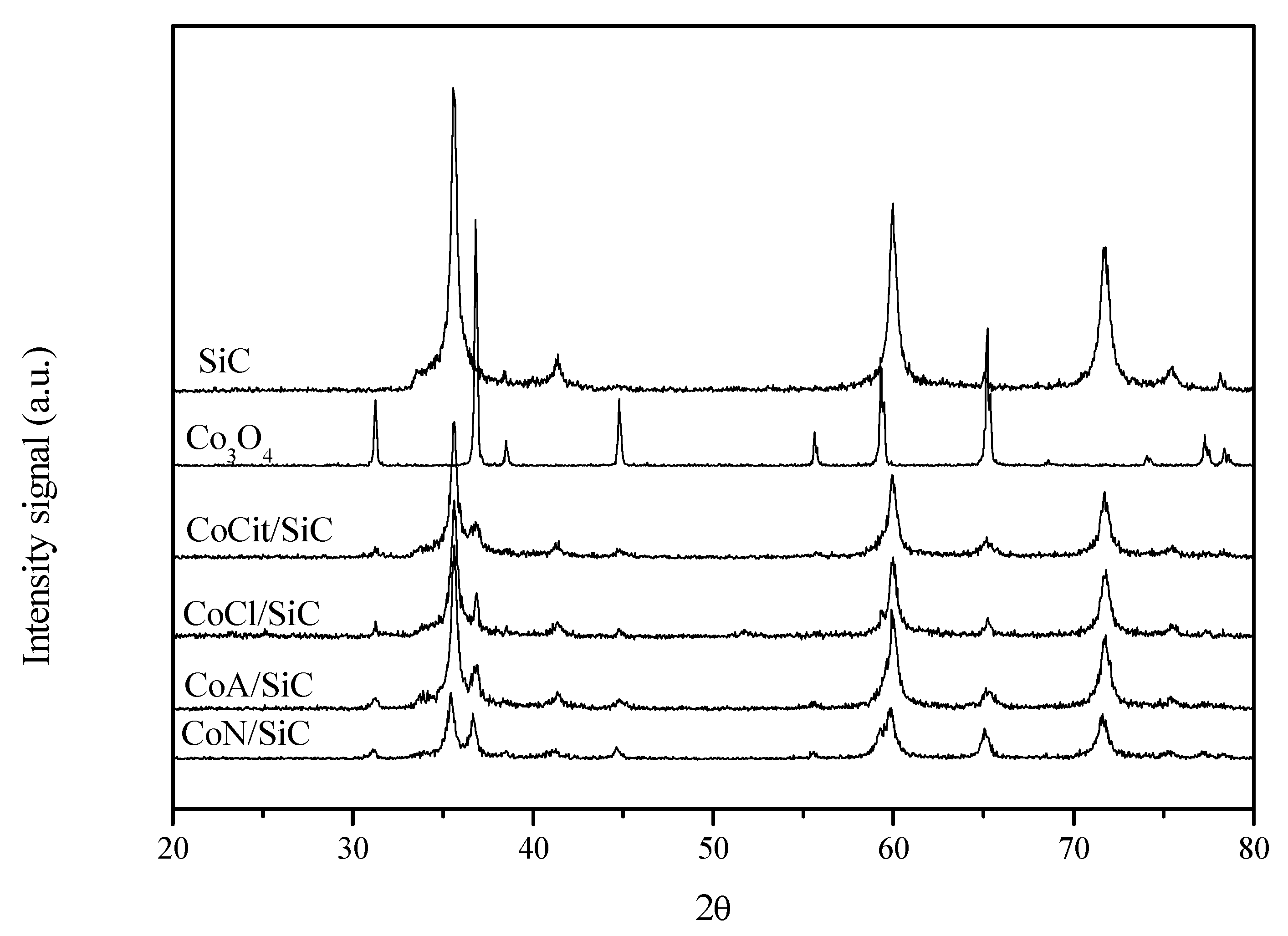
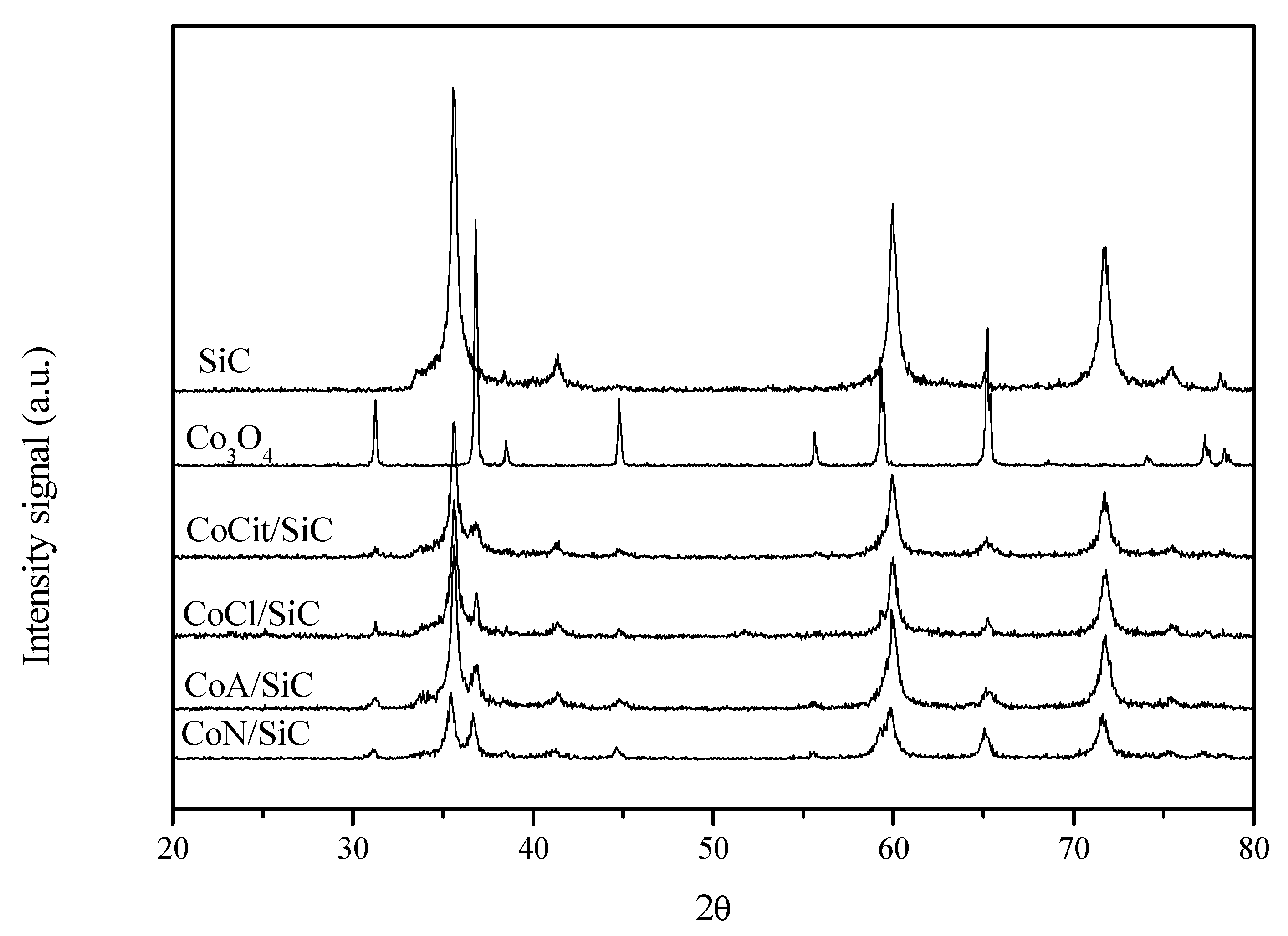
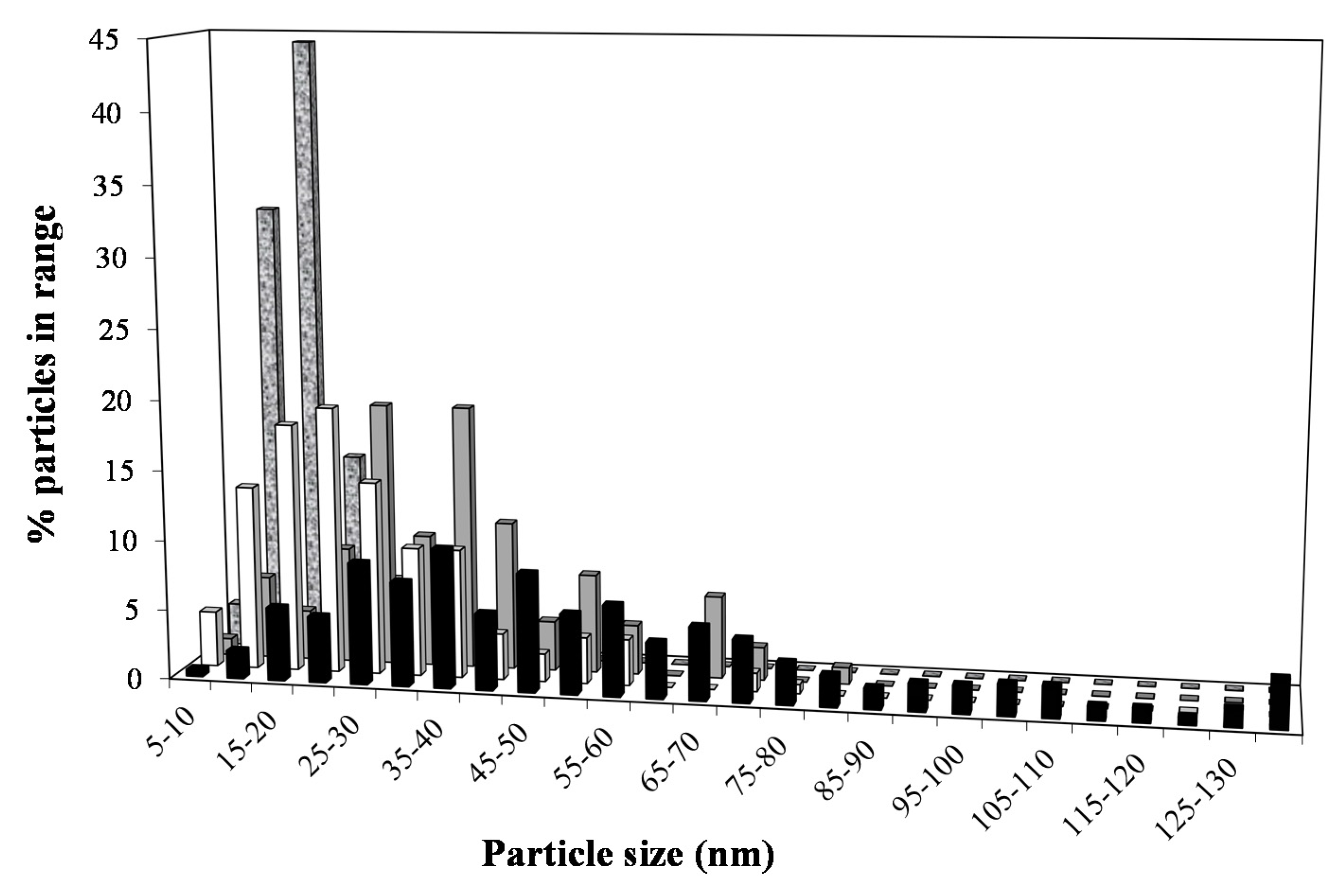
2.3. XRD and TEM
2.4. Cobalt Oxide Reducibility
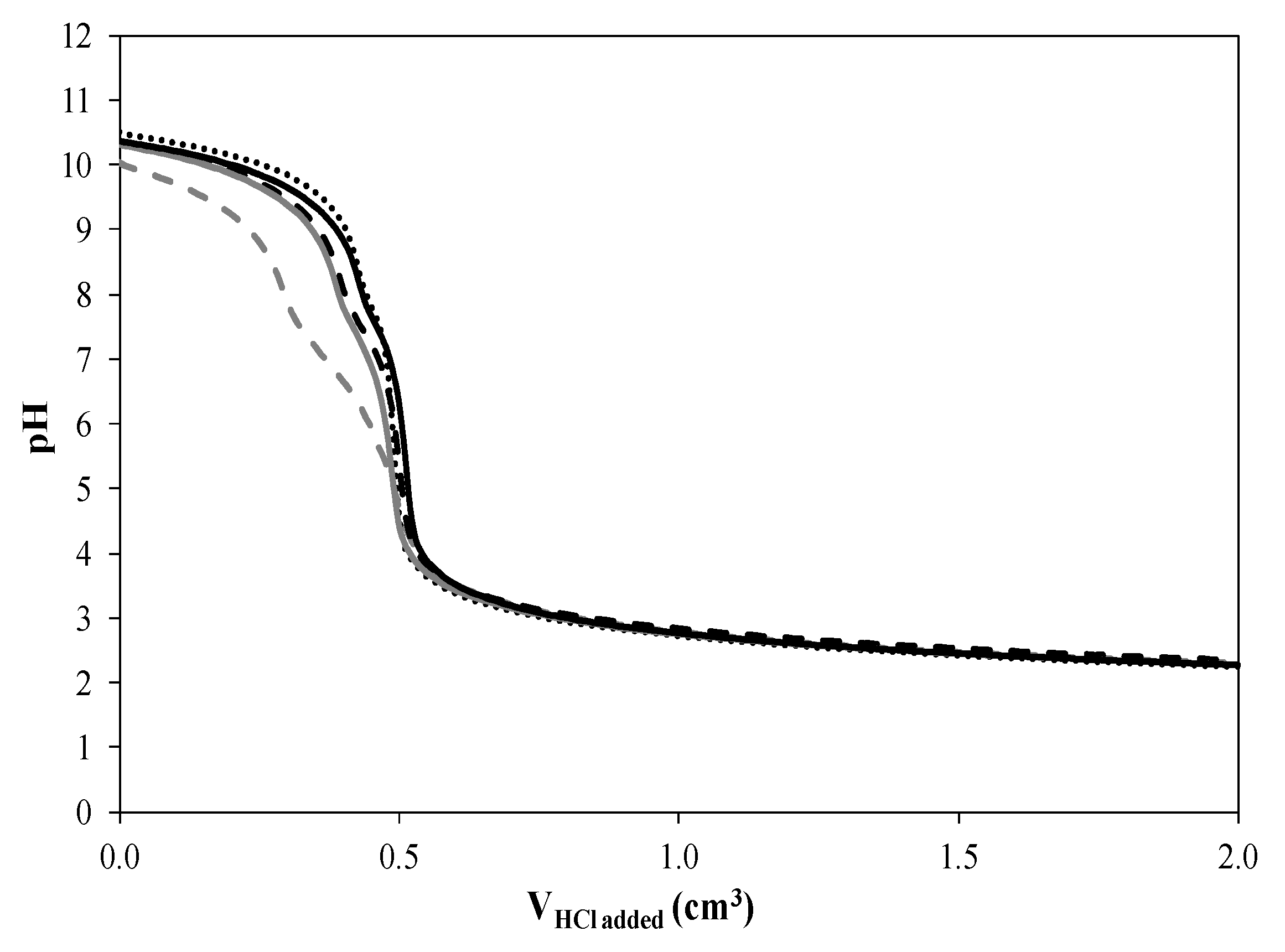
2.5. Acid-Base Titrations
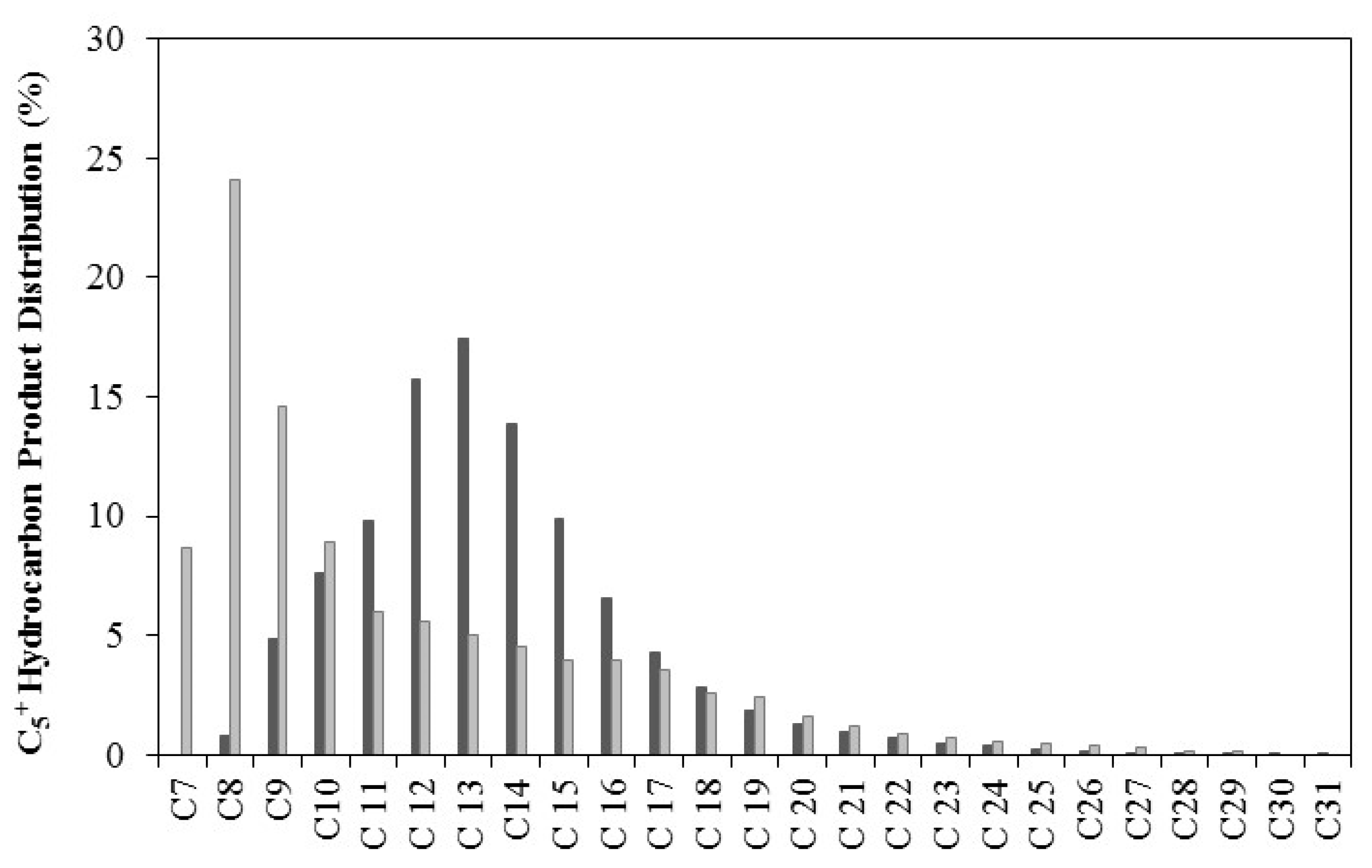
2.6. Fischer–Tropsch Synthesis
3. Experimental Section
3.1. Synthesis of Com/SiC Catalyst
3.2. Catalyst Characterization
3.2.1. Thermal Analysis (TG/DTG)
3.2.2. Atomic Absorption (AA)
3.2.3. Textural Characteristics
3.2.4. X-ray Powder Diffraction
3.2.5. Transmission Electron Microscopy
3.2.6. Temperature Programmed Reduction
3.2.7. O2 Pulse
3.2.8. Titrations
3.3. Activity Test
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Borg, Ø.; Eri, S.; Blekkan, E.A.; Storsæter, S.; Wigum, H.; Rytter, E.; Holmen, A. Fischer-Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables. J. Catal. 2007, 248, 89–100. [Google Scholar] [CrossRef]
- Jacobs, G.; Ji, Y.; Davis, B.H.; Cronauer, D.; Kropf, A.J.; Marshall, C.L. Fischer-Tropsch synthesis: Temperature programmed EXAFS/XANES investigation of the influence of support type, cobalt loading, and noble metal promoter addition to the reduction behavior of cobalt oxide particles. Appl. Catal. A 2007, 333, 177–191. [Google Scholar] [CrossRef]
- Martı́nez, A.N.; López, C.; Márquez, F.; Dı́az, I. Fischer-Tropsch synthesis of hydrocarbons over mesoporous Co/SBA-15 catalysts: The influence of metal loading, cobalt precursor, and promoters. J. Catal. 2003, 220, 486–499. [Google Scholar] [CrossRef]
- Iglesia, E. Design, synthesis, and use of cobalt-based Fischer-Tropsch synthesis catalysts. Appl. Catal. A 1997, 161, 59–78. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, S.L.; Fiato, R.A. Fischer-Tropsch synthesis on cobalt and ruthenium. Metal dispersion and support effects on reaction rate and selectivity. J. Catal. 1992, 137, 212–224. [Google Scholar] [CrossRef]
- Iglesia, E.; Reyes, S.C.; Madon, R.J.; Soled, S.L. Selectivity control and catalyst design in the Fischer-Tropsch synthesis: Sites, pellets, and reactors. Adv. Catal. 1993, 39, 221–302. [Google Scholar]
- Johnson, B.G.; Bartholomew, C.H.; Goodman, D.W. The role of surface structure and dispersion in CO hydrogenation on cobalt. J. Catal. 1991, 128, 231–247. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. Effects of support and dispersion on the CO hydrogenation activity/selectivity properties of cobalt. J. Catal. 1984, 85, 78–88. [Google Scholar] [CrossRef]
- Bessell, S. Support effects in cobalt-based Fischer-Tropsch catalysis. Appl. Catal. A 1993, 96, 253–268. [Google Scholar] [CrossRef]
- Clause, O.; Kermarec, M.; Bonneviot, L.; Villain, F.; Che, M. Nickel(II) ion-support interactions as a function of preparation method of silica-supported nickel materials. J. Am. Chem. Soc. 1992, 114, 4709–4717. [Google Scholar] [CrossRef]
- Lekhal, A.; Glasser, B.J.; Khinast, J.G. Influence of pH and ionic strength on the metal profile of impregnation catalysts. Chem. Eng. Sci. 2004, 59, 1063–1077. [Google Scholar] [CrossRef]
- Van de Water, L.G.A.; Bezemer, G.L.; Bergwerff, J.A.; Versluijs-Helder, M.; Weckhuysen, B.M.; de Jong, K.P. Spatially resolved UV-VIS microspectroscopy on the preparation of alumina-supported Co Fischer-Tropsch catalysts: Linking activity to Co distribution and speciation. J. Catal. 2006, 242, 287–298. [Google Scholar] [CrossRef]
- Rosynek, M.P.; Polansky, C.A. Effect of cobalt source on the reduction properties of silica-supported cobalt catalysts. Appl. Catal. 1991, 73, 97–112. [Google Scholar] [CrossRef]
- Niemelä, M.K.; Krause, A.O.I.; Vaara, T.; Lahtinen, J. Preparation and characterization of Co/SiO2, Co-Mg/SiO2 and Mg-Co/SiO2 catalysts and their activity in CO hydrogenation. Top. Catal. 1995, 2, 45–57. [Google Scholar] [CrossRef]
- Niemelä, M.K.; Krause, A.O.I. The long-term performance of Co/SiO2 catalysts in CO hydrogenation. Catal. Lett. 1996, 42, 161–166. [Google Scholar] [CrossRef]
- Van de Loosdrecht, J.; Van der Haar, M.; Van der Kraan, A.M.; Van Dillen, A.J.; Geus, J.W. Preparation and properties of supported cobalt catalysts for Fischer-Tropsch synthesis. Appl. Catal. A Gen. 1997, 150, 365–376. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Takeuchi, K.; Hanaoka, T.; Arakawa, H.; Sugi, Y. Hydrogenation of carbon monoxide over highly dispersed cobalt catalysts derived from cobalt(II) acetate. Catal. Today 1996, 28, 251–259. [Google Scholar] [CrossRef]
- Sun, S.; Tsubaki, N.; Fujimoto, K. The reaction performances and characterization of Fischer-Tropsch synthesis Co/SiO2 catalysts prepared from mixed cobalt salts. Appl. Catal. A 2000, 202, 121–131. [Google Scholar] [CrossRef]
- Schanke, D.; Vada, S.; Blekkan, E.A.; Hilmen, A.M.; Hoff, A.; Holmen, A. Study of Pt-promoted cobalt CO hydrogenation catalysts. J. Catal. 1995, 156, 85–95. [Google Scholar] [CrossRef]
- Lacroix, M.; Dreibine, L.; De Tymowski, B.; Vigneron, F.; Edouard, D.; Bégin, D.; Nguyen, P.; Pham, C.; Savin-Poncet, S.; Luck, F.; et al. Silicon carbide foam composite containing cobalt as a highly selective and re-usable Fischer-Tropsch synthesis catalyst. Appl. Catal. A 2011, 397, 62–72. [Google Scholar] [CrossRef]
- De Tymowski, B.; Liu, Y.; Meny, C.; Lefèvre, C.; Begin, D.; Nguyen, P.; Pham, C.; Edouard, D.; Luck, F.; Pham-Huu, C. Co-Ru/SiC impregnated with ethanol as an effective catalyst for the Fischer-Tropsch synthesis. Appl. Catal. A 2012, 419–420, 31–40. [Google Scholar] [CrossRef]
- Díaz, J.A.; Calvo-Serrano, M.; De la Osa, A.R.; García-Minguillán, A.M.; Romero, A.; Giroir-Fendler, A.; Valverde, J.L. β-silicon carbide as a catalyst support in the Fischer-Tropsch synthesis: Influence of the modification of the support by a pore agent and acidic treatment. Appl. Catal. A 2014, 475, 82–89. [Google Scholar] [CrossRef]
- De la Osa, A.R.; De Lucas, A.; Díaz-Maroto, J.; Romero, A.; Valverde, J.L.; Sánchez, P. FTS fuels production over different Co/SiC catalysts. Catal. Today 2012, 187, 173–182. [Google Scholar] [CrossRef]
- De la Osa, A.R.; De Lucas, A.; Sánchez-Silva, L.; Díaz-Maroto, J.; Valverde, J.L.; Sánchez, P. Performing the best composition of supported Co/SiC catalyst for selective FTS diesel production. Fuel 2012, 95, 587–598. [Google Scholar] [CrossRef]
- Lee, B.; Koo, H.; Park, M.-J.; Lim, B.; Moon, D.; Yoon, K.; Bae, J. Deactivation behavior of Co/SiC Fischer-Tropsch catalysts by formation of filamentous carbon. Catal. Lett. 2013, 143, 18–22. [Google Scholar] [CrossRef]
- Lee, J.S.; Jung, J.S.; Moon, D.J. The effect of cobalt loading on Fischer-Tropsch synthesis over silicon carbide supported catalyst. J. Nanosci. Nanotechnol. 2015, 15, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lu, X.; Liu, X.; Hildebrandt, D.; Glasser, D. Heat transfer study with and without Fischer-Tropsch reaction in a fixed bed reactor with TiO2, SiO2, and SiC supported cobalt catalysts. Chem. Eng. J. 2014, 247, 75–84. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; Koeken, A.C.J.; Bitter, J.H.; Davidian, T.; Ruitenbeek, M.; Dugulan, A.I.; De Jong, K.P. Effect of precursor on the catalytic performance of supported iron catalysts for the Fischer-Tropsch synthesis of lower olefins. Catal. Today 2013, 215, 95–102. [Google Scholar] [CrossRef]
- Khodakov, A.Y. Enhancing cobalt dispersion in supported Fischer-Tropsch catalysts via controlled decomposition of cobalt precursors. Braz. J. Phys. 2009, 39, 171–175. [Google Scholar] [CrossRef]
- Mishra, S.K.; Kanungo, S.B. Thermal dehydration and decomposition of cobalt chloride hydrate (CoCl2·xH2O). J. Therm. Anal. 1992, 38, 2437–2454. [Google Scholar] [CrossRef]
- Yuan, X.; Lü, J.; Yan, X.; Hu, L.; Xue, Q. Preparation of ordered mesoporous silicon carbide monoliths via preceramic polymer nanocasting. Microporous Mesoporous Mater. 2011, 142, 754–758. [Google Scholar] [CrossRef]
- Jacobson, N.S.; Myers, D.L. Active oxidation of SiC. Oxid. Met. 2011, 75, 1–25. [Google Scholar] [CrossRef]
- Labuschagne, J.; Meyer, R.; Chonco, Z.H.; Botha, J.M.; Moodley, D.J. Application of water-tolerant Co/β-SiC catalysts in slurry phase Fischer-Tropsch synthesis. Catal. Today 2016. [Google Scholar] [CrossRef]
- Kolasinski, K.W. Surface Science Foundations of Catalysis and Nanoscience; John Wiley and Sons: Chichester, UK, 2007. [Google Scholar]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Nguyen, P.; Pham, C. Innovative porous SiC-based materials: From nanoscopic understandings to tunable carriers serving catalytic needs. Appl. Catal. A 2011, 391, 443–454. [Google Scholar] [CrossRef]
- Ledoux, M.J.; Pham-Huu, C. Silicon carbide a novel catalyst support for heterogeneous catalysis. Cattech 2001, 5, 226–246. [Google Scholar] [CrossRef]
- Lee, S.-H.; Yun, S.-M.; Kim, S.; Park, S.-J.; Lee, Y.-S. Characterization of nanoporous β-SiC fiber complex prepared by electrospinning and carbothermal reduction. Res. Chem. Intermed. 2010, 36, 731–742. [Google Scholar] [CrossRef]
- Cook, K.M.; Poudyal, S.; Miller, J.T.; Bartholomew, C.H.; Hecker, W.C. Reducibility of alumina-supported cobalt Fischer-Tropsch catalysts: Effects of noble metal type, distribution, retention, chemical state, bonding, and influence on cobalt crystallite size. Appl. Catal. A 2012, 449, 69–80. [Google Scholar] [CrossRef]
- Panpranot, J.; Kaewkun, S.; Praserthdam, P.; Goodwin, J., Jr. Effect of cobalt precursors on the dispersion of cobalt on MCM-41. Catal. Lett. 2003, 91, 95–102. [Google Scholar] [CrossRef]
- Jones, R.D.; Bartholomew, C.H. Improved flow technique for measurement of hydrogen chemisorption on metal catalysts. Appl. Catal. 1988, 39, 77–88. [Google Scholar] [CrossRef]
- Belambe, A.R.; Oukaci, R.; Goodwin, J.G., Jr. Effect of pretreatment on the activity of a Ru-promoted Co/Al2O3 Fischer-Tropsch catalyst. J. Catal. 1997, 166, 8–15. [Google Scholar] [CrossRef]
- Terörde, R.J.A.M.; Van den Brink, P.J.; Visser, L.M.; Van Dillen, A.J.; Geus, J.W. Selective oxidation of hydrogen sulfide to elemental sulfur using iron oxide catalysts on various supports. Catal. Today 1993, 17, 217–224. [Google Scholar] [CrossRef]
- Sun, S.; Fujimoto, K.; Yoneyama, Y.; Tsubaki, N. Fischer-Tropsch synthesis using Co/SiO2 catalysts prepared from mixed precursors and addition effect of noble metals. Fuel 2002, 81, 1583–1591. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, J.-G.; Fang, K.-G.; Sun, Y.-H. The deactivation of Co/SiO2 catalyst for Fischer-Tropsch synthesis at different ratios of H2 to CO. Fuel Process. Technol. 2006, 87, 609–616. [Google Scholar] [CrossRef]
- Solomonik, I.G.; Gryaznov, K.O.; Skok, V.F.; Mordkovich, V.Z. Formation of surface cobalt structures in SiC-supported Fischer-Tropsch catalysts. RSC Adv. 2015, 5, 78586–78597. [Google Scholar] [CrossRef]
- Rodrigues, E.L.; Bueno, J.M.C. Co/SiO2 catalysts for selective hydrogenation of crotonaldehyde II: Influence of the Co surface structure on selectivity. Appl. Catal. A 2002, 232, 147–158. [Google Scholar] [CrossRef]
- Bechara, R.; Balloy, D.; Dauphin, J.-Y.; Grimblot, J. Influence of the characteristics of γ-aluminas on the dispersion and the reducibility of supported cobalt catalysts. Chem. Mater. 1999, 11, 1703–1711. [Google Scholar] [CrossRef]
- Ernst, B.; Bensaddik, A.; Hilaire, L.; Chaumette, P.; Kiennemann, A. Study on a cobalt silica catalyst during reduction and Fischer-Tropsch reaction: In situ EXAFS compared to XPS and XRD. Catal. Today 1998, 39, 329–341. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Lynch, J.; Bazin, D.; Rebours, B.; Zanier, N.; Moisson, B.; Chaumette, P. Reducibility of cobalt species in silica-supported Fischer-Tropsch catalysts. J. Catal. 1997, 168, 16–25. [Google Scholar] [CrossRef]
- Keyvanloo, K.; Fisher, M.J.; Hecker, W.C.; Lancee, R.J.; Jacobs, G.; Bartholomew, C.H. Kinetics of deactivation by carbon of a cobalt Fischer-Tropsch catalyst: Effects of CO and H2 partial pressures. J. Catal. 2015, 327, 33–47. [Google Scholar] [CrossRef]
- Bao, A.; Liew, K.; Li, J. Fischer-Tropsch synthesis on CaO-promoted Co/Al2O3 catalysts. J. Mole. Cataly. A 2009, 304, 47–51. [Google Scholar] [CrossRef]
- De la Osa, A.R.; De Lucas, A.; Valverde, J.L.; Romero, A.; Monteagudo, I.; Coca, P.; Sánchez, P. Influence of alkali promoters on synthetic diesel production over Co catalyst. Catal. Today 2011, 167, 96–106. [Google Scholar] [CrossRef]
- Dry, M.E.; Oosthuizen, G.J. The correlation between catalyst surface basicity and hydrocarbon selectivity in the Fischer-Tropsch synthesis. J. Catal. 1968, 11, 18–24. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Ren, J.; Sun, Y. Chemical treatment of γ-Al2O3 and its influence on the properties of Co-based catalysts for Fischer-Tropsch synthesis. Appl. Catal. A 2003, 243, 121–133. [Google Scholar] [CrossRef]
- Koo, H.-M.; Lee, B.S.; Park, M.-J.; Moon, D.J.; Roh, H.-S.; Bae, J.W. Fischer-Tropsch synthesis on Cobalt/Al2O3-modified SiC catalysts: Effect of cobalt-alumina interactions. Catal. Sci. Technol. 2014, 4, 343–351. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Ren, J.; Li, Y.; Sun, Y. Support effect of Co/Al2O3 catalysts for Fischer-Tropsch synthesis. Fuel 2003, 82, 581–586. [Google Scholar] [CrossRef]
- Courty, P.; Ajot, H.; Marcilly, C.; Delmon, B. Oxydes mixtes ou en solution solide sous forme très divisée obtenus par décomposition thermique de précurseurs amorphes. Powder Technol. 1973, 7, 21–38. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen. Mathematisch-Physikalische Klasse 1918, 2, 98–100. [Google Scholar]
- Klug, H.P.; Alexander, L.E. X-ray Diffraction Procedures for Polycrystalline Amorphous Materials, 2nd ed.; Wiley: New York, NY, USA, 1974; p. 992. [Google Scholar]
- Van ’t Blik, H.F.J.; Koningsberger, D.C.; Prins, R. Characterization of supported cobalt and Cobalt-Rhodium catalysts. III. Temperature-programmed reduction (TPR), oxidation (TPO), and EXAFS of CoRh SiO2. J. Catal. 1986, 97, 210–218. [Google Scholar] [CrossRef]
- Datye, A.K.; Xu, Q.; Kharas, K.C.; McCarty, J.M. Particle size distributions in heterogeneous catalysts: What do they tell us about the sintering mechanism? Catal. Today 2006, 111, 59–67. [Google Scholar] [CrossRef]
- Mustard, D.G.; Bartholomew, C.H. Determination of metal crystallite size and morphology in supported nickel catalysts. J. Catal. 1981, 67, 186–206. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Chemistry of nickel-alumina catalysts. J. Catal. 1976, 45, 41–53. [Google Scholar] [CrossRef]
- Song, D.; Li, J. Effect of catalyst pore size on the catalytic performance of silica supported cobalt Fischer-Tropsch catalysts. J. Mol. Catal. A 2006, 247, 206–212. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer-Tropsch synthesis: Support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A 2002, 233, 263–281. [Google Scholar] [CrossRef]
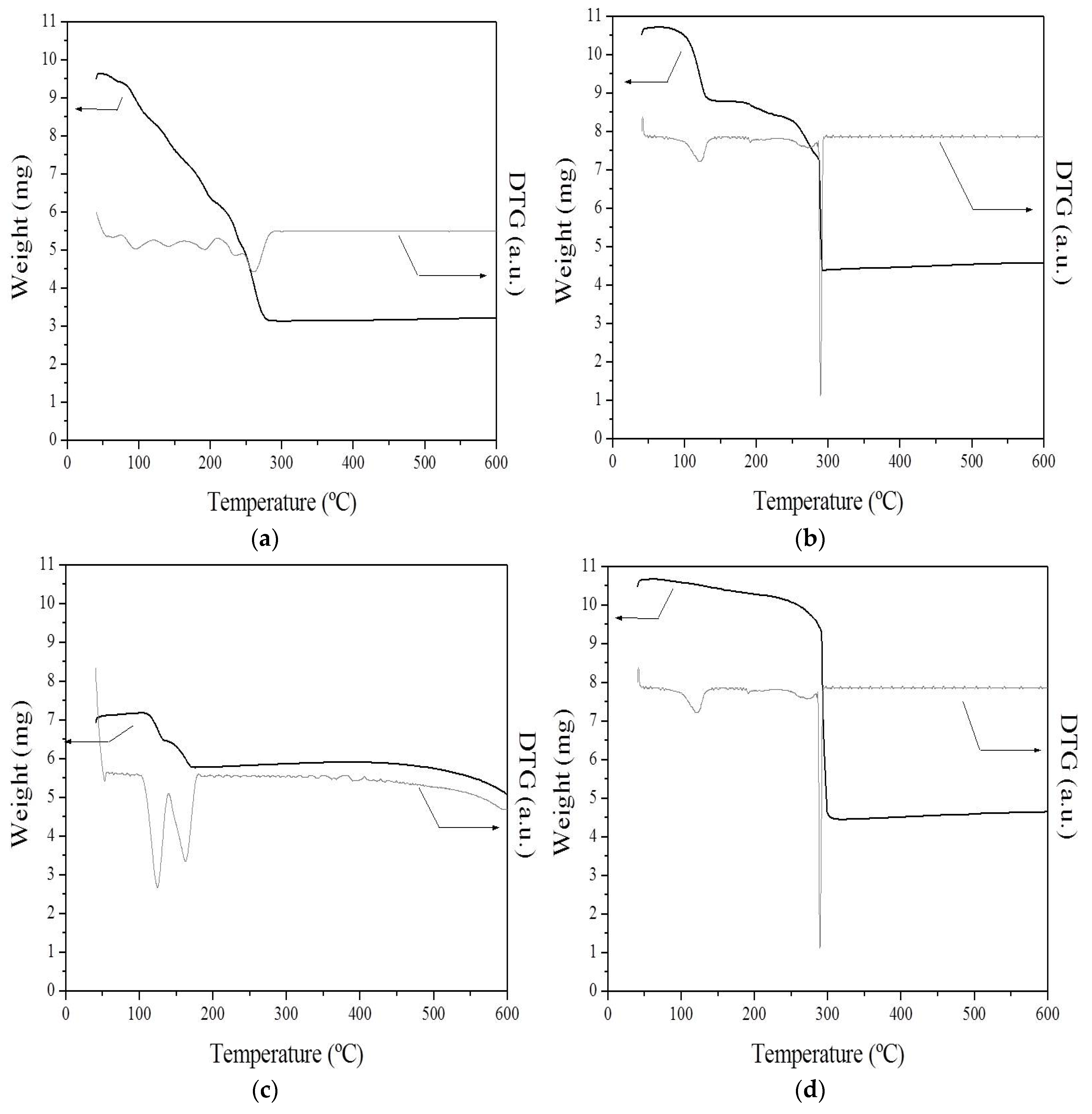

 CoN/SiC;
CoN/SiC;  CoA/SiC;
CoA/SiC;  CoCl/SiC; and
CoCl/SiC; and  CoCit/SiC.
CoCit/SiC.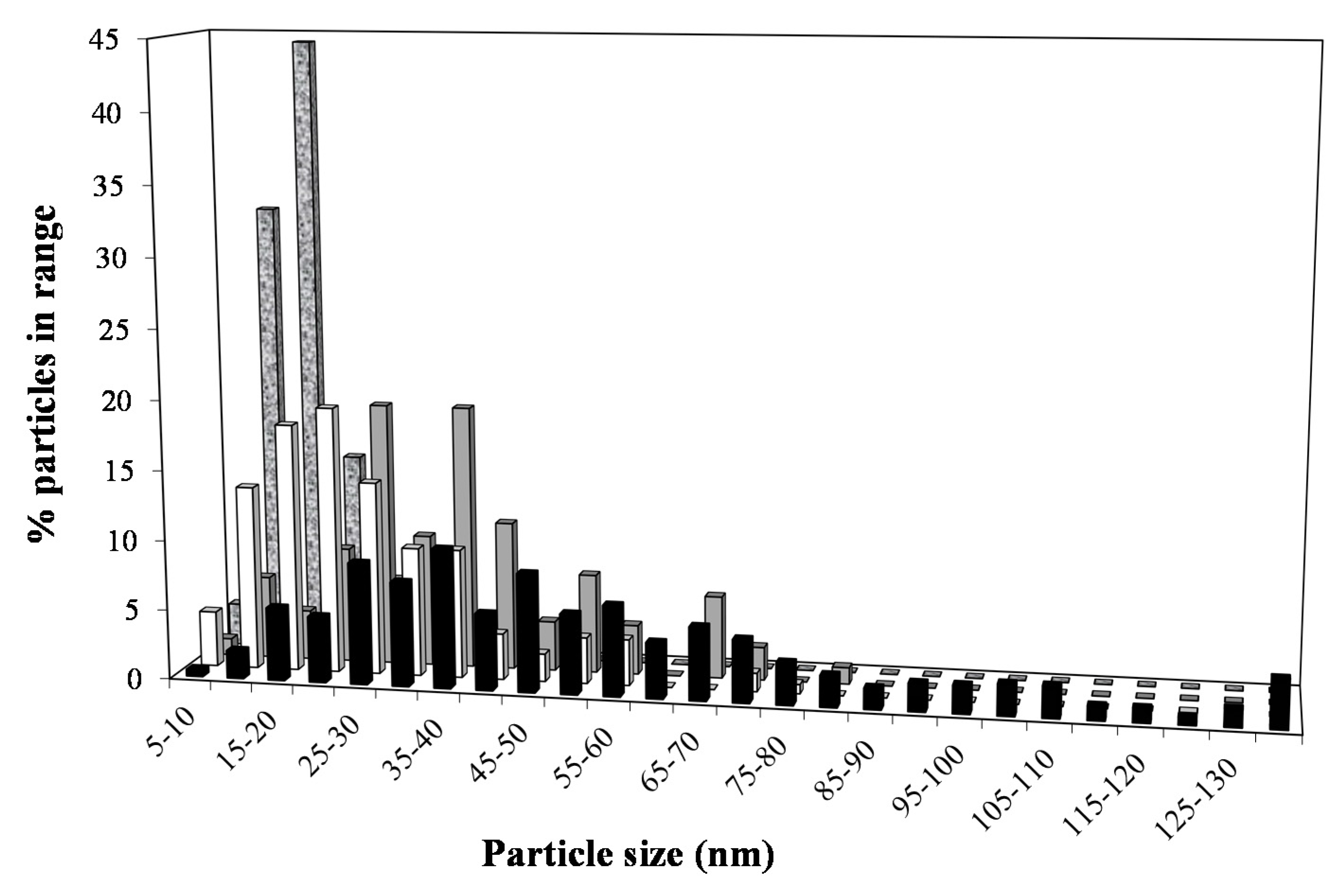
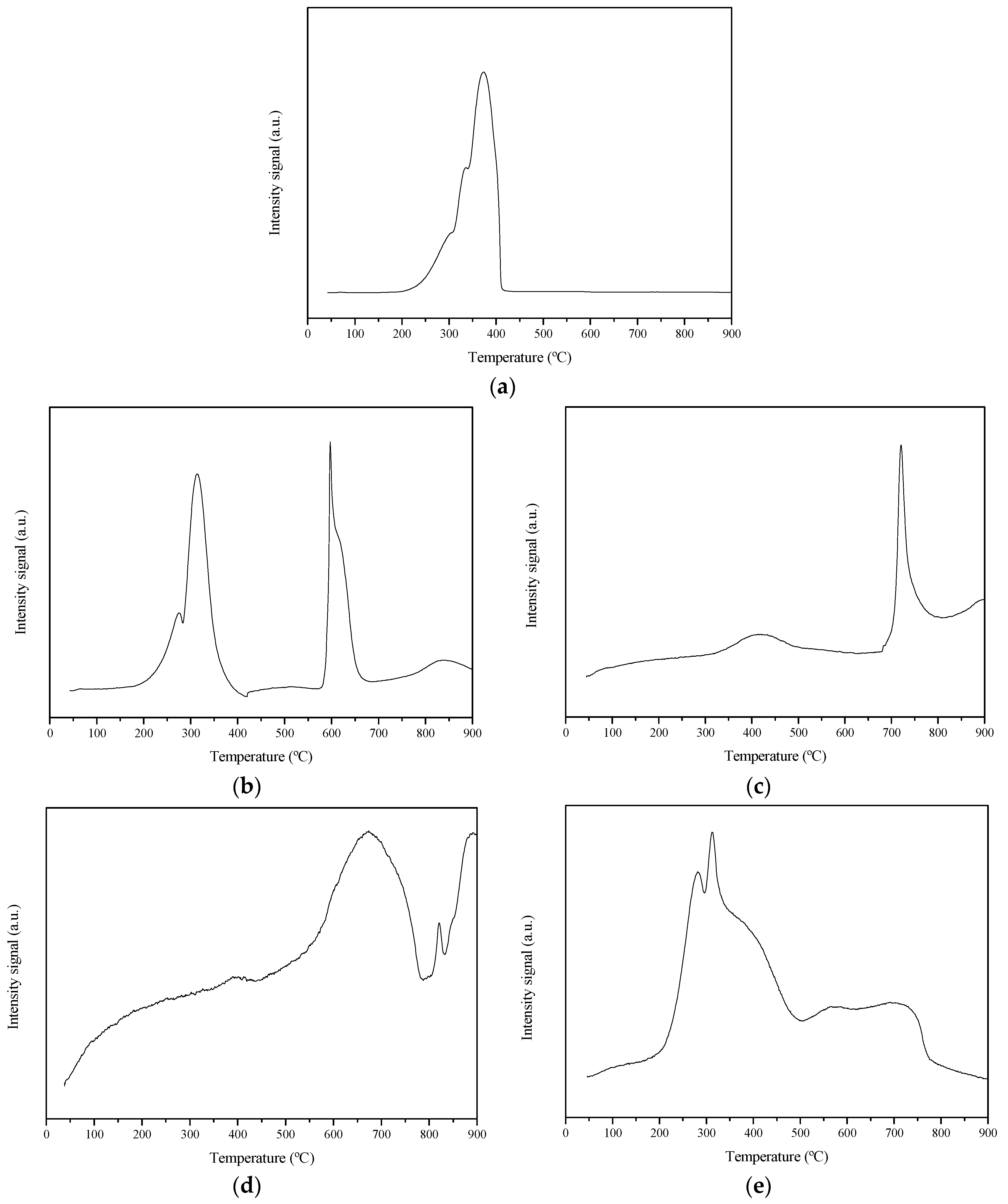
 Reference;
Reference;  CoN/SiC;
CoN/SiC;  CoA/SiC;
CoA/SiC;  CoCl/SiC; and
CoCl/SiC; and 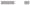 CoCit/SiC.
CoCit/SiC.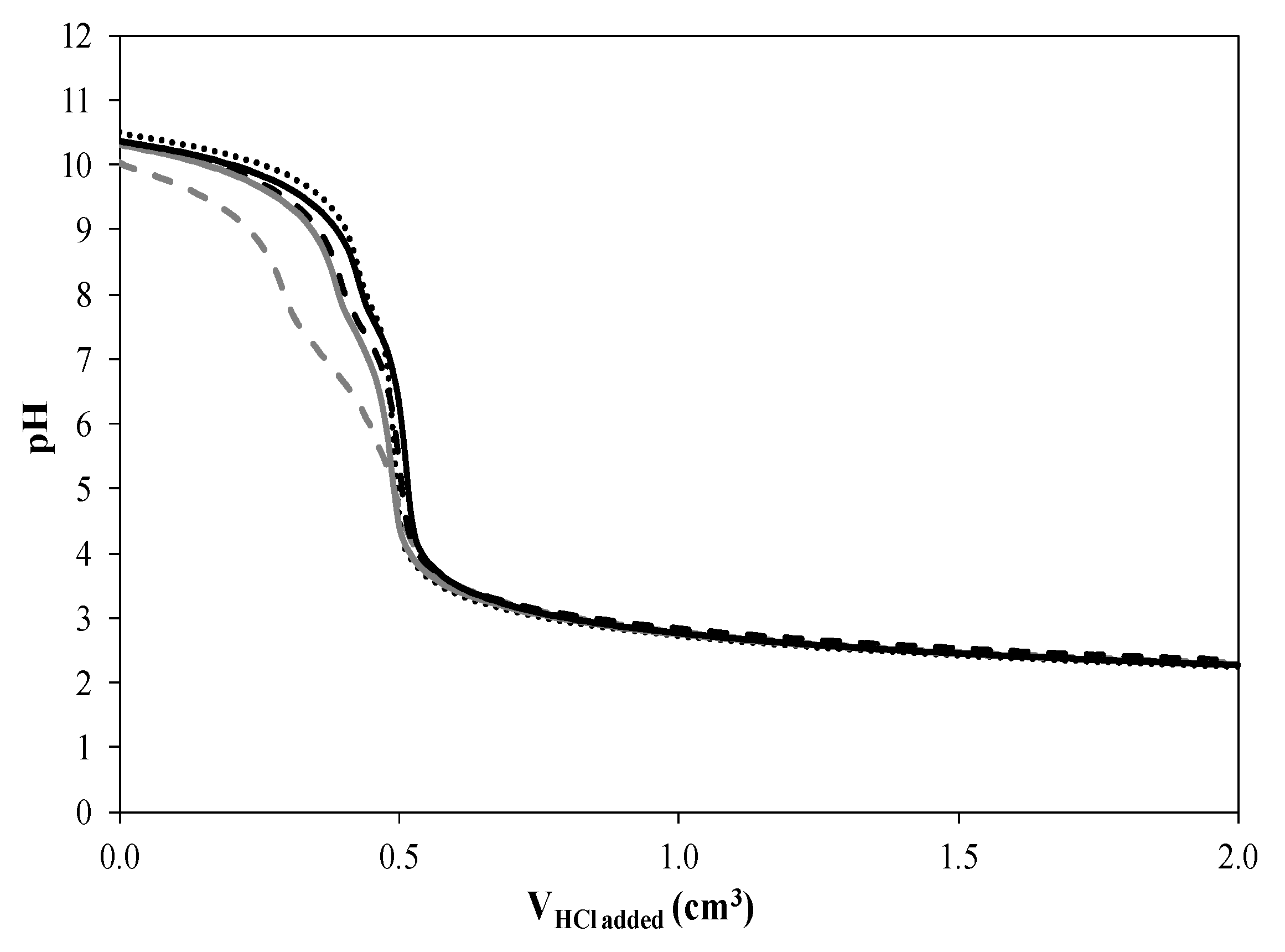
 CoCit/SiC; and
CoCit/SiC; and  CoN/SiC. Reaction conditions: 20 bar; 250 °C; 6000 Ncm3/g·h; H2/CO: 2.
CoCit/SiC; and CoN/SiC. Reaction conditions: 20 bar; 250 °C; 6000 Ncm3/g·h; H2/CO: 2.
CoN/SiC. Reaction conditions: 20 bar; 250 °C; 6000 Ncm3/g·h; H2/CO: 2.
CoCit/SiC; and CoN/SiC. Reaction conditions: 20 bar; 250 °C; 6000 Ncm3/g·h; H2/CO: 2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Wt % Co ± 0.1 | dCo3O4 (nm) 1 | dCo0 (nm) 11 | D (%) 12 | D (%) 22 | dCo0 (nm) 2 | Degree of Reduction (%) 3 ± 2 | BET Area (m2/g) 4 ± 1.1 | Pore Diameter (nm) 5 ± 0.3 | Total Pore Volume (cm3/g) 6 | Basicity (cm3/g) 7 ± 0.03 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CoN/SiC | 9.9 | 45.0 ± 5.8 | 33.8 | 2.8 | 1.7 | 56.3 ± 10.1 | 74.1 | 21.5 | 7.2 | 0.039 | 0.26 |
| CoCl/SiC | 7.2 | 75.9 ± 8.7 | 56.9 | 1.7 | 2.6 | 37.1 ± 5.2 | 41.5 | 22.1 | 7.3 | 0.055 | −0.53 |
| CoA/SiC | 10.9 | 34.4 ± 5.2 | 25.8 | 3.7 | 3.6 | 27.0 ± 5.5 | 58.5 | 24.8 | 8.6 | 0.106 | −1.21 |
| CoCit/SiC | 8.9 | 21.4 ± 4.6 | 16.1 | 5.9 | 5.7 | 17.0 ± 2.1 | 41.8 | 34.9 | 8.6 | 0.151 | −4.19 |
| Catalyst | T (°C) | FTS Rate (mol/molCo·h) ± 0.4 | WGS Rate (mol/molCo·h) ± 0.03 | Conversion (%) ± 0.5 | Selectivity (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| CO | H2 | CO2 ± 0.03 | C1–C4 ± 0.4 | C2 OR 1 | C3 OR 1 | C5+ ± 0.4 | ||||
| CoN/SiC | 220.00 | 3.47 | 0.02 | 7.40 | 24.10 | 0.50 | 6.20 | 0.36 | 1.00 | 93.30 |
| 235.00 | 31.50 | 0.21 | 67.20 | 80.20 | 0.70 | 5.30 | 0.09 | 0.73 | 94.10 | |
| 250.00 | 34.81 | 0.41 | 74.90 | 85.70 | 0.70 | 9.30 | 0.10 | 0.72 | 90.00 | |
| CoCit/SiC | 220.00 | 5.69 | 0.04 | 24.50 | 29.80 | 0.70 | 9.60 | 0.09 | 0.74 | 89.80 |
| 235.00 | 17.22 | 0.74 | 76.50 | 89.20 | 4.10 | 25.00 | 0.01 | 0.19 | 70.90 | |
| 250.00 | 19.85 | 1.69 | 92.30 | 97.50 | 7.80 | 26.50 | 0.00 | 0.11 | 65.70 | |
| Catalyst | C5+ Hydrocarbon Distribution (wt %) | Diesel (vol %) | Diesel Yield (%) | α | ||||
|---|---|---|---|---|---|---|---|---|
| Gasoline (C7–C10) | Kerosene (C11–C14) | Diesel (C15–C18) | Lubricants (C19–C20) | Waxes (C20+) | ||||
| CoN/SiC | 13.3 | 56.8 | 23.5 | 3.2 | 3.2 | 17.2 | 12.9 | 0.90 |
| CoCit/SiC | 56.3 | 21.1 | 14.1 | 4.1 | 4.8 | 5.9 | 5.4 | 0.76 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De la Osa, A.R.; Romero, A.; Dorado, F.; Valverde, J.L.; Sánchez, P. Influence of Cobalt Precursor on Efficient Production of Commercial Fuels over FTS Co/SiC Catalyst. Catalysts 2016, 6, 98. https://doi.org/10.3390/catal6070098
De la Osa AR, Romero A, Dorado F, Valverde JL, Sánchez P. Influence of Cobalt Precursor on Efficient Production of Commercial Fuels over FTS Co/SiC Catalyst. Catalysts. 2016; 6(7):98. https://doi.org/10.3390/catal6070098
Chicago/Turabian StyleDe la Osa, Ana Raquel, Amaya Romero, Fernando Dorado, José Luis Valverde, and Paula Sánchez. 2016. "Influence of Cobalt Precursor on Efficient Production of Commercial Fuels over FTS Co/SiC Catalyst" Catalysts 6, no. 7: 98. https://doi.org/10.3390/catal6070098
APA StyleDe la Osa, A. R., Romero, A., Dorado, F., Valverde, J. L., & Sánchez, P. (2016). Influence of Cobalt Precursor on Efficient Production of Commercial Fuels over FTS Co/SiC Catalyst. Catalysts, 6(7), 98. https://doi.org/10.3390/catal6070098