
Structural and Biochemical Characterization of a Cyanobacterial PP2C Phosphatase Reveals Insights into Catalytic Mechanism and Substrate Recognition


Abstract
:
1. Introduction
2. Results and Discussion
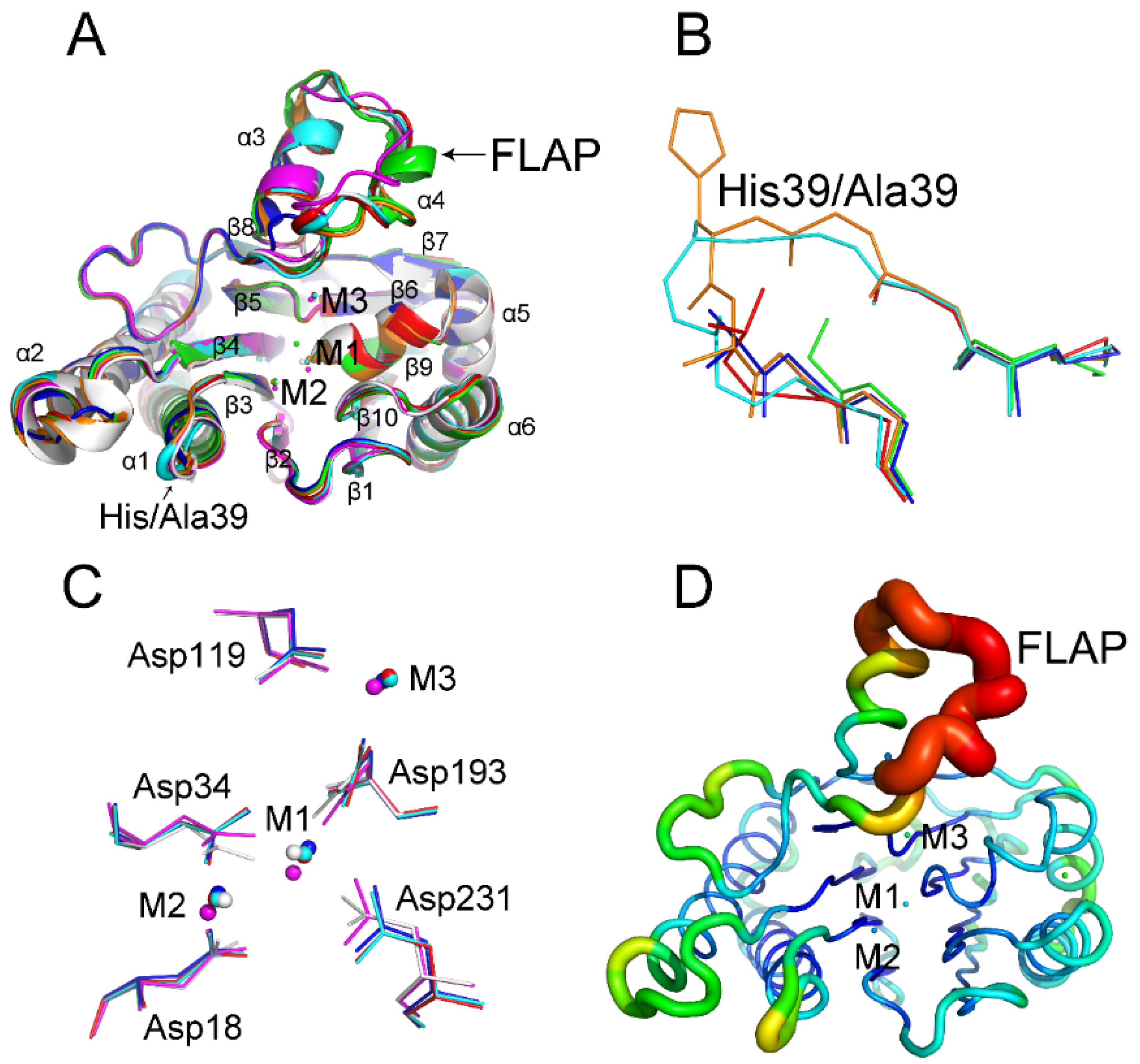
2.1. His39 Determines tPphA Substrate Specificity
2.2. New Insights into the Catalytic Center of tPphA
2.3. Relationship between the FLAP Subdomain and M3
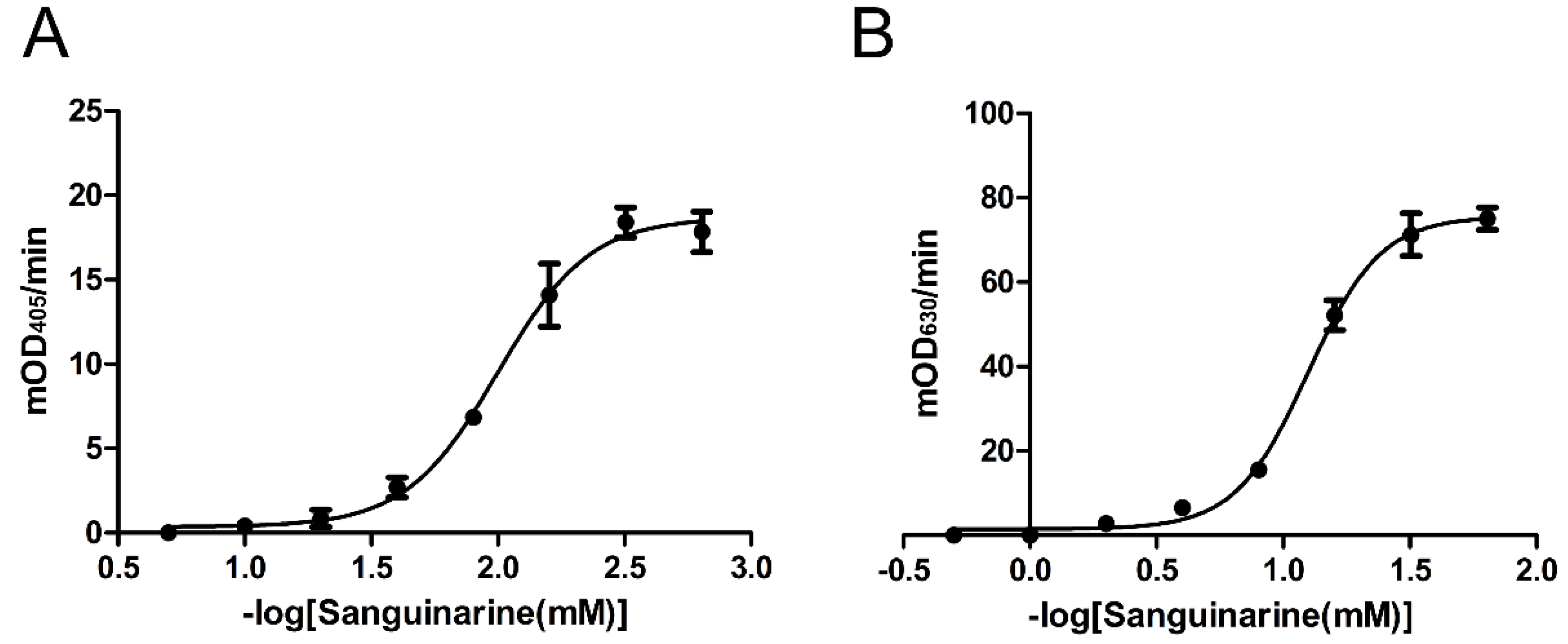
2.4. Inhibition Effects of Nine Chemicals on tPphA Activity
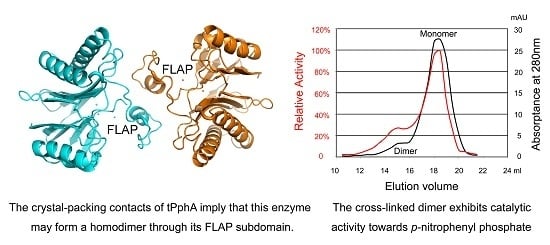
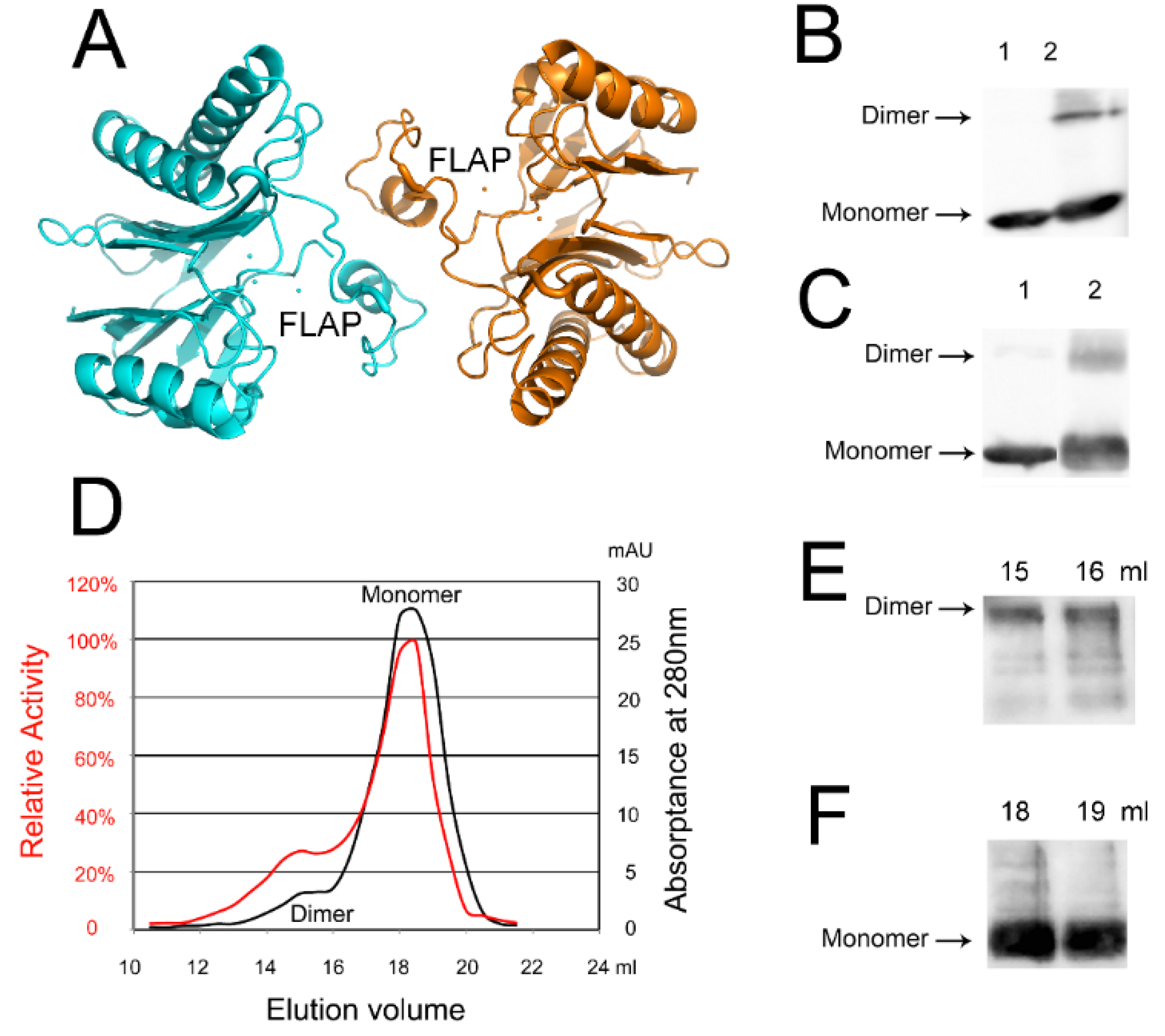
2.5. An Active tPphA Dimer
3. Experimental Section
3.1. Overexpression and Purification of PP2C Phosphatases
3.2. H39A Crystallization, X-ray Data Collection and Structure Determination
3.3. pNPP Assay
3.4. Casein Assay
3.5. tPphA Cross-Linking Reaction
3.6. Size Exclusion Chromatography
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Woude, G.F.V.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. PNAS 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed]
- Schweighofer, A.; Hirt, H.; Meskiene, I. Plant PP2C phosphatases: emerging functions in stress signaling. Trends Plant Sci. 2004, 9, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Arino, J.; Casamayor, A.; Gonzalez, A. Type 2C protein phosphatases in fungi. Eukaryot. cell 2011, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Irmler, A.; Forchhammer, K. A PP2C-type phosphatase dephosphorylates the PII signaling protein in the cyanobacterium Synechocystis PCC 6803. PNAS 2001, 98, 12978–12983. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Forchhammer, K. Determinants for substrate specificity of the bacterial PP2C protein phosphatase tPphA from Thermosynechococcus elongatus. FEBS J. 2013, 280, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Melcher, K.; Ng, L.M.; Zhou, X.E.; Soon, F.F.; Xu, Y.; Suino-Powell, K.M.; Park, S.Y.; Weiner, J.J.; Fujii, H.; Chinnusamy, V.; et al. A gate-latch-lock mechanism for hormone signalling by abscisic acid receptors. Nature 2009, 462, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Pullen, K.E.; Ng, H.L.; Sung, P.Y.; Good, M.C.; Smith, S.M.; Alber, T. An alternate conformation and a third metal in PstP/Ppp, the M. tuberculosis PP2C-Family Ser/Thr protein phosphatase. Structure 2004, 12, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Wehenkel, A.; Bellinzoni, M.; Schaeffer, F.; Villarino, A.; Alzari, P.M. Structural and binding studies of the three-metal center in two mycobacterial PPM Ser/Thr protein phosphatases. J. Mol. Boil. 2007, 374, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Rantanen, M.K.; Lehtio, L.; Rajagopal, L.; Rubens, C.E.; Goldman, A. Structure of Streptococcus agalactiae serine/threonine phosphatase. The subdomain conformation is coupled to the binding of a third metal ion. FEBS J. 2007, 274, 3128–3137. [Google Scholar] [CrossRef] [PubMed]
- Bellinzoni, M.; Wehenkel, A.; Shepard, W.; Alzari, P.M. Insights into the catalytic mechanism of PPM Ser/Thr phosphatases from the atomic resolution structures of a mycobacterial enzyme. Structure 2007, 15, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, C.; Fokina, O.; Kloft, N.; Grune, T.; Becker, S.; Sheldrick, G.M.; Forchhammer, K. Structural analysis of the PP2C phosphatase tPphA from Thermosynechococcus elongatus: A flexible flap subdomain controls access to the catalytic site. J. Mol. Boil. 2008, 376, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Schlicker, C.; Forchhammer, K. A third metal is required for catalytic activity of the signal-transducing protein phosphatase M tPphA. J. Boil. Chem. 2011, 286, 13481–13488. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Boil. 2014, 10, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Tanoue, K.; Durell, S.R.; Chatterjee, D.K.; Jenkins, L.M.; Appella, D.H.; Appella, E. Optimization of a cyclic peptide inhibitor of Ser/Thr phosphatase PPM1D (Wip1). Biochemistry 2011, 50, 4537–4549. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nguyen, T.A.; Moon, S.H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Cha, H.; Lee, M.O.; Mazur, S.J.; Appella, E.; Fornace, A.J., Jr. Regulation of the Wip1 phosphatase and its effects on the stress response. Front. Biosci. 2012, 17, 1480–1498. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Bulavin, D.V. Wip1-dependent signaling pathways in health and diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 307–325. [Google Scholar] [PubMed]
- Cao, M.; Liu, X.; Zhang, Y.; Xue, X.; Zhou, X.E.; Melcher, K.; Gao, P.; Wang, F.; Zeng, L.; Zhao, Y.; et al. An ABA-mimicking ligand that reduces water loss and promotes drought resistance in plants. Cell Res. 2013, 23, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.; Merlot, S.; Giraudat, J. The Arabidopsis ABSCISIC ACID-INSENSITIVE2 (ABI2) and ABI1 genes encode homologous protein phosphatases 2C involved in abscisic acid signal transduction. Plant Cell 1997, 9, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Leube, M.P.; Grill, E.; Amrhein, N. ABI1 of Arabidopsis is a protein serine/threonine phosphatase highly regulated by the proton and magnesium ion concentration. FEBS Lett. 1998, 424, 100–104. [Google Scholar] [CrossRef]
- Gosti, F.; Beaudoin, N.; Serizet, C.; Webb, A.A.; Vartanian, N.; Giraudat, J. ABI1 protein phosphatase 2C is a negative regulator of abscisic acid signaling. Plant Cell 1999, 11, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Vlad, F.; Rubio, S.; Rodrigues, A.; Sirichandra, C.; Belin, C.; Robert, N.; Leung, J.; Rodriguez, P.L.; Lauriere, C.; Merlot, S. Protein phosphatases 2C regulate the activation of the Snf1-related kinase OST1 by abscisic acid in Arabidopsis. Plant Cell 2009, 21, 3170–3184. [Google Scholar] [CrossRef] [PubMed]
- Aburai, N.; Yoshida, M.; Ohnishi, M.; Kimura, K. Sanguinarine as a potent and specific inhibitor of protein phosphatase 2C in vitro and induces apoptosis via phosphorylation of p38 in HL60 cells. Biosci. Biotechnol. Biochem. 2010, 74, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, P.; Hallock, S.; Brown, R.E.; Loose, D.S.; Buja, L.M. A model for cardiomyocyte cell death: Insights into mechanisms of oncosis. Exp. Mol. Pathol. 2013, 94, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Adhami, V.M.; Aziz, M.H.; Mukhtar, H.; Ahmad, N. Activation of prodeath Bcl-2 family proteins and mitochondrial apoptosis pathway by sanguinarine in immortalized human HaCaT keratinocytes. Clin. Cancer Res. 2003, 9, 3176–3182. [Google Scholar]
- Sun, M.; Lou, W.; Chun, J.Y.; Cho, D.S.; Nadiminty, N.; Evans, C.P.; Chen, J.; Yue, J.; Zhou, Q.; Gao, A.C. Sanguinarine suppresses prostate tumor growth and inhibits survivin expression. Genes Cancer 2010, 1, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Holy, J.; Lamont, G.; Perkins, E. Disruption of nucleocytoplasmic trafficking of cyclin D1 and topoisomerase II by sanguinarine. BMC Cell Boil. 2006, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Malikova, J.; Zdarilova, A.; Hlobilkova, A.; Ulrichova, J. The effect of chelerythrine on cell growth, apoptosis, and cell cycle in human normal and cancer cells in comparison with sanguinarine. Cell Boil. Toxicol. 2006, 22, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Pica, F.; Balestrieri, E.; Serafino, A.; Sorrentino, R.; Gaziano, R.; Moroni, G.; Moroni, N.; Palmieri, G.; Mattei, M.; Garaci, E. Antitumor effects of the benzophenanthridine alkaloid sanguinarine in a rat syngeneic model of colorectal cancer. Anti-Cancer Drugs 2012, 23, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, S.; Selke, D.; Fischer, D.; Baumann, A.; Muller, F.; Thanos, S. Protein phosphatase type-2C isozymes present in vertebrate retinae: purification, characterization, and localization in photoreceptors. J. Neurosci. Res. 1998, 51, 328–338. [Google Scholar] [CrossRef]
- Klumpp, S.; Selke, D.; Hermesmeier, J. Protein phosphatase type 2C active at physiological Mg2+: Stimulation by unsaturated fatty acids. FEBS Lett. 1998, 437, 229–232. [Google Scholar] [CrossRef]
- Wang, Y.; Santini, F.; Qin, K.; Huang, C.Y. A Mg2+-dependent, Ca2+-inhibitable serine/threonine protein phosphatase from bovine brain. J. Biol. Chem. 1995, 270, 25607–25612. [Google Scholar] [CrossRef] [PubMed]
- Signorile, A.; Sardanelli, A.M.; Nuzzi, R.; Papa, S. Serine (threonine) phosphatase(s) acting on cAMP-dependent phosphoproteins in mammalian mitochondria. FEBS Lett. 2002, 512, 91–94. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kapatral, V.; Xu, W.; Chakrabarty, A.M. Characterization of a Hank's type serine/threonine kinase and serine/threonine phosphoprotein phosphatase in Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6615–6622. [Google Scholar] [PubMed]
- Takezawa, D. Characterization of a novel plant PP2C-like protein Ser/Thr phosphatase as a calmodulin-binding protein. J. Biol. Chem. 2003, 278, 38076–38083. [Google Scholar] [CrossRef] [PubMed]
- Bevilaqua, L.R.; Cammarota, M.; Dickson, P.W.; Sim, A.T.; Dunkley, P.R. Role of protein phosphatase 2C from bovine adrenal chromaffin cells in the dephosphorylation of phospho-serine 40 tyrosine hydroxylase. J. Neurochem. 2003, 85, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Newton, A.C. Biochemical characterization of the phosphatase domain of the tumor suppressor PH domain leucine-rich repeat protein phosphatase. Biochemistry 2014, 53, 3971–3981. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Noguchi, Y. Substrate-dependent metal preference of PPM1H, a cancer-associated protein phosphatase 2C: Comparison with other family members. Biometals 2009, 22, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Marley, A.E.; Sullivan, J.E.; Carling, D.; Abbott, W.M.; Smith, G.J.; Taylor, I.W.; Carey, F.; Beri, R.K. Biochemical characterization and deletion analysis of recombinant human protein phosphatase 2C alpha. Biochem. J. 1996, 320, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Schenk, G.; Elliott, T.W.; Leung, E.; Carrington, L.E.; Mitic, N.; Gahan, L.R.; Guddat, L.W. Crystal structures of a purple acid phosphatase, representing different steps of this enzyme's catalytic cycle. BMC Struct. Boil. 2008, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cho, H.S.; Kim, R.; Jancarik, J.; Yokota, H.; Nguyen, H.H.; Grigoriev, I.V.; Wemmer, D.E.; Kim, S.H. Structural characterization of the reaction pathway in phosphoserine phosphatase: Crystallographic "snapshots" of intermediate states. J. Mol. Boil. 2002, 319, 421–431. [Google Scholar] [CrossRef]
- Lopez-Gallego, F.; Guisan, J.M.; Betancor, L. Glutaraldehyde-mediated protein immobilization. Methods Mol. Boil. 2013, 1051, 33–41. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Potterton, E.; Briggs, P.; Turkenburg, M.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr.rd 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.7.2. Schrödinger LLC, 2015. Available online: www.pymol.org (accessed on 25 April 2016).
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P.; Mannaerts, G.P. Inorganic and organic phosphate measurements in the nanomolar range. Anal. Biochem. 1987, 161, 45–48. [Google Scholar] [CrossRef]
- Ekman, P.; Jager, O. Quantification of subnanomolar amounts of phosphate bound to seryl and threonyl residues in phosphoproteins using alkaline hydrolysis and malachite green. Anal. Biochem. 1993, 214, 138–141. [Google Scholar] [CrossRef] [PubMed]
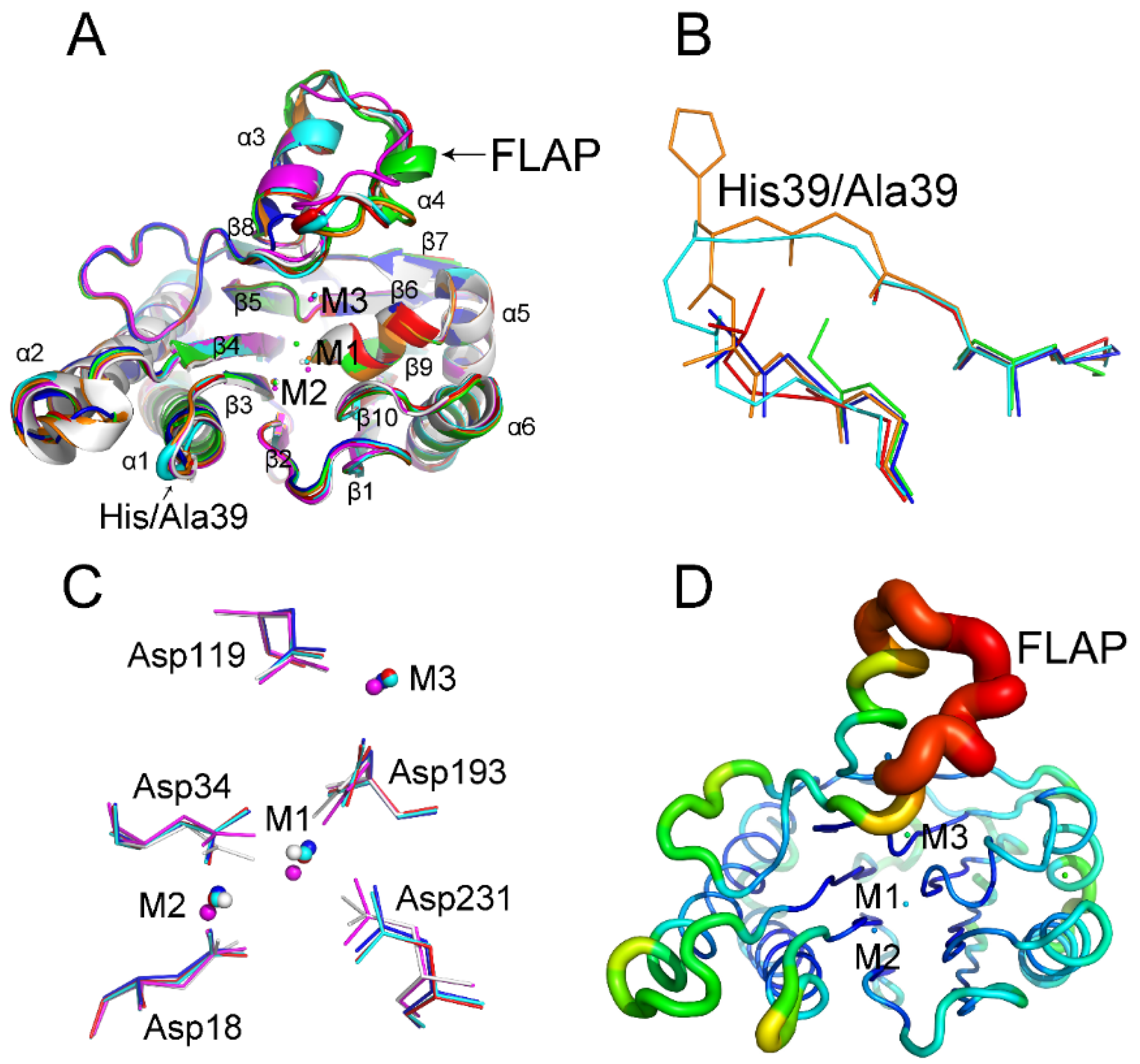
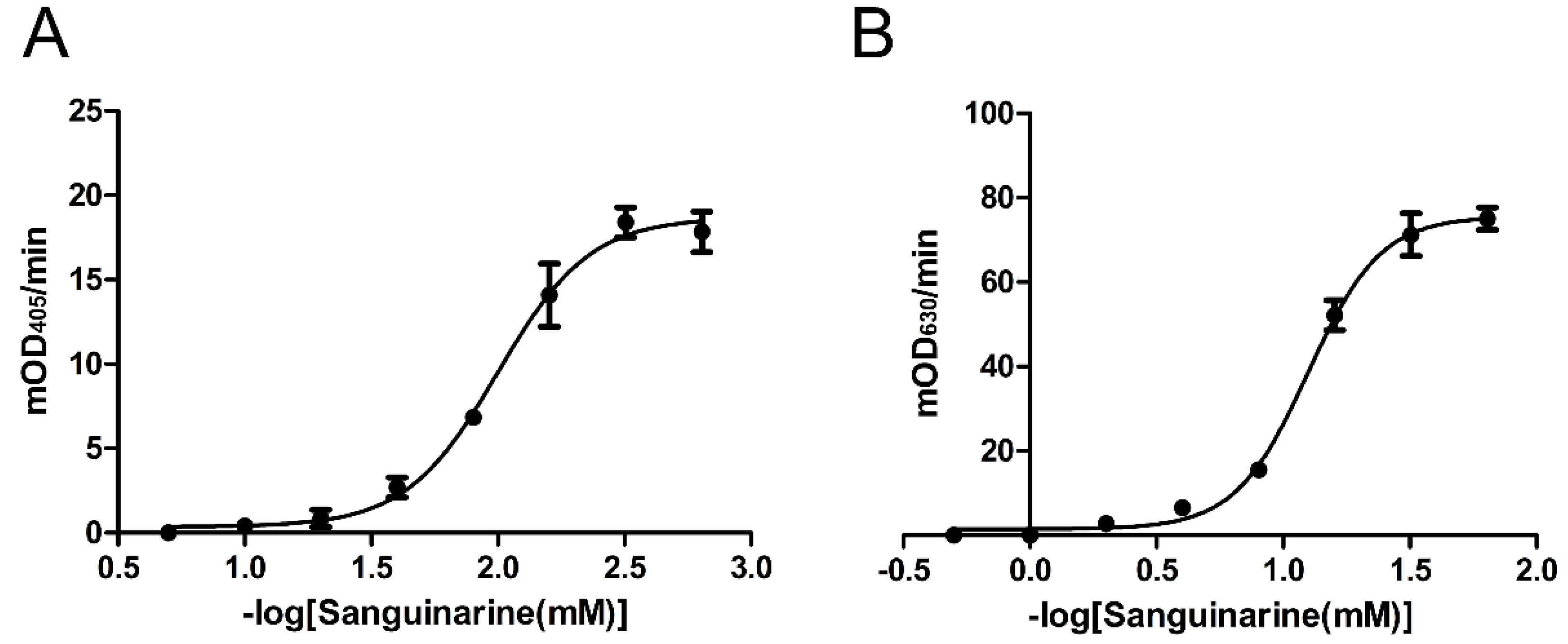
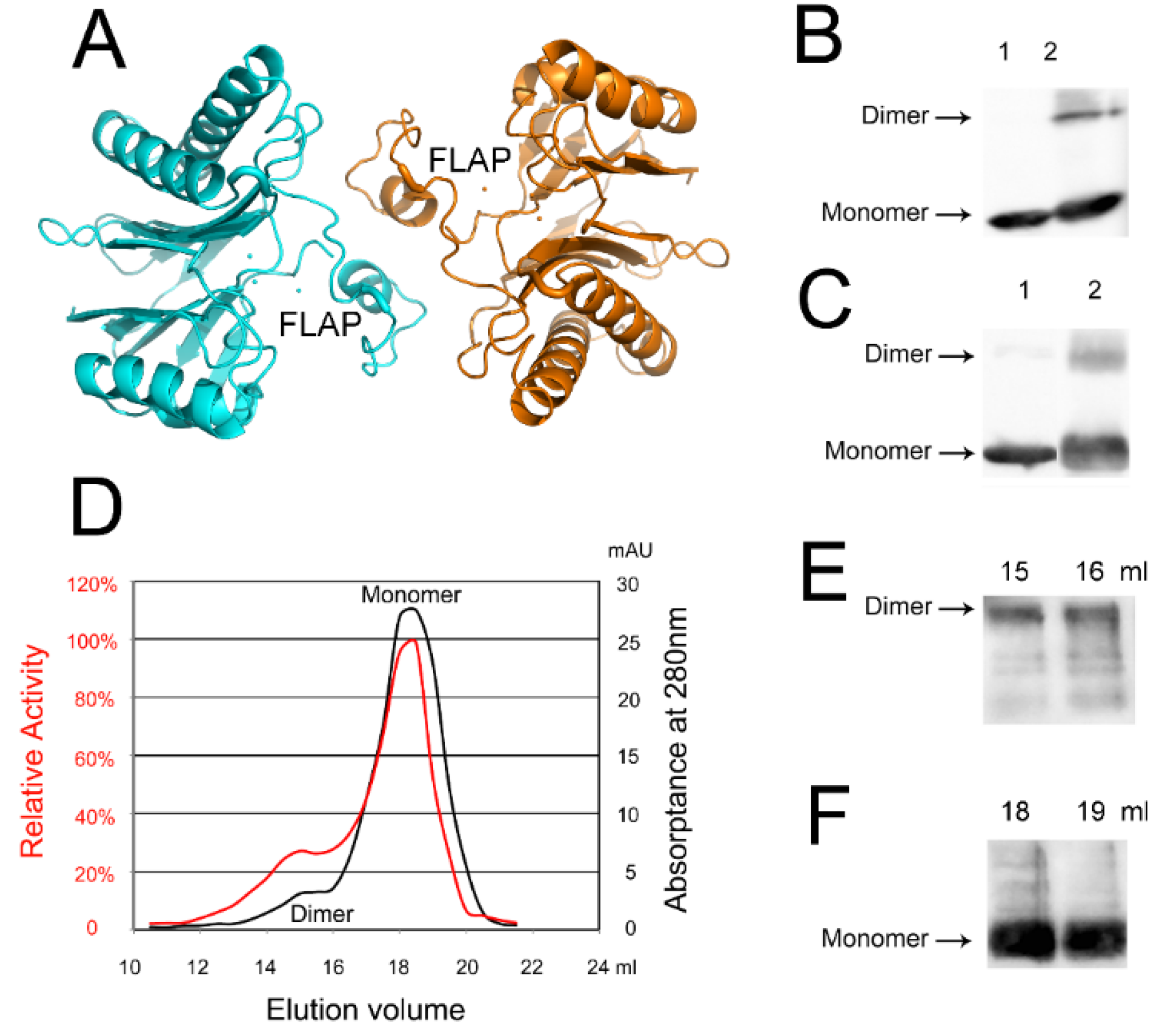

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | PDB Number | |
|---|---|---|
| 5D2U | 5ITI | |
| Wavelength (Å) | 0.9794 | 0.9794 |
| Resolution (Å) | 19.23–1.8 (1.84–1.8) | 19.23–1.95 (2.0–1.95) |
| Space group | C2221 | C2221 |
| Unit cell parameters (a, b, c) (Å) | 38.04 153.87 82.76 | 38.01 153.82 82.93 |
| No. of measured reflections | 144931 (8901) | 116108 (8160) |
| No. of unique reflections | 23018 (1344) | 18220 (1267) |
| Completeness (%) | 99.9 (100.0) | 99.9 (100.0) |
| Multiplicity | 6.3 (6.6) | 6.4 (6.3) |
| Rmerge (%) b | 7.0 (97.1) | 6.3 (79.6) |
| Rpim (%) c | 3.2 (43.0) | 2.9 (36.6) |
| <I/δ(I)> | 14.4 (2.1) | 15.7 (2.5) |
| Refine | ||
| Resolution limits (Å) | 19.23–1.8 | 19.23–1.95 |
| Rmodel (%) d | 21.3 | 21.5 |
| Rfree (%) e | 24.8 | 26.8 |
| No. of waters | 112 | 87 |
| Rmsd bond lengths (Å) | 0.0085 | 0.0077 |
| Rmsd bond angles (°) | 1.335 | 0.958 |
| Average B factors, all atoms (Å2) | 37.0 | 40.9 |
| B factor of metal ions: M1/M2/M3 | 31.8/28.9/42.7 | 32.85/32.18/48.72 |
| Ramachandran plot f residues in favored regions (%) | 93.48 | 93.9 |
| Ramachandran outliers (%) | 0 | 0 |
| Cα RMSD | Wt-tPphA PDB: 2J86_A | Wt-tPphA PDB: 2J86_B | Wt-tPphA PDB: 5ITI | H39A PDB: 5D2U | D119A PDB: 2XZV | D193A PDB: 2Y09 |
|---|---|---|---|---|---|---|
| Wt-tPphA PDB: 2J86_A | - | 2.223 | 2.858 | 2.928 | 3.343 | 2.449 |
| Wt-tPphA PDB: 2J86_B | 2.223 | - | 2.244 | 2.199 | 2.734 | 1.635 |
| Wt-tPphA PDB: 5ITI | 2.858 | 2.244 | - | 0.681 | 1.844 | 1.084 |
| H39A PDB: 5D2U | 2.928 | 2.199 | 0.681 | - | 1.802 | 1.215 |
| D119A PDB: 2XZV | 3.343 | 2.734 | 1.844 | 1.802 | - | 0.707 |
| D193A PDB: 2Y09 | 2.449 | 1.635 | 1.084 | 1.215 | 0.707 | - |
| Enzyme Variants | Km (μM) | Kcat (s−1) | Kcat/Km (s−1/M) |
|---|---|---|---|
| tPphA | 0.85 ± 0.16 | 1.12 ± 0.26 | 1.36 × 106 ± 0.31 × 106 |
| H39A | 1.23 ± 0.20 | 1.69 ± 0.11 | 1.37 × 106 ± 0.09 × 106 |
| IC50 (mM) | pNPP Assay | Casein Assay | ||
|---|---|---|---|---|
| tPphA | H39A | tPphA | H39A | |
| Sanguinarine | 0.010 ± 0.002 | 0.008 ± 0.001 | 0.08 ± 0.03 | 0.07 ± 0.02 |
| NiSO4 | 0.04 ± 0.01 | 0.08 ± 0.02 | 0.21 ± 0.02 | 0.15 ± 0.01 |
| CaCl2 | 17.66 ± 1.57 | 10.69 ± 1.64 | 19.48 ± 3.86 | 11.86 ± 4.26 |
| MgCl2 | 10.98 ± 1.11 | 9.64 ±0.93 | - | - |
| EDTA | 0.93 ± 0.05 | 1.21 ± 0.06 | 2.29 ± 0.09 | 2.50 ± 0.18 |
| NaF | 21.08 ± 3.58 | 23.73 ± 2.59 | 1.24 ± 0.15 | 32.52 ± 3.84 |
| AlF3 | 67.52 ± 5.69 | 156.34 ± 5.23 | 76.49 ± 2.69 | 63.32 ± 5.93 |
| Sodium phenyl phosphate | 8.65 ± 1.31 | 7.37 ± 1.58 | - | - |
| Glycerol-phosphate | 17.18 ± 0.83 | 25.93 ± 3.60 | 12.36 ± 2.65 | 4.52 ± 1.89 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, Y.; Yuan, Y.; Wang, Y.; Gao, J.; Hu, Y.; Feng, S.; Su, J. Structural and Biochemical Characterization of a Cyanobacterial PP2C Phosphatase Reveals Insights into Catalytic Mechanism and Substrate Recognition. Catalysts 2016, 6, 60. https://doi.org/10.3390/catal6050060
Si Y, Yuan Y, Wang Y, Gao J, Hu Y, Feng S, Su J. Structural and Biochemical Characterization of a Cyanobacterial PP2C Phosphatase Reveals Insights into Catalytic Mechanism and Substrate Recognition. Catalysts. 2016; 6(5):60. https://doi.org/10.3390/catal6050060
Chicago/Turabian StyleSi, Yunlong, Ye Yuan, Yue Wang, Jin Gao, Yanbo Hu, Shiqiong Feng, and Jiyong Su. 2016. "Structural and Biochemical Characterization of a Cyanobacterial PP2C Phosphatase Reveals Insights into Catalytic Mechanism and Substrate Recognition" Catalysts 6, no. 5: 60. https://doi.org/10.3390/catal6050060
APA StyleSi, Y., Yuan, Y., Wang, Y., Gao, J., Hu, Y., Feng, S., & Su, J. (2016). Structural and Biochemical Characterization of a Cyanobacterial PP2C Phosphatase Reveals Insights into Catalytic Mechanism and Substrate Recognition. Catalysts, 6(5), 60. https://doi.org/10.3390/catal6050060