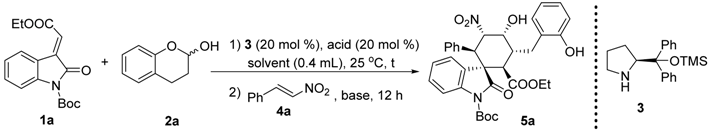
3.2. General Procedure for the One-Pot, [2+2+2] Tandem Reaction
To a mixture of 3 (0.005 mmol, 0.05 equiv) and p-nitrobenzoic acid (0.005 mmol, 0.05 equiv) in 0.4 mL EtOH (with 5% H2O) was added olefinic oxindole 1 (0.1 mmol, 1 equiv), lactol 2 (0.12 mmol, 1.2 equiv) subsequently. The reaction was stirred at room temperature for 3 h, after which nitroolefin 4 was added followed by the addition of TEA (0.04 mmol 0.4 equiv). The reaction was kept in 25 °C for another 12 h. The product 5 was isolated by chromatography.
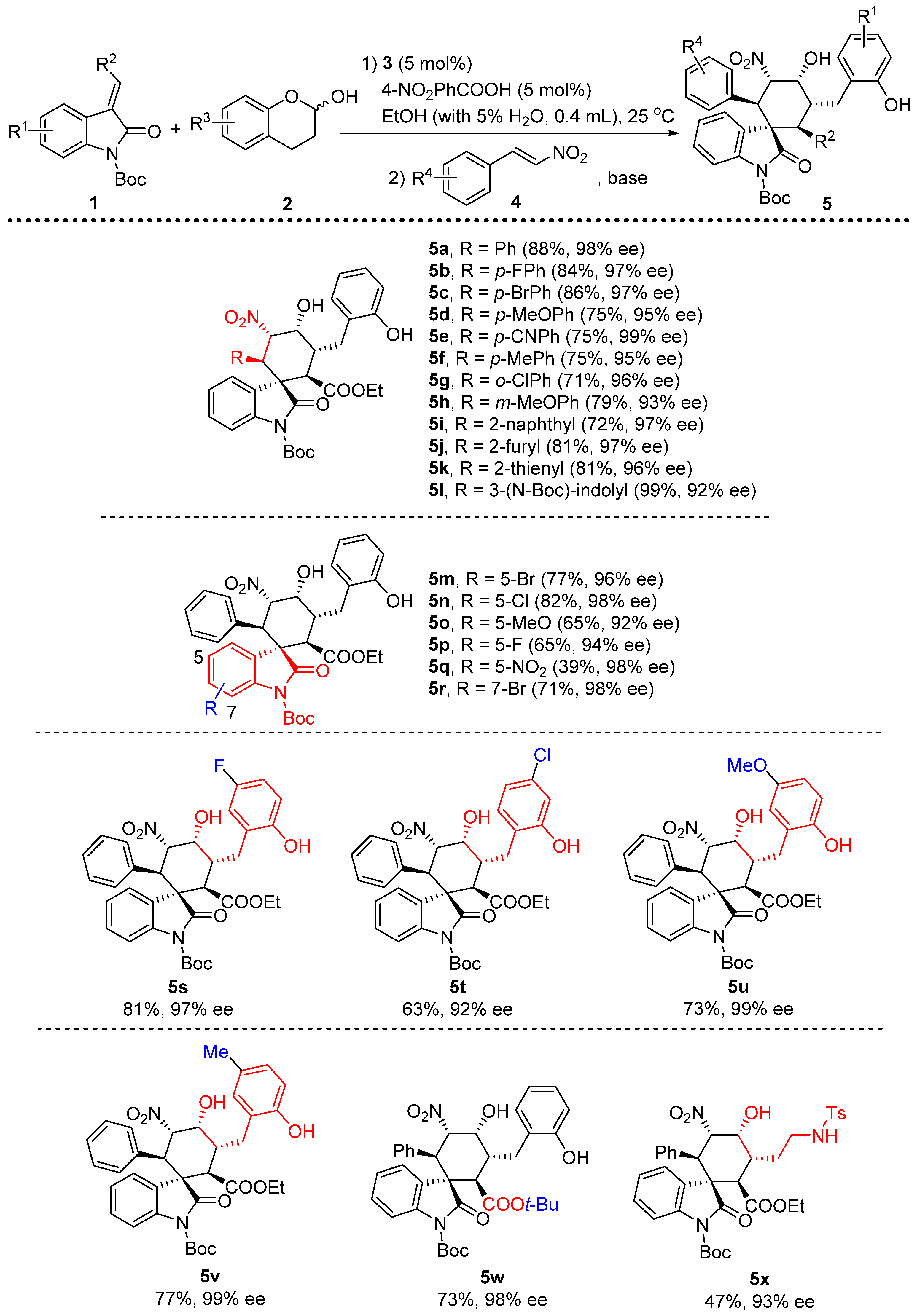
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5a): 88% yield; [α] = +52.6 (c = 1.0 in CHCl3); 98% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.86 min, tminor = 9.11 min]; 1H NMR (500 MHz, CDCl3) δ 7.48 (dd, J = 7.3, 1.2 Hz, 1H), 7.40 (dd, J = 7.9, 0.9 Hz, 1H), 7.22–7.12 (m, 4H), 7.04–6.84 (m, 6H), 6.71–6.58 (m, 1H), 6.03 (dd, J = 12.3, 2.9 Hz, 1H), 4.40 (s, 1H), 4.13 (d, J = 12.3 Hz, 1H), 3.92–3.83 (m, 2H), 3.66 (d, J = 11.9 Hz, 1H), 3.40–3.34 (m, 1H), 3.08–3.02 (m, 1H), 2.46 (dd, J = 13.7, 4.4 Hz, 1H), 1.55 (s, 9H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.83, 171.38, 153.74, 148.43, 139.34, 133.02, 131.73, 129.03, 128.34, 127.86, 127.57, 124.39, 123.83, 122.83, 121.62, 115.82, 114.75, 85.94, 84.23, 68.24, 61.03, 54.68, 51.06, 46.62, 39.50, 29.42, 28.04, 13.62. ESI-HRMS: calcd. for C34H35N2O9 [M − H]− 615.2343, found 615.2340.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-6-(4-fluorophenyl)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5b): 84% yield; [α] = +21.5 (c = 1.0 in CHCl3); 97% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.39 min, tminor = 8.38 min]; 1H NMR (500 MHz, CD3OD) δ 7.62 (d, J = 7.2 Hz, 1H), 7.39–7.35 (m, 1H), 7.29–7.19 (m, 2H), 7.14–7.04 (m, 2H), 6.84–6.52 (m, 5H), 5.87 (dd, J = 12.2, 2.7 Hz, 1H), 4.37 (s, 1H), 4.17 (d, J = 12.2 Hz, 1H), 3.79–3.71 (m, 2H), 3.58 (d, J = 12.2 Hz, 1H), 3.52–3.45 (m, 1H), 2.85–2.78 (m, 1H), 2.61 (dd, J = 13.4, 4.4 Hz, 1H), 1.55 (s, 8H), 0.84 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, CD3OD) δ 175.42, 171.43, 162.95, 161.33, 155.38, 148.48, 139.28, 131.03, 130.31, 128.87, 127.93, 127.58, 124.66, 124.49, 123.33, 119.41, 114.71, 114.38, 86.90, 84.36, 68.61, 60.64, 54.83, 51.35, 45.49, 38.12, 30.25, 26.93, 12.66. ESI-HRMS: calcd. for C34H34FN2O9 [M − H]− 633.2248, found 633.2243.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-2-(4-bromophenyl)-4-hydroxy-5-(2-hydroxybenzyl)-3-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,6-dicarboxylate (5c): 86% yield; [α] = +65.7(c = 1.0 in CHCl3); 97% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.50 min, tminor = 9.20 min]; 1H NMR (500 MHz, CD3OD) δ 7.64–7.59 (m, 1H), 7.37 (dd, J = 7.7, 1.1 Hz, 1H), 7.30–7.21 (m, 2H), 7.18–6.95 (m, 4H), 6.79 (dd, J = 13.7, 7.4 Hz, 2H), 6.64–6.31 (m, 1H), 5.86 (dd, J = 12.1, 2.8 Hz, 1H), 4.38 (s, 1H), 4.15 (d, J = 12.1 Hz, 1H), 3.79–3.71 (m, 2H), 3.58 (d, J = 12.2 Hz, 1H), 3.53–3.44 (m, 1H), 2.85–2.78 (m, 1H), 2.62 (dd, J = 13.4, 4.4 Hz, 1H), 1.55 (s, 7H), 0.84 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ 175.37, 171.41, 163.55, 155.39, 148.39, 139.28, 133.69, 131.03, 130.62, 128.97, 127.80, 127.59, 124.65, 124.54, 123.35, 121.31, 119.41, 114.71, 114.47, 86.64, 84.44, 68.62, 60.66, 54.71, 51.26, 45.72, 38.13, 30.26, 26.94, 12.66. ESI-HRMS: calcd. for C34H34BrN2O9 [M − H]− 693.1448, found 693.1451.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-6-(4-methoxyphenyl)-5-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5d): 75% yield; [α] = +56.3 (c = 1.0 in CHCl3); 95% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.76 min, tminor = 12.08 min]; 1H NMR (500 MHz, CDCl3) δ 7.47–7.42 (m, 2H), 7.22–7.12 (m, 4H), 6.93 (dd, J = 10.7, 4.1 Hz, 1H), 6.84 (d, J = 7.7 Hz, 1H), 6.57–6.40 (m, 3H), 5.98 (dd, J = 12.3, 2.7 Hz, 1H), 4.37 (s, 1H), 4.10–4.02 (m, 2H), 3.92–3.82 (m, 2H), 3.62 (s, 3H), 3.38–3.29 (m, 1H), 3.07–2.99 (m, 1H), 2.45 (dd, J = 13.7, 4.4 Hz, 1H), 1.57 (s, 8H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.91, 171.29, 158.97, 153.79, 148.45, 139.40, 131.71, 129.00, 128.35, 127.70, 124.75, 124.37, 123.79, 122.73, 121.63, 115.91, 114.87, 113.33, 86.17, 84.18, 68.08, 61.05, 54.98, 54.73, 51.07, 45.98, 39.39, 28.06, 13.62. ESI-HRMS: calcd. for C35H37N2O10 [M − H]− 645.2448, found 645.2450.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-6-(4-cyanophenyl)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5e): 75% yield; [α] = +83.2 (c = 1.0 in CHCl3); 99% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 10.29 min, tminor = 19.55 min]; 1H NMR (500 MHz, CD3OD) δ 7.65 (d, J = 7.0 Hz, 1H), 7.48–7.18 (m, 6H), 7.15–7.05 (m, 2H), 6.80 (dd, J = 13.9, 7.5 Hz, 2H), 5.94 (dd, J = 12.1, 2.8 Hz, 1H), 4.43 (s, 1H), 4.28 (d, J = 12.1 Hz, 1H), 3.80–3.70 (m, 2H), 3.60 (d, J = 12.1 Hz, 1H), 3.53–3.45 (m, 1H), 2.84–2.79 (m, 1H), 2.63 (dd, J = 13.4, 4.4 Hz, 1H), 1.55 (s, 9H), 0.84 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, CD3OD) δ 175.00, 171.27, 163.55, 155.39, 148.35, 140.46, 139.16, 131.03, 129.15, 127.62, 127.39, 124.64, 124.61, 123.47, 119.43, 117.80, 114.72, 114.42, 111.26, 86.42, 84.57, 68.64, 60.72, 54.69, 51.41, 46.24, 38.14, 30.24, 26.93, 12.66. ESI-HRMS: calcd. For C35H34N3O9 [M − H]− 640.2295, found 640.2291.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-(p-tolyl)spiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5f): 75% yield; [α] = +59.7 (c = 1.0 in CHCl3); 95% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.92 min, tminor = 9.40 min]; 1H NMR (500 MHz, CD3OD) δ 7.60 (d, J = 7.3 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.28–7.17 (m, 2H), 7.14–7.04 (m, 2H), 6.85–6.67 (m, 4H), 5.90–5.84 (m, 1H), 4.35 (s, 1H), 4.11 (d, J = 12.2 Hz, 1H), 3.79–3.69 (m, 2H), 3.57 (d, J = 12.1 Hz, 1H), 3.53–3.45 (m, 1H), 2.82 (dd, J = 13.4, 10.1 Hz, 1H), 2.60 (dd, J = 13.4, 4.4 Hz, 1H), 2.12 (s, 3H), 1.54 (s, 8H), 0.83 (t, J = 7.1 Hz, 3H).13C NMR (125 MHz, CD3OD) δ 175.48, 171.40, 155.24, 148.43, 139.24, 137.05, 130.91, 128.55, 128.11, 128.02, 127.44, 124.59, 124.22, 123.16, 119.30, 114.60, 114.26, 86.86, 84.05, 68.50, 60.47, 54.74, 51.30, 45.72, 38.05, 30.14, 26.85, 19.50, 12.55. ESI-HRMS: calcd. For C35H37N2O9 [M − H]− 629.2499, found 629.2498.
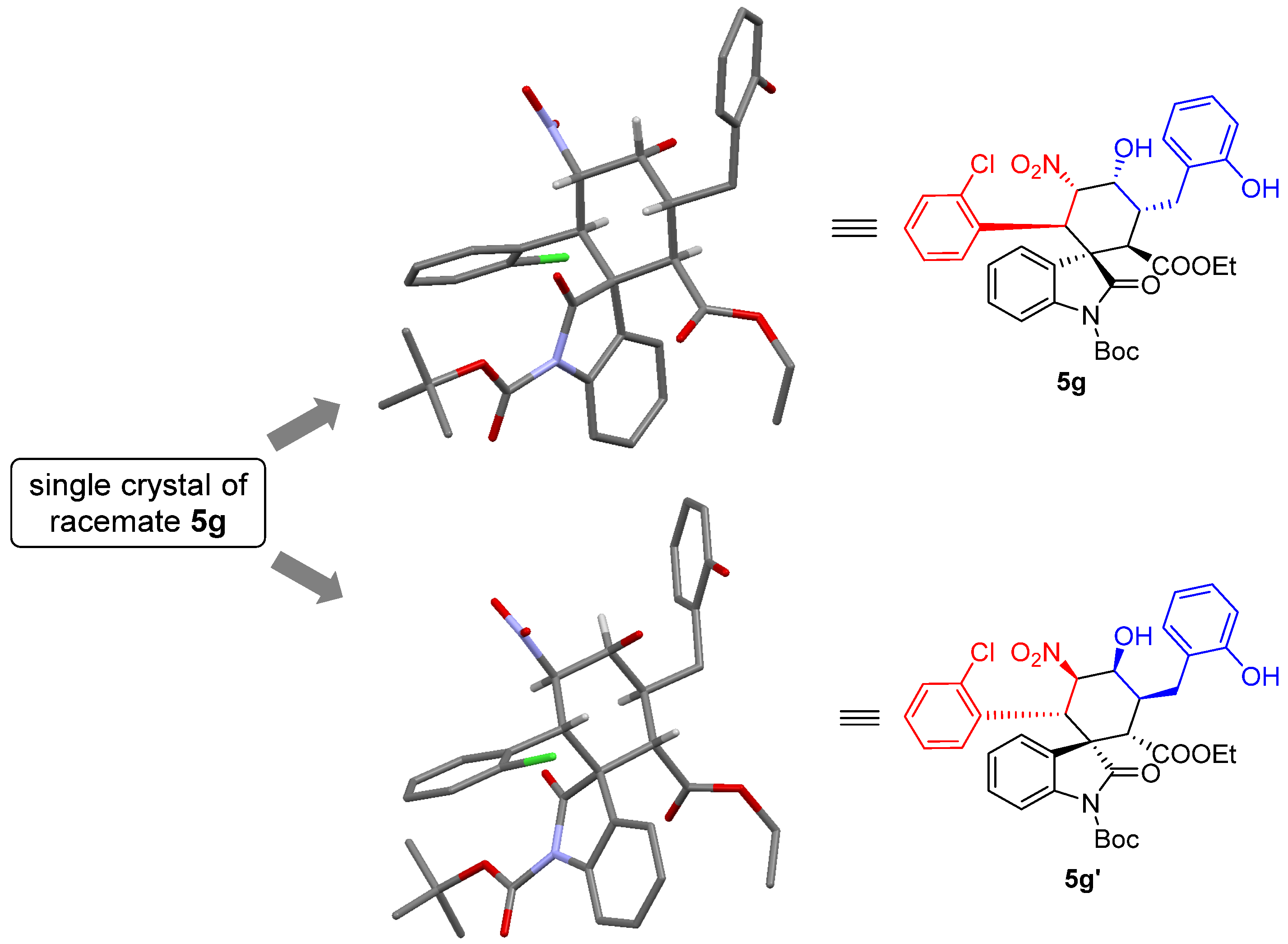
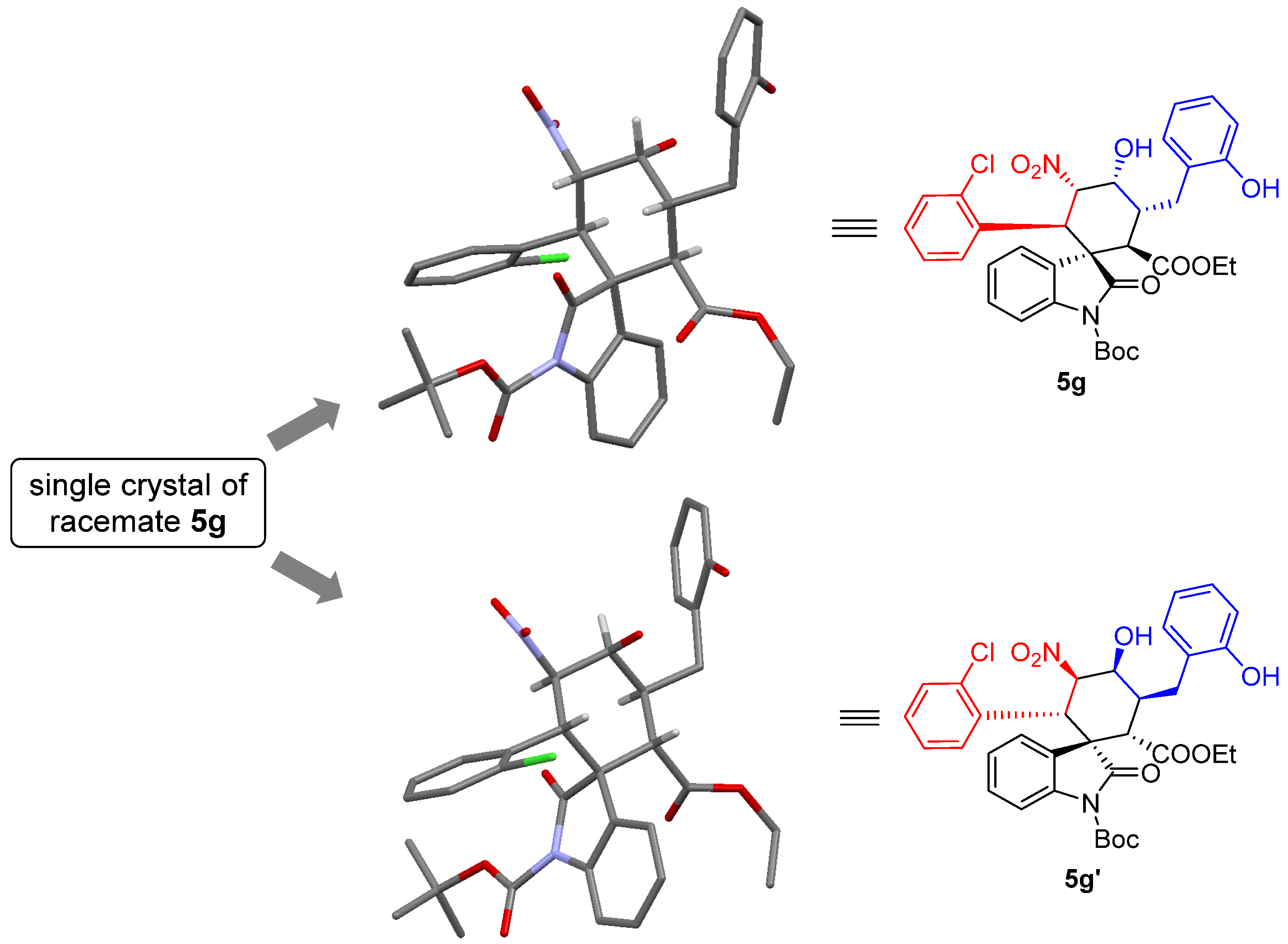
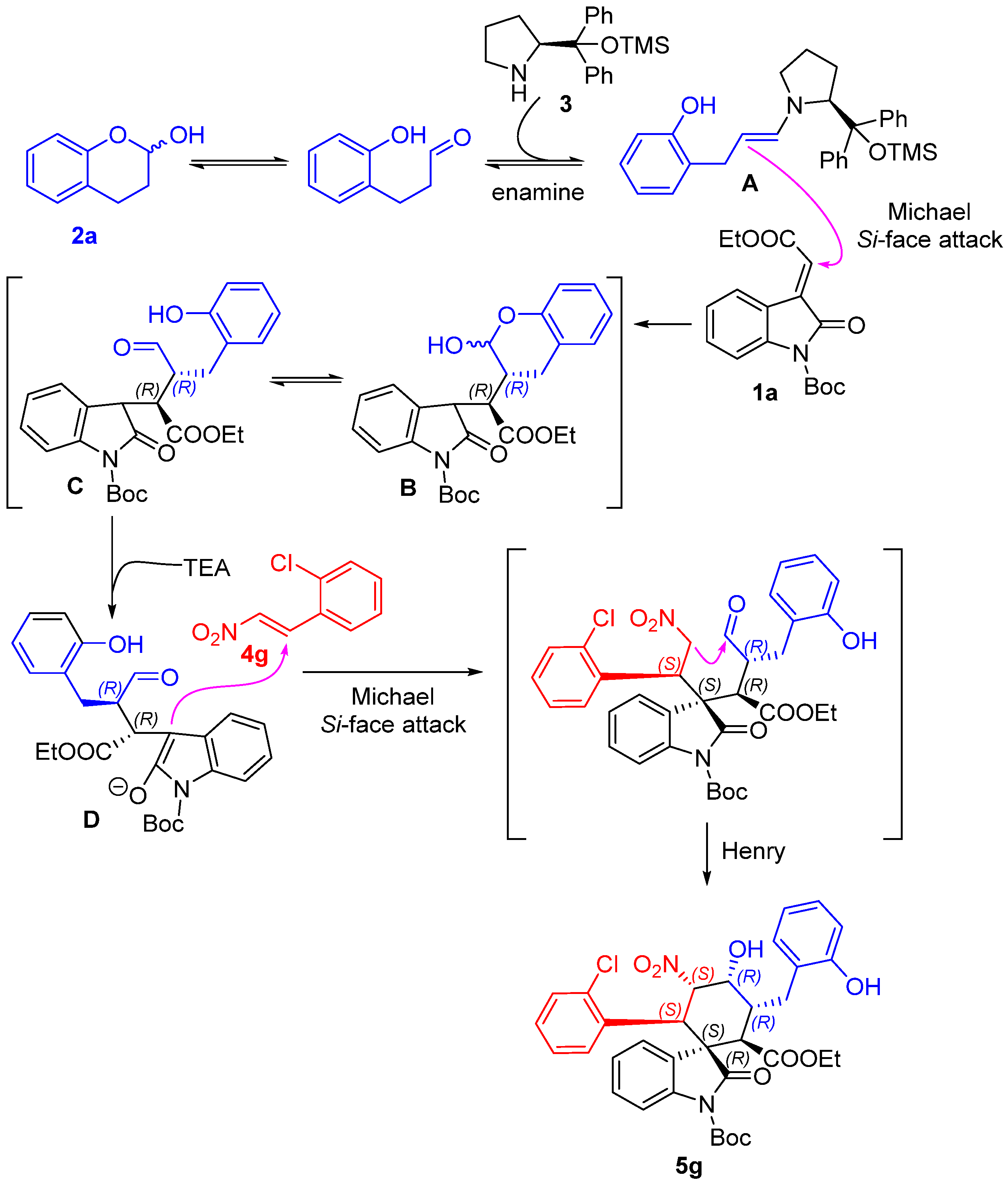
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-6-(2-chlorophenyl)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5g): 71% yield; [α] = +27.8 (c = 1.0 in CHCl3); 96% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.83 min, tminor = 10.62 min]; 1H NMR (500 MHz, CD3OD) δ 7.68–7.64 (m, 1H), 7.34 (dd, J = 12.0, 4.8 Hz, 2H), 7.20–7.01 (m, 7H), 6.80 (dd, J = 13.2, 7.2 Hz, 2H), 5.83 (dd, J = 12.0, 2.7 Hz, 1H), 5.01 (d, J = 12.0 Hz, 1H), 4.38 (s, 1H), 3.78–3.69 (m, 2H), 3.62 (d, J = 12.1 Hz, 1H), 3.58–3.51 (m, 1H), 2.86–2.81 (m, 1H), 2.61 (dd, J = 13.4, 4.3 Hz, 1H), 1.59 (s, 9H), 0.83 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ 175.83, 171.35, 163.55, 155.37, 148.61, 138.93, 135.87, 132.98, 131.03, 129.64, 128.88, 128.69, 127.57, 126.97, 126.39, 125.14, 124.71, 123.77, 119.42, 114.71, 113.72, 87.64, 84.43, 68.68, 60.63, 54.60, 51.89, 40.53, 38.13, 30.23, 27.00, 12.64. ESI-HRMS: calcd. for C34H34ClN2O9 [M − H]− 649.1953, found 649.1957.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-6-(3-methoxyphenyl)-5-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5h): 79% yield; [α] = +57.2 (c = 1.0 in CHCl3); 93% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.33 min, tminor = 7.09 min]; 1H NMR (500 MHz, CDCl3) δ 7.46 (t, J = 8.3 Hz, 2H), 7.23–7.10 (m, 4H), 6.98–6.80 (m, 3H), 6.57 (d, J = 8.2 Hz, 1H), 6.38 (s, 1H), 6.00 (d, J = 12.3 Hz, 1H), 4.38 (s, 1H), 4.14–4.07 (m, 1H), 3.92–3.81 (m, 2H), 3.65 (d, J = 11.9 Hz, 1H), 3.58–3.42 (m, 3H), 3.36 (t, J = 10.5 Hz, 1H), 3.04 (t, J = 12.8 Hz, 1H), 2.46 (dd, J = 13.6, 3.8 Hz, 1H), 1.55 (s, 8H), 0.90–0.84 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 174.69, 171.27, 153.67, 148.47, 139.52, 134.40, 131.73, 129.07, 128.81, 128.36, 127.68, 124.29, 123.79, 122.76, 121.70, 115.84, 114.88, 85.91, 84.23, 68.20, 61.08, 54.91, 54.60, 51.06, 46.65, 39.43, 29.38, 27.97, 13.62. ESI-HRMS: calcd. for C35H37N2O10 [M − H]− 645.2448, found 645.24544.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-6-(naphthalen-2-yl)-5-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5i): 72% yield; [α] = +109.8 (c = 1.0 in CHCl3); 97% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.93 min, tminor = 9.89 min]; 1H NMR (500 MHz, CDCl3) δ 7.68–7.51 (m, 3H), 7.40–7.27 (m, 3H), 7.24–7.09 (m, 4H), 6.96 (t, J = 7.4 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.77–6.56 (m, 1H), 6.30 (s, 1H), 6.21–6.13 (m, 1H), 4.44 (s, 1H), 4.30 (d, J = 12.2 Hz, 1H), 4.06 (s, 1H), 3.94–3.81 (m, 2H), 3.71 (d, J = 11.9 Hz, 1H), 3.43 (t, J = 10.5 Hz, 1H), 3.07 (t, J = 12.8 Hz, 1H), 2.49 (dd, J = 13.7, 4.1 Hz, 1H), 1.39 (s, 8H), 0.90–0.85 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 174.82, 171.26, 153.69, 148.26, 139.35, 132.73, 132.64, 131.75, 129.10, 128.38, 127.97, 127.52, 127.31, 126.08, 125.87, 124.38, 123.81, 122.82, 121.73, 115.87, 114.82, 86.18, 84.16, 68.26, 61.09, 54.77, 51.21, 41.95, 39.48, 29.42, 27.87, 13.62. ESI-HRMS: calcd. for C38H37N2O9 [M − H]− 665.2499, found 665.2494.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-6-(furan-2-yl)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2'-oxospiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5j): 81% yield; [α] = +42.7 (c = 1.0 in CHCl3); 97% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 9.20 min, tminor = 11.41 min]; 1H NMR (500 MHz, CDCl3) δ 7.57 (d, J = 7.7 Hz, 1H), 7.45–7.41 (m, 1H), 7.28–7.19 (m, 2H), 7.16–7.11 (m, 2H), 7.00 (d, J = 1.0 Hz, 1H), 6.93 (dd, J = 15.4, 8.0 Hz, 1H), 6.86–6.81 (m, 1H), 6.00 (dd, J = 3.2, 1.8 Hz, 1H), 5.89 (dd, J = 12.1, 2.9 Hz, 1H), 5.72 (d, J = 3.3 Hz, 1H), 4.37 (s, 1H), 4.30 (d, J = 12.1 Hz, 1H), 3.93–3.83 (m, 2H), 3.56 (d, J = 11.9 Hz, 1H), 3.33–3.24 (m, 1H), 3.05–2.97 (m, 1H), 2.42 (dd, J = 13.6, 4.4 Hz, 1H), 1.59 (s, 8H), 0.88 (q, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.40, 171.13, 153.59, 148.71, 148.24, 142.04, 139.57, 131.76, 129.19, 128.34, 127.73, 124.60, 123.76, 122.79, 121.72, 115.73, 114.86, 110.12, 108.17, 85.06, 84.31, 67.94, 61.12, 53.76, 50.91, 41.10, 39.40, 29.25, 28.05, 13.63. ESI-HRMS: calcd. for C32H33N2O10 [M − H]− 605.2135, found 605.2136.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-(thiophen-2-yl)spiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5k): 81% yield; [α] = +46.6 (c = 1.0 in CHCl3); 96% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.15 min, tminor = 8.72 min]; 1H NMR (500 MHz, CDCl3) δ 7.54–7.50 (m, 1H), 7.45 (dd, J = 8.3, 4.9 Hz, 1H), 7.24 (dd, J = 5.6, 3.4 Hz, 2H), 7.14 (t, J = 6.7 Hz, 2H), 6.93 (dd, J = 13.5, 6.2 Hz, 2H), 6.84 (d, J = 8.4 Hz, 1H), 6.71–6.65 (m, 2H), 5.93 (dd, J = 12.0, 2.7 Hz, 1H), 4.40–4.34 (m, 2H), 3.93–3.83 (m, 2H), 3.61 (d, J = 11.9 Hz, 1H), 3.35–3.27 (m, 1H), 3.05–2.98 (m, 1H), 2.43 (dd, J = 13.6, 4.4 Hz, 1H), 1.57 (s, 9H), 0.88 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.78, 171.19, 153.64, 148.55, 139.72, 135.62, 131.74, 129.32, 128.37, 127.84, 126.13, 125.63, 125.23, 124.68, 123.73, 122.71, 121.69, 115.76, 114.96, 87.16, 84.32, 68.11, 61.13, 54.99, 51.06, 42.04, 39.34, 29.29, 28.04, 13.63. ESI-HRMS: calcd. for C32H33N2O9S [M − H]− 621.1907, found 621.1909.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-2-(1-(tert-butoxycarbonyl)-1H-indol-3-yl)-4-hydroxy-5-(2-hydroxybenzyl)-3-nitro-2’-oxospiro[cyclohexane-1,3’-indoline]-1’,6-dicarboxylate (5l): 99% yield; [α] = +53.7 (c = 1.0 in CHCl3); 92% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.46 min, tminor = 8.30 min]; 1H NMR (500 MHz, CDCl3) δ 7.81 (s, 1H), 7.55 (d, J = 7.4 Hz, 1H), 7.49 (s, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 9.1 Hz, 1H), 7.15 (dd, J = 12.1, 7.6 Hz, 2H), 7.09 (t, J = 7.5 Hz, 2H), 7.04–6.92 (m, 3H), 6.83 (d, J = 7.9 Hz, 1H), 6.31 (s, 1H), 5.90 (d, J = 12.2 Hz, 1H), 4.52 (d, J = 12.2 Hz, 1H), 4.36 (s, 1H), 4.06 (d, J = 6.9 Hz, 1H), 3.92–3.81 (m, 2H), 3.68 (d, J = 11.9 Hz, 1H), 3.33 (t, J = 9.9 Hz, 1H), 3.06 (t, J = 12.8 Hz, 1H), 2.45 (dd, J = 13.6, 3.9 Hz, 1H), 1.60 (d, J = 2.5 Hz, 15H), 0.88 (t, J = 7.1 Hz, 4H). 13C NMR (125 MHz, CDCl3) δ 175.29, 171.20, 153.59, 149.22, 148.69, 139.31, 131.75, 130.00, 129.19, 128.37, 127.50, 124.36, 124.31, 123.79, 123.30, 122.86, 122.06, 121.73, 118.65, 115.79, 114.87, 114.75, 114.58, 87.64, 84.34, 83.95, 68.01, 61.10, 54.67, 51.58, 39.42, 36.67, 29.27, 28.10, 28.01, 13.61. ESI-HRMS: calcd. for C41H44N3O11 [M − H]− 754.2976, found 754.2977.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-5’-bromo-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5m): 77% yield; [α] = +33.8 (c = 1.0 in CHCl3); 96% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.27 min, tminor = 10.20 min]; 1H NMR (500 MHz, CDCl3) δ 7.61 (s, 1H), 7.31 (q, J = 8.7 Hz, 2H), 7.16 (t, J = 7.2 Hz, 2H), 7.08–6.93 (m, 4H), 6.84 (d, J = 8.2 Hz, 1H), 6.30 (s, 1H), 5.97 (dd, J = 12.2, 2.0 Hz, 1H), 4.38 (s, 1H), 4.08 (d, J = 12.1 Hz, 1H), 3.99–3.84 (m, 2H), 3.61 (d, J = 11.9 Hz, 1H), 3.33 (dd, J = 11.6, 9.2 Hz, 1H), 3.03 (dd, J = 24.0, 11.7 Hz, 1H), 2.47 (dd, J = 13.6, 3.9 Hz, 1H), 1.55 (s, 8H), 0.94–0.86 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 173.91, 171.20, 153.61, 148.19, 138.51, 132.60, 132.01, 131.76, 129.89, 128.44, 128.09, 128.07, 125.88, 123.67, 121.76, 85.70, 84.59, 68.08, 61.21, 54.72, 50.82, 46.58, 39.49, 29.36, 28.01, 13.69. ESI-HRMS: calcd. for C34H34BrN2O9 [M − H]− 693.1448, found 693.1446.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-5’-chloro-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5n): 82% yield; [α] = +103.8 (c = 1.0 in CHCl3); 98% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.99 min, tminor = 11.37 min]; 1H NMR (500 MHz, CDCl3) δ 7.48 (d, J = 1.7 Hz, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.16 (t, J = 8.0 Hz, 3H), 7.08–6.92 (m, 4H), 6.84 (d, J = 7.8 Hz, 1H), 6.29 (s, 1H), 5.98 (dd, J = 12.2, 2.2 Hz, 1H), 4.37 (d, J = 13.9 Hz, 1H), 4.09 (d, J = 12.5 Hz, 2H), 3.97–3.85 (m, 2H), 3.62 (d, J = 11.9 Hz, 1H), 3.38–3.30 (m, 1H), 3.04 (t, J = 12.8 Hz, 1H), 1.55 (s, 8H), 0.92 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.05, 171.18, 153.62, 137.99, 132.63, 131.75, 129.88, 129.56, 129.09, 128.43, 128.07, 123.67, 123.02, 121.75, 116.11, 115.82, 85.73, 84.57, 68.09, 61.20, 54.77, 50.83, 46.57, 39.49, 29.37, 28.02, 13.68. ESI-HRMS: calcd. for C34H34ClN2O9 [M − H]− 649.1953, found 649.1957.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-5’-methoxy-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5o): 65% yield; [α] = +88.8 (c = 1.0 in CHCl3); 92% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.45 min, tminor = 15.19 min]; 1H NMR (500 MHz, CDCl3) δ 7.26 (d, J = 2.5 Hz, 1H), 7.15 (t, J = 8.1 Hz, 2H), 7.05–6.91 (m, 5H), 6.85 (d, J = 7.8 Hz, 1H), 6.38 (s, 1H), 6.02 (dd, J = 12.3, 2.2 Hz, 1H), 4.38 (s, 1H), 4.11–4.04 (m, 2H), 3.64 (d, J = 11.9 Hz, 1H), 3.40–3.30 (m, 1H), 3.04 (t, J = 12.8 Hz, 1H), 2.47 (dd, J = 13.7, 4.2 Hz, 1H), 2.39 (s, 3H), 1.54 (s, 7H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.90, 171.25, 153.73, 148.46, 137.00, 134.04, 132.94, 131.74, 129.53, 128.36, 127.85, 127.45, 123.81, 123.24, 121.68, 115.87, 114.56, 85.98, 84.00, 68.18, 61.02, 54.66, 51.05, 46.68, 39.41, 29.42, 28.05, 21.12, 13.61.. ESI-HRMS: calcd. for C35H37N2O10 [M − H]− 645.2448, found 645.2448.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-5’-fluoro-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5p): 65% yield; [α] = +47.8 (c = 1.0 in CHCl3); 94% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 5.31 min, tminor = 11.33 min]; 1H NMR (500 MHz, CDCl3) δ 7.41 (dd, J = 8.9, 4.4 Hz, 1H), 7.21 (dd, J = 7.5, 2.3 Hz, 1H), 7.16 (t, J = 7.4 Hz, 2H), 7.07–6.93 (m, 4H), 6.90–6.82 (m, 2H), 6.30 (s, 1H), 5.99 (dd, J = 12.2, 2.4 Hz, 1H), 4.38 (s, 1H), 4.08 (dd, J = 12.5, 5.1 Hz, 2H), 3.96–3.85 (m, 2H), 3.60 (d, J = 11.9 Hz, 1H), 3.35 (td, J = 11.9, 2.8 Hz, 1H), 3.08–3.00 (m, 1H), 2.46 (dd, J = 13.7, 4.3 Hz, 1H), 1.55 (s, 8H), 0.92 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.80, 171.22, 153.59, 147.88, 144.77, 144.37, 132.40, 131.76, 129.50, 128.49, 128.32, 128.23, 125.37, 123.58, 121.80, 118.39, 115.69, 115.00, 85.50, 68.04, 65.90, 61.40, 54.68, 50.75, 46.46, 39.67, 29.37, 27.98, 13.77. ESI-HRMS: calcd. for C34H34FN2O9 [M − H]− 633.2248, found 633.2243.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-5,5’-dinitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5q): 39% yield; [α] = +136.5 (c = 1.0 in CHCl3); 98% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 6.39 min, tminor = 15.73 min]; 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 2.0 Hz, 1H), 8.12 (dd, J = 9.0, 2.1 Hz, 1H), 7.61 (d, J = 9.0 Hz, 1H), 7.17 (t, J = 7.5 Hz, 2H), 7.10–6.93 (m, 4H), 6.84 (d, J = 7.9 Hz, 1H), 5.94 (dd, J = 12.2, 2.5 Hz, 1H), 4.41 (s, 1H), 4.18 (d, J = 12.2 Hz, 2H), 3.90 (q, J = 7.1 Hz, 2H), 3.73 (t, J = 11.1 Hz, 1H), 3.34 (td, J = 11.8, 2.7 Hz, 1H), 3.07 (t, J = 12.8 Hz, 1H), 2.48 (dd, J = 13.6, 4.3 Hz, 1H), 1.58 (s, 9H), 0.91 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.34, 171.16, 160.75, 158.81, 153.62, 148.35, 135.39, 132.68, 131.75, 129.57, 129.51, 128.42, 128.07, 128.03, 123.68, 121.76, 116.32, 116.25, 115.82, 115.76, 115.58, 110.37, 110.17, 85.75, 84.43, 68.09, 61.21, 54.90, 50.90, 46.60, 39.51, 29.35, 28.03, 13.68. ESI-HRMS: calcd. for C34H34N3O11 [M − H]− 660.2193, found 660.2195.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-7’-bromo-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5r): 71% yield; [α] = +21.3 (c = 1.0 in CHCl3); 98% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.60 min, tminor = 10.12 min]; 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.4 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.16 (t, J = 6.9 Hz, 2H), 7.10–6.93 (m, 5H), 6.83 (d, J = 8.2 Hz, 1H), 6.09 (s, 1H), 6.04 (dd, J = 12.2, 2.2 Hz, 1H), 4.37 (s, 1H), 4.14 (d, J = 12.2 Hz, 1H), 4.01–3.85 (m, 3H), 3.58 (d, J = 11.9 Hz, 1H), 3.27 (dd, J = 11.9, 9.4 Hz, 1H), 3.01 (t, J = 12.8 Hz, 1H), 2.44 (dd, J = 13.7, 4.1 Hz, 1H), 1.56 (s, 8H), 0.96–0.90 (m, 3H).13C NMR (125 MHz, CDCl3) δ 174.54, 170.50, 153.60, 150.83, 146.72, 138.28, 133.79, 132.48, 131.74, 131.16, 128.42, 128.14, 125.25, 123.65, 122.14, 121.77, 115.87, 106.21, 85.77, 85.15, 68.00, 61.40, 55.68, 51.28, 46.38, 39.38, 29.27, 27.66, 13.61. ESI-HRMS: calcd. for C34H34BrN2O9 [M − H]− 693.1448, found 693.1449.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-3-(5-fluoro-2-hydroxybenzyl)-4-hydroxy-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5s): 81% yield; [α] = +51.2 (c = 1.0 in CHCl3); 97% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.53 min, tminor = 7.64 min]; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 7.1 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.23–7.14 (m, 2H), 7.06–6.77 (m, 7H), 6.65 (s, 1H), 6.05 (dd, J = 12.3, 2.0 Hz, 1H), 4.39 (s, 1H), 4.14–4.04 (m, 2H), 3.92–3.81 (m, 2H), 3.65 (d, J = 11.9 Hz, 1H), 3.41–3.32 (m, 1H), 3.02 (t, J = 12.7 Hz, 1H), 2.43 (dd, J = 13.6, 4.2 Hz, 1H), 1.55 (s, 8H), 0.86 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.79, 171.22, 158.28, 156.37, 149.92, 149.91, 148.40, 139.33, 132.77, 129.10, 127.95, 127.90, 127.44, 125.52, 125.46, 124.42, 122.79, 117.78, 117.60, 116.86, 116.79, 114.79, 114.60, 85.91, 84.34, 68.16, 61.19, 54.63, 50.94, 46.68, 39.20, 29.67, 28.03, 13.58. ESI-HRMS: calcd. for C34H34FN2O9 [M − H]− 633.2248, found 633.2245.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-3-(4-chloro-2-hydroxybenzyl)-4-hydroxy-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5t): 63% yield; [α] = +33.9 (c = 1.0 in CHCl3); 92% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.36 min, tminor = 7.11 min]; 1H NMR (500 MHz, CDCl3) δ 7.47 (t, J = 8.8 Hz, 1H), 7.38 (d, J = 7.9 Hz, 1H), 7.23–7.15 (m, 2H), 7.10–6.85 (m, 7H), 6.06–6.00 (m, 1H), 4.38 (d, J = 21.5 Hz, 1H), 4.16–4.03 (m, 2H), 3.93–3.79 (m, 2H), 3.65 (d, J = 11.9 Hz, 1H), 3.36 (td, J = 11.8, 2.8 Hz, 1H), 3.06–2.96 (m, 1H), 2.45 (dd, J = 13.7, 4.3 Hz, 1H), 1.55 (s, 8H), 0.86 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 175.04, 171.39, 154.83, 148.37, 139.26, 133.16, 132.83, 132.43, 129.11, 127.93, 127.48, 124.49, 122.85, 122.63, 121.45, 116.27, 114.80, 85.92, 84.48, 68.18, 61.22, 54.70, 50.99, 46.65, 39.20, 29.14, 28.02, 13.59. ESI-HRMS: calcd. for C34H34ClN2O9 [M − H]− 649.1953, found 649.1951.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxy-5-methoxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5u): 73% yield; [α] = +71.4 (c = 1.0 in CHCl3); 99% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 6.91 min, tminor = 13.76 min]; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 7.2 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.22–7.13 (m, 2H), 7.06–6.88 (m, 4H), 6.78 (d, J = 9.2 Hz, 1H), 6.70 (d, J = 7.0 Hz, 2H), 6.12 (s, 1H), 6.02 (dt, J = 11.8, 5.9 Hz, 1H), 4.39 (s, 1H), 4.20 (s, 1H), 4.11 (d, J = 12.3 Hz, 1H), 3.91–3.83 (m, 2H), 3.79 (s, 3H), 3.65 (d, J = 11.9 Hz, 1H), 3.37 (td, J = 11.7, 3.4 Hz, 1H), 3.03 (t, J = 12.8 Hz, 1H), 2.42 (dd, J = 13.6, 4.4 Hz, 1H), 1.56 (s, 8H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.79, 171.30, 154.30, 148.41, 147.48, 139.40, 132.94, 129.04, 127.88, 127.55, 124.74, 124.35, 122.76, 116.69, 116.28, 114.77, 113.92, 85.89, 84.17, 68.24, 61.06, 55.83, 54.63, 51.00, 46.64, 39.45, 29.72, 28.04, 13.63.ESI-HRMS: calcd. for C35H37N2O10 [M − H]− 645.2448, found 645.2450.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxy-5-methylbenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5v): 77% yield; [α] = +71.8 (c = 1.0 in CHCl3); 99% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 4.77 min, tminor = 8.59 min]; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 7.2 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.23–7.13 (m, 2H), 7.06–6.86 (m, 5H), 6.73 (d, J = 8.0 Hz, 1H), 6.10 (s, 1H), 6.03 (dd, J = 12.3, 2.2 Hz, 1H), 4.38 (s, 1H), 4.11 (d, J = 12.4 Hz, 2H), 3.91–3.84 (m, 2H), 3.64 (d, J = 11.9 Hz, 1H), 3.38–3.29 (m, 1H), 3.01 (t, J = 12.8 Hz, 1H), 2.40 (dd, J = 13.6, 4.2 Hz, 1H), 2.28 (s, 3H), 1.56 (s, 8H), 0.87 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.79, 171.30, 154.30, 148.41, 147.48, 139.40, 132.94, 129.04, 127.88, 127.55, 124.74, 124.35, 122.76, 116.69, 116.28, 114.77, 113.92, 85.89, 84.17, 68.24, 61.06, 55.83, 54.63, 51.00, 46.64, 39.45, 29.72, 28.04, 13.63. ESI-HRMS: calcd. for C35H37N2O9 [M − H]− 629.2499, found 629.2495.
di-tert-butyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-hydroxybenzyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3’-indoline]-1’,2-dicarboxylate (5w): 73% yield; [α] = +48.8 (c = 1.0 in CHCl3); 98% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 3.84 min, tminor = 8.05 min]; 1H NMR (500 MHz, CDCl3) δ 7.49 (d, J = 7.2 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.24–7.12 (m, 4H), 7.04–6.91 (m, 4H), 6.86–6.80 (m, 2H), 6.03 (dd, J = 12.3, 2.5 Hz, 1H), 4.38 (s, 1H), 4.18–4.09 (m, 2H), 3.51 (d, J = 11.9 Hz, 1H), 3.35–3.27 (m, 1H), 3.04 (t, J = 12.8 Hz, 1H), 2.49 (dd, J = 13.7, 4.0 Hz, 1H), 1.55 (s, 9H), 1.11 (d, J = 8.4 Hz, 9H). 13C NMR (125 MHz, CDCl3) δ 174.65, 170.20, 153.86, 148.49, 139.42, 133.09, 131.72, 128.93, 128.29, 127.83, 127.72, 124.31, 124.01, 123.03, 121.53, 115.82, 114.65, 85.97, 84.06, 81.98, 68.16, 54.74, 51.92, 46.67, 39.50, 29.29, 28.04, 27.30. ESI-HRMS: calcd. for C36H39N2O9 [M − H]− 643.2656, found 643.2651.
1’-(tert-butyl)2-ethyl(1S,2R,3R,4R,5S,6R)-4-hydroxy-3-(2-((4-methylphenyl)sulfonamido)ethyl)-5-nitro-2’-oxo-6-phenylspiro[cyclohexane-1,3'-indoline]-1’,2-dicarboxylate (5x): 47% yield; [α] = +32.6 (c = 1.0 in CHCl3); 93% ee, determined by chiral HPLC analysis [Daicel Chiralcel IC, n-hexane/i-PrOH = 85/15, 1.0 mL/min, λ = 201 nm, tmajor = 11.21 min, tminor = 8.98 min]; 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.2 Hz, 2H), 7.43–7.40 (m, 1H), 7.38–7.30 (m, 3H), 7.19–7.11 (m, 2H), 7.07–6.92 (m, 3H), 6.07 (dd, J = 12.3, 2.2 Hz, 1H), 4.89 (t, J = 6.0 Hz, 1H), 4.58 (s, 1H), 4.05 (d, J = 12.3 Hz, 1H), 3.80–3.66 (m, 2H), 3.49 (d, J = 12.0 Hz, 1H), 3.29 (d, J = 3.0 Hz, 1H), 3.16 (t, J = 10.0 Hz, 1H), 3.10–2.97 (m, 2H), 2.43 (s, 3H), 1.97–1.81 (m, 1H), 1.55 (s, 8H), 0.76 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.76, 170.94, 148.31, 143.70, 139.27, 136.32, 132.56, 129.86, 129.04, 128.00, 127.89, 127.28, 127.18, 124.33, 122.83, 114.73, 86.40, 84.19, 68.15, 61.04, 54.40, 50.49, 46.67, 40.41, 35.37, 29.04, 28.04, 21.53, 13.46. ESI-HRMS: calcd. for C36H40N3O10S [M − H]− 706.2434, found 706.2437.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}